

Abstract
Substance P (SP) is a prototypical neuropeptide with roles in pain and inflammation. Numerous mechanisms regulate endogenous SP levels, including the differential expression of SP mRNA and the controlled secretion of SP from neurons. Proteolysis has long been suspected to regulate extracellular SP concentrations but data in support of this hypothesis is scarce. Here, we provide evidence that proteolysis controls SP levels in the spinal cord. Using peptidomics to detect and quantify endogenous SP fragments, we identify the primary SP cleavage site as the C-terminal side of the ninth residue of SP. If blocking this pathway increases SP levels, then proteolysis controls SP concentration. We performed a targeted chemical screen using spinal cord lysates as a proxy for the endogenous metabolic environment and identified GM6001 (galardin, ilomastat) as a potent inhibitor of the SP 1–9-producing activity present in the tissue. Administration of GM6001 to mice results in a greater-than-three-fold increase in the spinal cord levels of SP, which validates the hypothesis that proteolysis controls physiological SP levels.
Introduction
A member of the tachykinin family of neuropeptides, substance P (SP) is an amidated undecapeptide (Fig. 1) that is widely expressed in the central and peripheral nervous systems [1] of mammals and functions as a neurotransmitter and neuromodulator [2]. It participates in a host of fundamentally and biomedically important physiological processes, including pain transmission [3]–[5], inflammation [6], [7], sleep [8], learning and memory [9], [10], depression and affective mood disorders [11]–[13], opioid dependence [14]–[16] and apoptosis [17], [18]. This broad function profile has driven interest in uncovering the mechanisms that control SP's activity.
Figure 1. C-terminal processing is the primary mode of SP degradation.

A) An integrated approach that combines chemical screening and peptide profiling provides a new strategy to determine whether proteolysis plays a role in the regulation of endogenous SP levels. B) Initial experiments begin in tissue lysates and the data clearly shows that SP is processed by membrane proteases to generate a series of C-terminally truncated fragments, while the soluble proteome has little impact on SP processing.
Several mechanisms have been definitively shown to regulate SP. These include the differential expression of SP mRNA [19]–[21] and the controlled release of SP from neuron terminals [22]. In view of the well-established role of proteolysis in regulating the activity of certain other bioactive peptides, such as GLP-1 [23] and PHI-27 [24], researchers have postulated that proteolysis of SP in the extracellular space also controls SP levels. A multitude of in vitro and pseudo in vivo studies indicate SP-degrading activity is abundant in mammalian nervous tissue lend plausibility to this hypothesis [25], [26]. However, one cannot conclude from the mere presence of SP-degrading activity in SP-containing tissues that proteolysis controls SP levels because it is possible that the enzymes responsible for the observed activities do not physically contact endogenous SP in the cell or are otherwise prevented from acting on the peptide (e.g., through protein-protein interactions that are not recapitulated in the test tube). Furthermore, even if one or more of the observed activities acts on SP in vivo, it doesn't follow that they control the peptide's levels: the cleavage events may destroy only a small fraction of the total SP present in the extracellular space and therefore not appreciably impact SP concentration. Thus, metabolic studies are not sufficient to establish proteolysis as a mechanism of SP regulation.
One way to definitively determine that proteolysis regulates SP would be to block a proteolytic pathway in vivo and show that SP levels change as a result. With this goal, many researchers have sought to identify the enzymes responsible for the SP-degrading activities observed in the aforementioned in vitro studies [27]–[33], the idea being that targeted pharmacological or genetic knockdown studies could then be used to probe an SP-degrading pathway. However, to date, no enzyme has been proven to degrade SP in vivo and no studies have shown that blocking a proteolytic pathway can modulate SP levels.
Recognizing that enzyme identification approaches are very time consuming given their reliance on extensive biochemical purification and confirmatory studies, we wondered whether there is an easier path to evaluating the hypothesis that proteolysis regulates SP. To this end, we devised a strategy that couples in vivo peptidomics with in vitro chemical screens to rapidly discover physiologically relevant proteolytic pathways and identify probes that can be used to block them. Focusing our efforts on the spinal cord, where SP executes its most widely studied function of transmitting pain signals from the periphery into the CNS [34], we used this method to determine that a major endogenous SP-degrading pathway cleaves SP at the C-terminal side of residue nine and identify a peptidase inhibitor (GM6001) capable of blocking this pathway. When we injected mice with this compound we observed a greater-than-three-fold increase in endogenous SP levels, thus proving that SP levels are controlled by proteolysis.
Materials and Methods
Compounds
Mouse SP was purchased from Anaspec, Inc. A protease inhibitor panel was obtained from Sigma Aldrich Inc.
Peptide synthesis
Heavy-labeled SP1–7 (Pro containing five 13C and one 15N), SP1–9 (Phe containing eight 2H), and SP (Leu containing ten 2H) were synthesized manually using FMOC chemistry for solid-phase peptide synthesis. Crude peptides were purified by RP-HPLC (Shimadzu) using a C18 column (150×20 mm, 10 μm particle size, Higgins Analytical). The HPLC gradient varied depending on the peptide (Mobile Phase A: 99% H2O, 1% Acetonitrile, 0.1% TFA; Mobile Phase B: 90% Acetonitrile, 10% H2O, 0.07% TFA). HPLC fractions were analyzed for purity by MALDI-TOF (Waters) using α-cyano-4-hydroxycinnamic acid as the matrix. Pure fractions were combined and lyophilized. Concentrations of the purified peptides were determined by UV-vis using the extinction coefficient for phenylalanine.
Animal studies
Wild type (C57BL/6) mice used in this study were either purchased (Jackson Labs) or taken from a breeding colony. Nep–/– mice were obtained from Craig Gerard at Children's Hospital (Boston, MA) and were on a C57BL/6 background [35]. Mice in these studies were not littermates from het x het crosses, but were obtained from separate colonies of Nep–/– and WT mice. All mice used in this study ranged from 3 to 6 months old. Animals were kept on a 12-h light, 12-h dark schedule and fed ad libitum. For spinal cord tissue collection, animals were euthanized with CO2, their tissue dissected, flash frozen with liquid N2, and stored at −80°C. All animal care and use procedures were in strict accordance with the standing committee on the use of animals in research and teaching at Harvard University and the National Institute of Health guidelines for the humane treatment of laboratory animals. The Harvard institutional Animal Care and Use Committee (IACUC) or ethics committee specifically approved this study and the protocol number is 26–06.
Isolation of physiological peptides from tissue
Tissue peptide isolation and fractionation were previously described [24], [36], [37]. Briefly, frozen spinal cords were placed in 500 µL of water and boiled for 10 minutes to inactivate any residual proteolytic activity prior to tissue homogenization. The aqueous fraction was separated and saved, and the tissue was dounce-homogenized in ice-cold 0.25% aqueous acetic acid. The aqueous fraction and the homogenate were combined and centrifuged at 20,000×g for 20 min at 4°C. The supernatant was then sent through a 10 kDa molecular weight cut-off filter (VWR Modified PES) to enrich the peptide pool and then a C18 Sep Pak cartridge (HLB 1cc; 30 mg, Oasis) to desalt the sample. The peptides were then eluted with 1 mL of 70:30 H2O/ACN and concentrated under vacuum using a speed vac prior to fractionation by strong cation exchange (SCX).
SCX was performed using a PolySULFOETHYL ATM column (200×2.1mm, 5 µm, 300 Å; PolyLC INC.) connected to an Agilent Technologies 1200 series LC. All runs were operated at 0.3 mL/min. The SCX buffers (prepared with MS quality water) consisted of: A) 7 mM KH2PO4, pH 2.6, 25% ACN (vol/vol); B) 40 mM KCl, 7 mM KH2PO4, pH 2.6, 25% ACN (vol/vol); C) 100 mM KCl, 7 mM KH2PO4, pH 2.6, 25% ACN (vol/vol); D) 600 mM KCl, 7 mM KH2PO4, pH 2.6, 25% ACN (vol/vol). Prior to the SCX runs, all samples (N = 4) were dissolved in 900 µL buffer A (1 mL sample loop). A step-gradient was applied that included 60 min with Buffer A, 60 min with Buffer B, 60 min with Buffer C, and 60 min with Buffer D, with 1 min transitions between the different buffer conditions. Fractions were collected separately for each of the different buffer conditions (e.g., a buffer A fraction, a buffer B fraction, and so on). Fraction C was isolated because SP and the primary peptide products cleaved at the C-terminus are expected to be +3 charged at pH 2.6. This fraction was applied to a C18 Sep Pak cartridge, washed with water to desalt the samples, and then eluted with 1 mL of 70:30 H2O/ACN and concentrated using a speed vac. It is important to note that the Met on SP becomes nearly 100% oxidized following SCX. The peptide samples were dissolved in 0.1% aqueous formic acid (50 mg tissue/20 µL), normalized according to the original tissue weight, prior to LC-MS analysis.
LC-MS/MS experiments to detect SP peptide fragments
Fractionated spinal cord samples (N = 4) were analyzed using a nano flow LC (Nano LC-2D, Eksigent Technologies) system coupled to a linear ion trap mass spectrometer (LTQ, ThermoFinnigan) following a 10 uL injection. The analytical column (Self-pack picofrit column, 75 μm ID, New Objective) was packed 15 cm with 3 μm C18 (Magic C18 AQ 200A 3U, Michrom Bioresources Inc). The trap column was obtained pre-packed from New Objective Inc. (Integrafrit sample trap, C18 5 μm, 100 μm column ID). The samples were trapped at an isocratic flow rate of 2 μl/min for 10 minutes and eluted at a flow rate of 300 nl/min via a mobile phase gradient of 2–50% B in 180 min (Mobile Phase A: 0.1% formic acid in water, Mobile Phase B: 0.1% formic acid in acetonitrile). The peptides were detected in the positive mode and the mass range for data acquisition was set from m/z 400–1600. The data were collected in Top 6 MS2 mode (N = 4) with the dynamic exclusion set for 30s, the exclusion size list set to 200, and the normalized collision energy for CID set to 35%. The capillary spray voltage was set to 2.5 kV. All experiments were repeated multiple times to ensure reproducibility.
Peptide identification was performed via two methods using the SEQUEST algorithm. The first method applied a differential modification of methionine to its sulfoxide. The uniprotmus_frc.fasta mouse database, concatenated to a reversed decoy database, served to estimate a false discovery rate (FDR). Peptides were accepted within 1 Da of the expected mass, meeting a series of custom filters on ScoreFinal (Sf), −10 Log P, and charge state that attained an average peptide FDR of <2% across data sets. Manual inspection of spectra, FDR calculation, and protein inference were performed in Proteomics Browser Suite 2.23 (ThermoFisher Scientific).
In the second method that has been previously described [24], [37], we used an algorithm written in-house that reveals related MSMS spectra (MuQuest; Harvard Proteomics Browser Suite). To analyze the spinal cord proteome to search for SP fragments, we used the MSMS data from in vitro membrane lysate experiments. MuQuest is then applied to compare the in vitro MSMS spectra with those of the in vivo data set to determine which in vitro MSMS spectra (which peptides) are present in the in vivo samples. The output files are filtered based on charge state, mass to charge values, and statistical scores. All SP related peptides that were identified in these data were confirmed by analysis of the extracted ion chromatograms (EICs) for these hits.
Isotope dilution MS (IDMS) to determine endogenous levels of SP and its N-terminal peptide fragments
The heavy-label versions of SP1–7, SP1–9, and SP were spiked into spinal cord samples (N = 4) at the beginning of the peptide isolation process. After fine tuning the amount of spiked peptides, it was determined that a final concentration of 100 fmol/μL of SP1–7 and SP and 25 fmol/μL of SP1–9 would lead to adequate measurements of the peptides using Top 6 MS/MS. A comparison was made of the integrated area for specific, corresponding fragments of the +2 charge state of the endogenous and heavy labeled peptides detected in the positive mode. The mass range for data acquisition was set from m/z 400–700. The peptide levels were measured in pmol peptide/g of tissue. All experiments were repeated multiple times to ensure reproducibility.
Monitoring N-terminal SP fragment formation in spinal cord lysates
Three mouse spinal cords were dounce homogenized in 1.1 mL phosphate buffered saline (PBS) on ice and then sonicated for 15 s at 4°C. Tissue debris was separated by centrifuging the sample at 5,000×g for 20 min at 4°C. The soluble fraction was collected after ultracentrifugation of the sample at 55,000×g for 1 h at 4°C. The membrane pellet that remained was washed 3x with 600 μL PBS. The pellet was then suspended in 100 μL PBS by sonication for 5 s at 4°C. The sample was ultracentrifugation at 55,000×g for 1 h at 4°C and the supernatant was separated as a wash membrane sample. The pellet was suspended in 100 μL of 1 mM sodium deoxycholate (Alfa Aesar) by sonication for 5 s at 4°C then stirred for 30 min at 4°C. Ultracentrifugation of the sample at 55,000×g for 1 h at 4°C separated the supernatant as a 1 mM deoxycholate-solubilized membrane fraction. The remaining pellet was washed 3x with 600 μL 1 mM deoxycholate solution. The pellet was subsequently suspended in 4, 12, and 24 mM deoxycholate following the same cycle. The final pellet was suspended in 100 μL PBS. The protein content in the soluble and membrane lysates was quantified by the Bradford assay. SP (100 μM) was incubated in 1 mg/mL soluble and membrane lysates diluted in 20 mM ammonium bicarbonate, pH 7.34 buffer for 1 h (determined to be the optimal incubation time) at 37°C. The reactions were quenched with neat formic acid and speed vac dried. The samples were dissolved in 0.1% formic acid (aq) and analyzed by MALDI-TOF MS for SP-degrading activity (i.e. formation of SP1–7 and SP1–9) using the method outlined in “MALDI-TOF MS and LC-MS/MS analysis of in vitro peptides” section. All in vitro degradation experiments were performed using the same bicarbonate buffer and quenching solutions. Experiments performed with lysates boiled for 5 minutes, served as a negative control.
Developing a candidate list for the SP degrading enzymes responsible for the formation of SP1–9
The MEROPS database was utilized to devise a candidate list for the enzymes that could cleave SP, forming SP1–9 in mice [38]. The candidate list was narrowed based on protein abundance using the following databases: www.brain-map.org; https:// www.nextbio.com/b/nextbio.nb.
Western blotting
Western blotting was used to detect endothelin-converting enzyme 2 (ECE2) (Proteintech Group Inc.; rabbit polyclonal) and pitrilysin (Proteintech Group Inc.; rabbit polyclonal) in the mouse spinal cord membrane lysates prepared by deoxycholate solubilization.
Protease inhibitor studies using general enzyme class inhibitors
The membrane fraction of spinal cord lysates (1 mg/mL) were pre-incubated at 37°C for 30 minutes separately with each of the following inhibitors (N = 4): 10 μM E-64 (cysteine protease), 1 mM iodoacetamide (cysteine protease), 1 mM o-phenanthroline (metalloprotease), 10 μM pepstatin A (aspartyl protease), 1 mM phenylmethylsulfonyl fluoride (PMSF, serine protease), 1 mM diisopropylfluorophosphate (serine protease), and vehicle (PBS with DMSO for 5% DMSO final concentration in reaction). After the pre-incubation with each inhibitor, SP was added to 100 μM final concentration. The reactions proceeded at 37°C for 1 h. Dried samples were dissolved in 0.1% formic acid (aq) and analyzed by LC-MS/MS for SP-degrading activity (i.e. formation of SP1–7 and SP1–9) using the method outlined in “MALDI-TOF MS and LC-MS/MS analysis of in vitro peptides” section.
Protease inhibitor studies using metalloprotease specific enzyme class inhibitors
The membrane fraction of spinal cord lysates (1 mg/mL) were pre-incubated at 37°C for 30 minutes separately with each of the following inhibitors (N = 4): SM-19712 (ECE1), phosphoramidon (neprilysin; ECE2), MMP9 inhibitor, TIMP2 (MMP inhibitor), GM6001 (MMP and neprilysin broad inhibitor), chymostatin (pitrilysin), captopril (ACE), enalaprilat (ACE), actinonin (Meprin 1A), and vehicle (PBS with DMSO for 5% DMSO final concentration in reaction). All inhibitors were present at 100 μM except for TIMP2, which was present at 4 μM. After the pre-incubation with each inhibitor, either SP or SP1–9 was added to 100 μM final concentration. The reactions proceeded at 37°C for 1 h. Dried samples were dissolved in 0.1% formic acid (aq) and analyzed by LC-MS/MS for SP or SP1–9-degrading activity. The same comparative reactions were performed with the deoxycholate-solubilized membrane fractions but only with the GM6001 inhibitor.
In vivo and in vitro comparative study of SP degradation in WT and Nep–/– mice spinal cord tissue
The levels of SP in WT and Nep–/– mice spinal cord tissue (N = 4) were compared by IDMS using Top 6 MS/MS as described previously. The degradation of SP was compared in 1 mg/mL WT and Nep–/– mice spinal cord membrane lysates using LC-MS/MS analysis following the above method.
MALDI-TOF MS and LC-MS/MS analysis of in vitro peptides
MALDI-TOF MS was performed with α-cyano-4-hydroxycinnamic acid as the matrix using 2 μL of a 50 μM reconstituted degradation reaction solution (based on initial SP quantities). Data were acquired on a Waters MALDI micro MX instrument operated in reflectron positive mode.
For LC-MS analysis, an Agilent 6220 LC-ESI-TOF instrument was used in the positive mode. A Bio-Bond C18 (5 μm, 150×2.1 mm) column was used together with a precolumn (C18, 3.5 μm, 2×20 mm). Following injection of 25 μL of 5 μM solutions, the samples were trapped at an isocratic flow rate of 0.1 ml/min for five minutes and eluted at a flow rate of 0.25 mL/min via a mobile phase gradient of 2–100% B in 40 min (Mobile Phase A: 0.1% formic acid in water, Mobile Phase B: 0.1% formic acid in acetonitrile). MS analysis was performed with an electrospray ionization (ESI) source. The capillary voltage was set at 4.0 kV and the fragmentor voltage to 100 V. The drying gas temperature was 350°C, the drying gas flow rate was 10 L/min, and the nebulizer pressure was 45 psi. Data was collected in the centroid mode using a mass range of 100–500 Da. The peptides were analyzed by mass extraction of the +3 charge state.
Endogenous GM6001 experiments
For GM6001 injection experiments, 3–4 month old female WT mice (N = 4) were fasted overnight. GM6001 was dissolved at a high concentration in DMSO. Intraperitoneal (i.p.) injections were performed with a 10 μL/g injection of either vehicle (5% DMSO, 95% saline) or 10 mg/mL GM6001 in 5% DMSO, 95% saline for a final dose of 100 mg/kg GM6001. Mice were allowed to return to their cages for three hours and then spinal cords were isolated as described in the ‘Animal studies’ section. IDMS was used to measure differences in the levels of SP in the inhibitor treated and untreated samples.
Data
All data will be made available upon request.
Results
Proteolysis of SP occurs primarily through C-terminal processing
To determine the candidate proteolytic pathway or pathways for SP degradation in the spinal cord, we separated tissue lysates from mouse spinal cords into membrane and soluble fractions, incubated them with full-length SP for varying lengths of time (15, 60, 240 min) and then analyzed the quenched reactions by MALDI mass spectrometry to identify any SP fragments that had been produced. No discernable fragments appeared in the soluble fraction, while ions corresponding to SP1–10, SP1–9, SP1–8, and SP1–7 were all produced in the membrane fraction (Fig. 1). These sequence assignments were validated by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [39]. The data indicates that the primary SP-degrading activity in spinal cord resides in the membrane fraction and that proteolysis of SP occurs through C-terminal processing, which is consistent with previous in vitro lysate experiments [32], [40], [41].
SP1–9 and SP1–7 are endogenous metabolites of SP
Lysate experiments are imperfect because the compartmentalization of a tissue is disrupted, which can lead to the production of SP fragments that are not physiologically relevant. Therefore, we complement these lysate experiments with LC-MS/MS experiments to determine which of these SP fragments, if any, are present in vivo [36]. SP fragment levels are considerably lower than that of full-length SP and therefore we included an offline strong cation exchange (SCX) fraction step prior to the LC-MS/MS to improve our sensitivity [37]. Using this workflow, we are able to detect SP, SP1–9 and SP1–7 in the spinal cord. We also performed an isotope dilution-mass spectrometry (IDMS) experiment to confirm these results. In these experiments, synthetic stable isotope-labeled versions of SP, SP1–10, SP1–9, SP1–8 and SP1–7 are added into the mixture at known concentrations during peptide extraction from spinal cords. These labeled synthetic compounds are easily distinguishable from endogenous SP by mass spectrometry, and the ratio of the stable isotope-labeled peptides to endogenous peptides can be used to calculate the absolute concentration of the SP peptides in the spinal cord. IDMS revealed that the endogenous levels of SP, SP1–9 and SP1–7 were 105.9±8.5 pmol/g, 2.1±0.5 pmol/g, and 1.6±0.5 pmol/g, respectively (Table 1). We did not detect SP1–10 or SP1–8 in any of these experiments, suggesting that these peptides are not generated in vivo.
Table 1. Absolute quantities of SP and SP fragments in the spinal cord as measured by isotope dilution mass spectrometry (IDMS).
| Peptides | SPC (pmol/g) |
| SP | 105.9±8.5 |
| SP1–10 | n.d. |
| SP1–9 | 2.1±0.5 |
| SP1–8 | n.d. |
| SP1–7 | 1.6±0.5 |
The primary SP-degrading enzyme is a metallopeptidase
To identify the enzyme class that mediates SP processing we relied on a screen using class-specific peptidase inhibitors coupled to quantitative MS analysis. The inhibitors used in this screen include compounds such as the metallopeptidase inhibitor O-phenanthroline [42] and PMSF, a serine peptidase inhibitor [43]. We assayed these compounds to test their ability to prevent the SP degradation and inhibit the production of SP1–9 and SP1–7 in mouse spinal cord lysates (Fig. 2). Lysate samples were heated (i.e. heat-treated) to denature all proteins and this served as a control for complete protease inactivation.
Figure 2. Metallopeptidase inhibitors potently block SP degradation in spinal cord lysates.
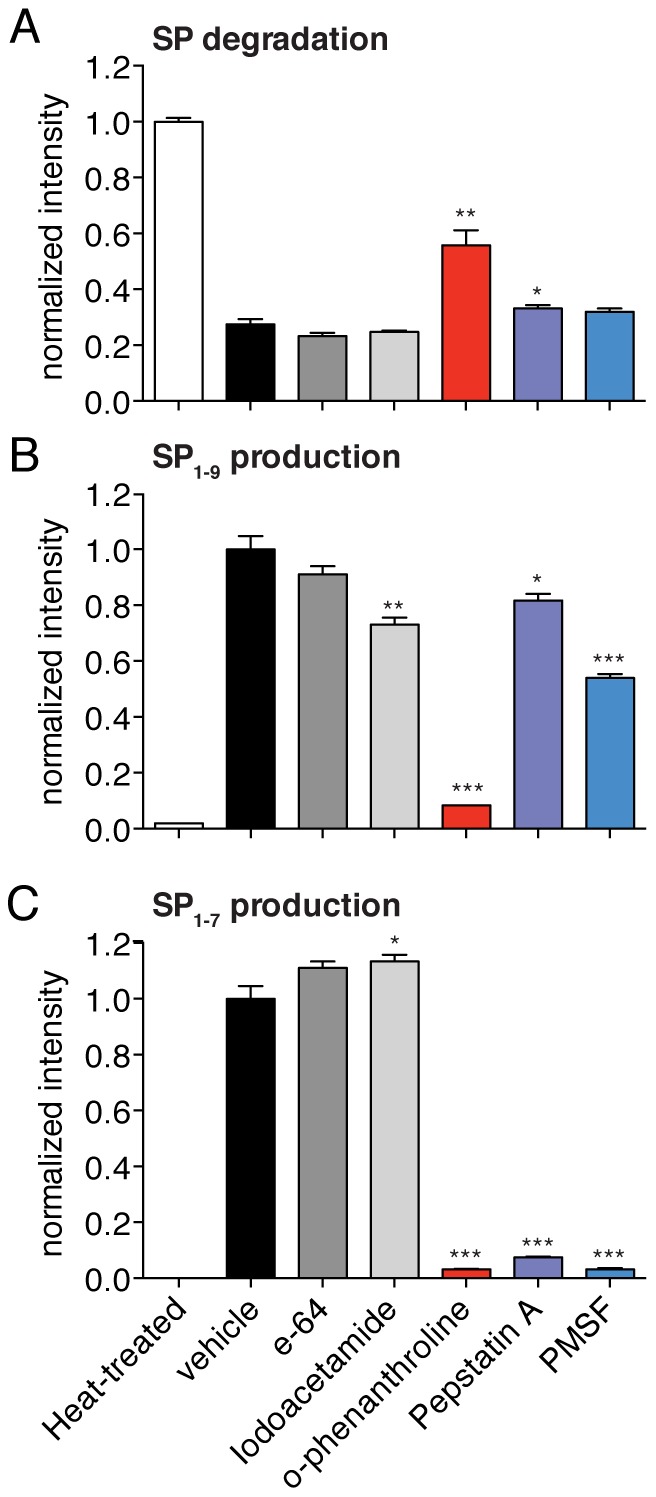
A) Different class-selective inhibitors were tested for their ability to slow SP degradation in spinal cord membrane lysates. The most effective compound at inhibiting SP degradation in this assay is O-phenanthroline, a metalloprotease inhibitor. B) O-phenanthroline was also the most potent inhibitor of SP1–9 production in these experiments. C) Multiple class-selective inhibitors regulate SP1-7 production including O-phenanthroline, pepstatin A and PMSF. (Statistical significance calculated by a Student's t-test; p-value <0.05, *; p-value <0.01, **; p-value <0.001, ***, N = 4).
O-phenanthroline inhibited the degradation of SP to the largest extent with SP values twice as high as the vehicle control, indicating that a metallopeptidase contributes the most to SP processing in these tissue lysates. Modest increases in SP levels were also seen for pepstatin and PMSF, but these differences were not statistically significant. O-phenanthroline was also the strongest inhibitor of SP1–9 production, and the overall pattern of SP1–9 production correlates with the inhibitor specificity for SP degradation. Specifically, O-phenanthroline was the best inhibitor but pepstatin and PMSF had a small effect on SP1–9 production. The correlation between SP1–9 production and SP degradation indicates that the conversion of SP to SP1–9 may be the key step in the conversion of SP in spinal cord lysates. By contrast, the inhibitor sensitivity of SP1–7 production is markedly different from that of SP degradation.
Contribution of reported SP-degrading enzymes to the production of SP1–9
Having characterized the conversion of SP to SP1–9 as the key step in SP degradation, we turned our attention toward the identification of a chemical inhibitor of this step. SP is often used as a model substrate for peptidases and proteases and therefore there are a number of metallopeptidases reported to cleave SP and produce SP1–9 [29], [44]–[48]; however, to our knowledge these enzymes have never been shown to control SP levels in vivo. We reasoned that inhibitors that target these metallopeptidases, even if they are not specific for these proteins, would provide a good chance of finding a compound that could influence SP processing in tissue lysates. The MEROPS database is an authoritative catalogue of all known peptidases and it includes detailed information on peptidase substrate specificity [38].
Using the MEROPS database, we identified eight mammalian peptidases reported to cleave SP to produce SP1–9 (Fig. 3). These enzymes are all membrane-associated metallopeptidases and are present in the mouse spinal cord [49], [50]. A total of seven metalloprotease inhibitors that target these enzymes were identified with the help of the MEROPS [38] and BRENDA databases [51] (see Materials and Methods). We do not believe any of these inhibitors are selective and therefore we are using these compounds as probes to inhibit the conversion of SP rather than to identify a specific enzyme. For example, the inhibitor actinonin only inhibits meprin 1A [52] but it is also a matrix metalloprotease (MMP) inhibitor, and phosphoramidon inhibits at least two of the candidate enzymes, NEP and ECE-2 (Fig. 3) [53], [54].
Figure 3. GM6001 is significantly more effective than other metallopeptidase inhibitors at preventing SP degradation and conversion of SP to SP1–9.
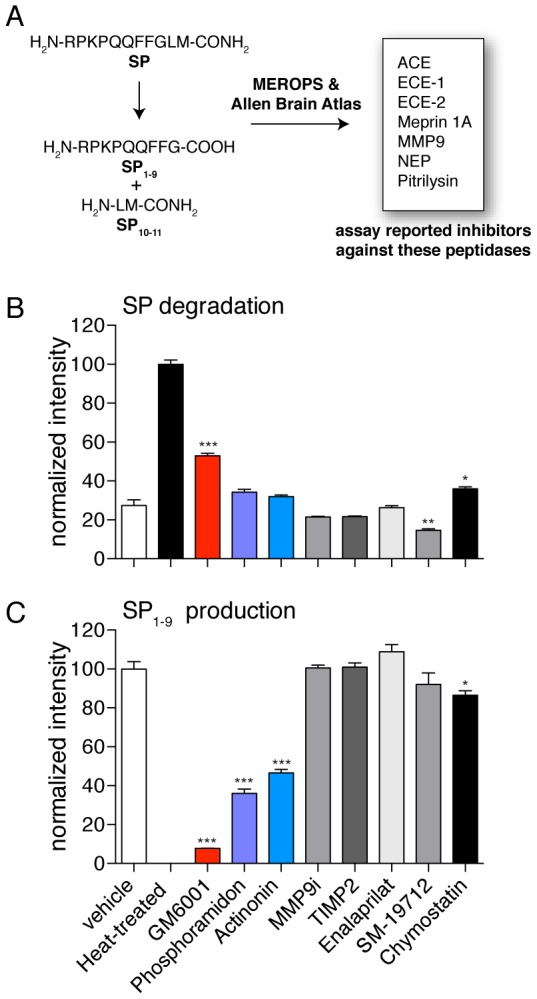
A) Utilizing the MEROPS and Allen Brain Map databases a number of candidate metallopeptidases in the nervous system that are capable of cleaving SP to produce SP1–9 are identified. Inhibitors against these peptidases were then used in lysates to evaluate their affect on SP degradation and SP1–9 production. B) The matrix metalloprotease (MMP) inhibitor GM6001 was the most effective compound at preventing SP degradation. C) GM6001 is also the best inhibitor of SP1–9 production. (Statistical significance calculated by a Student's t-test; p-value <0.05, *; p-value <0.01, **; p-value <0.001, ***, N = 4).
Each of these inhibitors was added to spinal cord membrane lysate along with SP. After incubation, the relative levels of SP and SP1–9 were assessed in the presence of each inhibitor. This assay revealed three compounds that could inhibit SP degradation and formation of SP1–9: phosphoramidon, actinonin and the MMP inhibitor GM6001 [55], [56]. GM6001 led to the least amount of SP degradation and strongly inhibited SP1–9 production (Fig. 3). Specifically, addition of GM6001 results in a nearly 2-fold increase in SP levels compared to a vehicle, and a greater than 10-fold decrease in SP1–9 production. In aggregate, this data demonstrates that GM6001 inhibits a key step in conversion from SP into SP1–9.
Based on the inhibitor sensitivities of SP degradation (Fig. 3) NEP and ECE-2 are viable SP-degrading enzyme candidates (Fig. S1). We tested this possibility through several experiments [35]. First, there was no difference in the processing activity between tissue lysates from wild type (NEP+/+) and NEP–/– mice [35], indicating that NEP is not the predominant SP-degrading activity in lysates. Furthermore, NEP–/– mice had the same spinal cord concentrations of SP as NEP+/+ mice to conclusively demonstrate that NEP does not regulate SP on its own (Fig. 4).
Figure 4. SP is regulated by proteolysis but not by NEP.

A) Phosphoramidon slows SP1–9 production in tissue lysates, which suggests that NEP might have a role in SP processing. Experiments in NEP+/+ and NEP–/– spinal cord lysates reveals no significant difference in SP degradation. B) Likewise, no difference in endogenous SP levels is observed in spinal cords from NEP+/+ and NEP–/– mice. C) Acute treatment of mice with GM6001 results in a 3-fold elevation of SP in the spinal cord to reveal a GM6001-sensitive pathway for SP regulation. (Statistical significance calculated by a Student's t-test; p-value <0.001, ***, N = 4).
To determine whether ECE-2 is responsible for the SP-degrading activity we detect in tissue lysates, we solubilized the proteome with different concentrations of deoxycholate and then tested these fractions for SP-degrading activity. In addition, we performed activity assays on each of these fractions to ascertain whether ECE-2 levels correlate with SP-degrading activity. These data clearly show that ECE-2 and SP-degrading activity are not connected and this data disfavors any role for ECE-2 in SP-degradation in tissue lysates (Fig. S2). Finally, additional MMP inhibitors (TIMP2 and MMP9i) were not active in protecting SP from degradation either (Fig. 3). These data indicate that none of the likely GM6001 targets is solely responsible for the C-terminal processing and that there may exist a yet unidentified GM6001-sensitive peptidase that regulates SP levels in vivo.
Small-molecule studies with GM6001 demonstrate that proteolysis regulates SP levels in the spinal cord
Typical approaches for testing enzymes or pathways for a role in peptide regulation in vivo require the generation of knockout mice [36]. By using a small-molecule inhibitor approach we circumvent the need for any knockout animals to greatly expedite testing the hypothesis that proteolysis regulates SP levels. Moreover, the use of a small molecule overcomes challenges of compensation by other enzymes that sometimes plague knockout studies [57]. Administration of GM6001 was followed by analysis of SP in the spinal cords after 3 hours. These experiments showed that SP levels were ∼3-fold higher in the mice treated with GM6001 versus the vehicle treated controls (Fig. 4). Thus, we discovered an endogenous GM6001-sensitive proteolytic pathway that regulates SP levels. More importantly, this data confirms the hypothesis that proteolysis controls endogenous SP levels.
Discussion
The hypothesis that SP is regulated by proteolysis originated with the earliest discoveries of potent SP-degrading activity in nervous tissue, which occurred over three decades ago [27], [58]. Subsequent work identified numerous peptidases [28], [31], [32], [40], [41], [48], [59]–[61] that cleave SP in vitro, but none of these enzymes has been shown to regulate SP in vivo. Believing that identifying a true SP-regulating enzyme may depend on clearly defining the products of endogenous SP metabolism, many researchers have conducted in vitro experiments in which the endogenous metabolic environment of the CNS is simulated [28], [30], [40], [41]. Typically these experiments involve incubating cell lysates with large amounts of synthetic peptide. Others have conducted pseudo-in vivo experiments where synthetic peptide is introduced into a living tissue and the excess metabolites are collected and analyzed [62]. In these studies, a multitude of fragments has been detected, among them SP1–9, SP1–8, SP1–7, SP5–11, SP6–11, SP7–11 and SP8–11. The data suggests an extremely complicated picture for SP proteolysis and does not present a clear path towards elucidating SP regulation.
Recently, we developed an advanced liquid chromatography-tandem mass spectrometry (LC-MS/MS) peptide profiling strategy to elucidate the proteolysis of bioactive peptides [24], [36]. The power of this approach lies in its ability to peer directly into the chemical milieu in which endogenous metabolism occurs. In applying this approach to SP we circumvent the epistemological worries one may have about the results of in vitro and pseudo-in vivo studies, which do not perfectly reconstitute endogenous metabolism. We profiled peptides from the mouse spinal cord and detected SP1–9 and SP1–7, indicating these are endogenous metabolites of SP. We confirmed these assignments and obtained the absolute concentrations of SP, SP1–9, and SP1–7 in tissues using isotope dilution-mass spectrometry [63]. Our results were consistent with previously reported concentrations of SP in the spinal cord (∼105.9 pmol/g) [64], and demonstrated that SP1–9 and SP1–7 are approximately 50-fold lower in concentration than SP (Table 1). We also performed in vitro degradation experiments and found that enzyme activity capable of producing SP1–9 and SP1–7 resides in the membrane fraction but not the soluble fraction of spinal cord lysate (Fig. 1). This finding is consistent with the notion that SP processing occurs outside the cell, where the SP is exposed to membrane-bound proteases. The combination of in vitro and in vivo peptide profiling indicates that SP1–9 and SP1–7 are the primary endogenous products from processing of SP in the spinal cord.
A series of lysate experiments with class selective inhibitors showed that SP is regulated by a metallopeptidase and that the levels of SP and SP1–9 are inversely correlated (Fig. 2). This indicates that SP degradation is coupled to SP1–9 production and that the conversion of SP to SP1–9 is a major means of SP degradation since the inhibition of this pathway affects SP levels. The results with SP1–7 were less clear, as several inhibitors affected SP1–7 production and there was no clear correlation between SP1–7 production and SP levels. We subjectedSP1–9 to degradation in spinal cord lysates and observed that SP1–7 was produced (Fig. S3). We reasoned that inhibition of SP1–9 production will reduce SP1–7 levels and therefore focused on the conversion of SP to SP1–9 as the key step in SP catabolism.
At this point, a typical approach would call for the identification of the enzyme responsible for this step followed by perturbation of the protein in vivo to verify the identification [36]. However, recognizing that the generation of knockout animals is very slow and often confounded by compensatory effects that mask the function of the enzyme in knockout animals [57] and that knowing the identity of the degrading enzyme is not necessary to evaluate whether SP is regulated by proteolysis, we opted to perform a small screen of metalloprotease inhibitors to identify a compound that could potentially be used to perturb the pathway in vivo. In this way we discovered that GM6001 potently blocks the conversion of SP to SP 1–9in tissue lysates (Fig. 3).
Fortuitously, GM6001 is bioavailable and had previously been shown to permeate the central nervous system (CNS) [65]. This allowed us to test whether GM6001 can influence SP levels in vivo. Administration of GM6001 to mice resulted in a 3-fold increase in SP levels in the spinal cord (p-value <0.001, Fig. 3). While this strategy does not identify any specific enzyme, it quickly reveals whether blocking proteolysis regulates SP levels and proves correct the long-standing hypothesis that SP levels are regulated by proteolysis. Moreover, given this data the enthusiasm for pursuing a SP degrading enzyme is increased and GM6001 can be used as a valuable tool in the discovery of such an enzyme. GM6001 was previously developed into an activity-based probe that targets metalloproteases [66] and this unbiased strategy can be applied to the discovery of a candidate SP-degrading enzyme.
Conclusions
We expect the strategy described herein to be applicable to all bioactive peptides [67] and we envision the power of the approach increasing with larger chemical libraries and faster high-throughput lysate assays. Thus, this work advances the methodology for the discovery of bioactive peptide regulatory pathways, provides a chemical inhibitor to study SP regulation in vivo and conclusively demonstrates that proteolysis plays a major role in the regulation of SP.
Supporting Information
Flow chart outlining the strategy used at each step to identify or eliminate a candidate SP-degrading enzyme. Ultimately this approach identified a GM6001-sensitive pathway in the spinal cord to validate the hypothesis that proteolysis regulates SP levels.
(PNG)
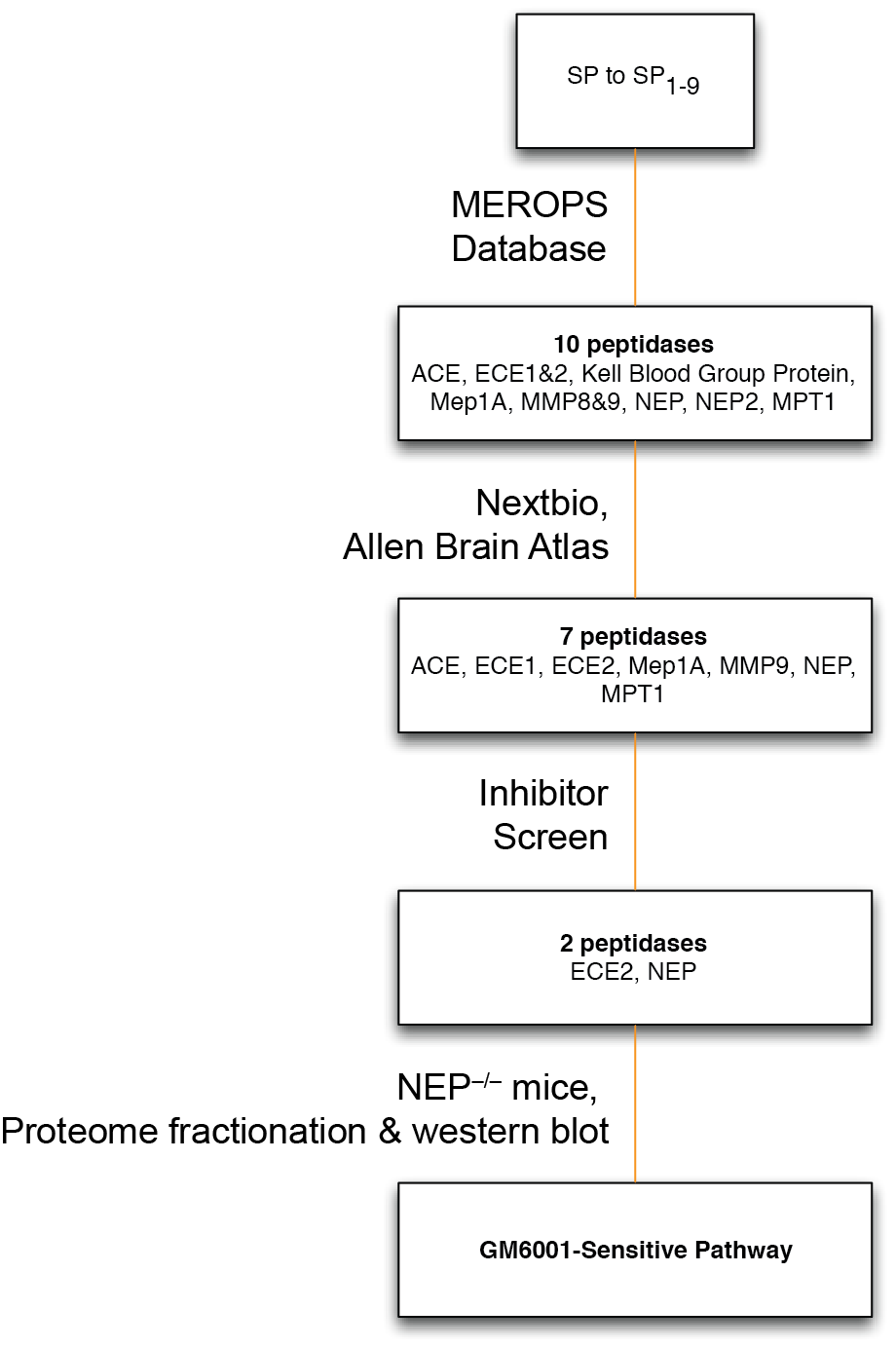{kind=link}
ECE2 is not responsible for SP-degrading activity in lysates. Fractionation of spinal cord lysate by successive solvation in the detergent deoxycholate coupled with an LCMS-based assay for SP 1–9 production shows that ECE2 abundance does not correlate with the activity of interest.
(PNG)
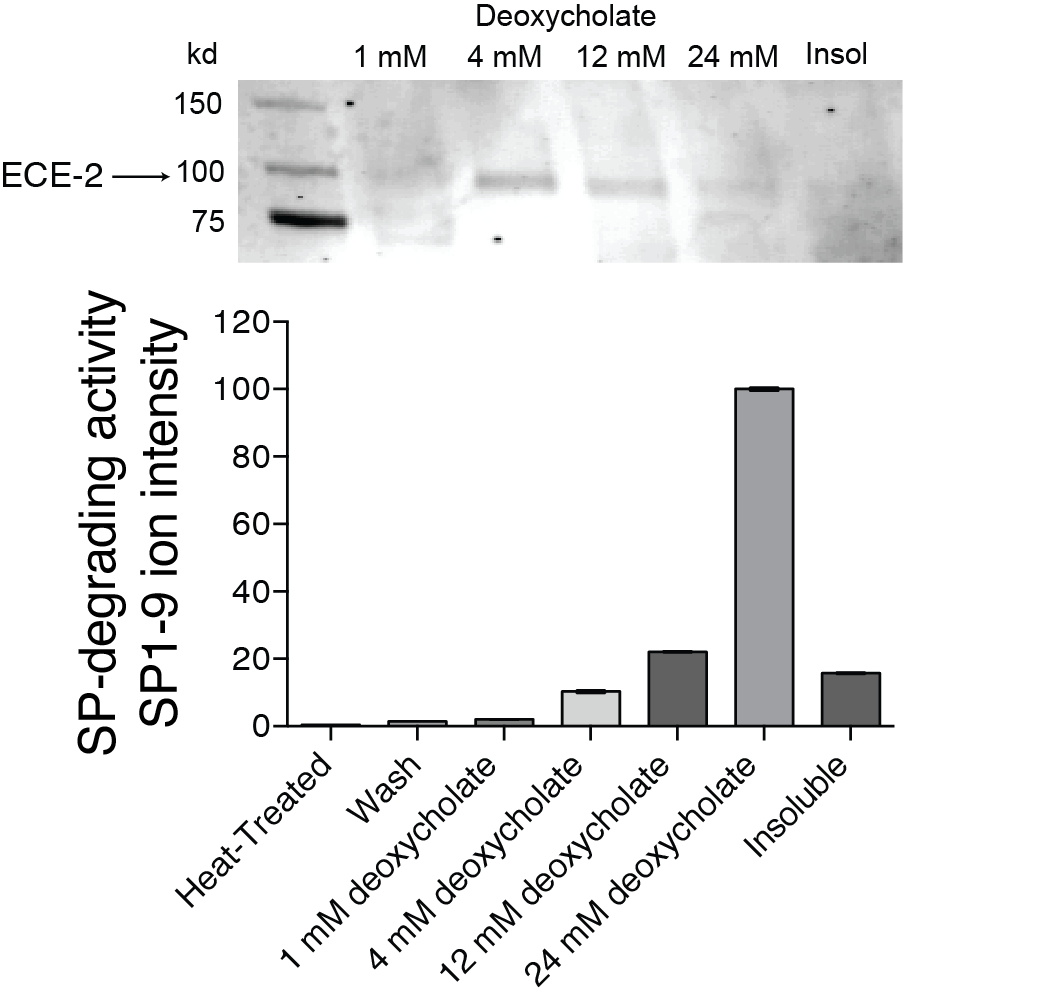{kind=link}
Existence of a proteolytic activity in spinal cord lysates that converts SP 1–9 to SP1–7. SP1–7 is produced when SP 1–9 is incubated in spinal cord lysate indicating the presence of a SP1–9 to SP1–7 activity.
(PNG)
{kind=link}
Funding Statement
This work was supported by a Forris Jewitt Moore Fellowship sponsored by Amherst College (AML), National Institutes of Health (NIH) Training Grant GM007598 (AJM and AML), Mary Fieser Postdoctoral Fellowship (ADT), Searle Scholar Award (to AS), a Burroughs Wellcome Fund Career Award in the Biomedical Sciences (AS), NIH Grant 1DP2OD002374 (AS), and the Sloan Foundation (AS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ribeiro-da-Silva A, Hokfelt T (2000) Neuroanatomical localisation of substance P in the CNS and sensory neurons. Neuropeptides 34: 256–271. [DOI] [PubMed] [Google Scholar]
- 2. Otsuka M, Yoshioka K (1993) Neurotransmitter functions of mammalian tachykinins. Physiological Reviews 73: 229–308. [DOI] [PubMed] [Google Scholar]
- 3. Hall ME, Stewart JM (1983) Substance P and antinociception. Peptides 4: 31–35. [DOI] [PubMed] [Google Scholar]
- 4. Hall ME, Stewart JM (1983) Substance P and behavior: opposite effects of N-terminal and C-terminal fragments. Peptides 4: 763–768. [DOI] [PubMed] [Google Scholar]
- 5. Yeomans D, Proudfit H (1992) Antinociception induced by microinjection of substance P into the A7 catecholamine cell group in the rat. Neuroscience 49: 681–691. [DOI] [PubMed] [Google Scholar]
- 6. Kolasinski SL, Haines KA, Siegel EL, Cronstein BN, Abramson SB (1992) Neuropeptides and inflammation. A somatostatin analog as a selective antagonist of neutrophil activation by substance P. Arthritis & Rheumatism 35: 369–375. [DOI] [PubMed] [Google Scholar]
- 7. Palmer J, Greenwood B (1993) Regional content of enteric substance P and vasoactive intestinal peptide during intestinal inflammation in the parasitized ferret. Neuropeptides 25: 95–103. [DOI] [PubMed] [Google Scholar]
- 8. Schlesinger K, Pelleymounter MA, Kamp Jvd, Bader DL, Stewart JM, et al. (1986) Substance P facilitation of memory: effects in an appetitively motivated learning task. Behavioral and neural biology 45: 230–239. [DOI] [PubMed] [Google Scholar]
- 9. Nagel JA, Welzl H, Bättig K, Huston JP (1993) Facilitation of tunnel maze performance by systemic injection of the neurokinin substance P. Peptides. 14: 85–95. [DOI] [PubMed] [Google Scholar]
- 10. Santangelo EM, Morato S, Mattioli R (2001) Facilitatory effect of substance P on learning and memory in the inhibitory avoidance test for goldfish. Neuroscience letters 303: 137–139. [DOI] [PubMed] [Google Scholar]
- 11. Berrettini WH, Rubinow DR, Nurnberger Jr JI, Simmons-Alling S, Post RM, et al. (1985) CSF substance P immunoreactivity in affective disorders. Biological psychiatry 20: 965–970. [DOI] [PubMed] [Google Scholar]
- 12. Lieb K, Treffurth Y, Berger M, Fiebich BL (2002) Substance p and affective disorders: new treatment opportunities by neurokinin 1 receptor antagonists? Neuropsychobiology 45: 2–6. [DOI] [PubMed] [Google Scholar]
- 13. Shirayama Y, Mitsushio H, Takashima M, Ichikawa H, Takahashi K (1996) Reduction of substance P after chronic antidepressants treatment in the striatum, substantia nigra and amygdala of the rat. Brain research 739: 70–78. [DOI] [PubMed] [Google Scholar]
- 14. Kreeger JS, Larson AA (1993) Substance P-(1–7), a substance P metabolite, inhibits withdrawal jumping in morphine-dependent mice. European journal of pharmacology 238: 111–115. [DOI] [PubMed] [Google Scholar]
- 15. Kreeger JS, Larson AA (1996) The substance P amino-terminal metabolite substance P (1–7), administered peripherally, prevents the development of acute morphine tolerance and attenuates the expression of withdrawal in mice. Journal of Pharmacology and Experimental Therapeutics 279: 662–667. [PubMed] [Google Scholar]
- 16. Nylander I, Sakurada T, Le Greves P, Terenius L (1991) Levels of dynorphin peptides, substance P and CGRP in the spinal cord after subchronic administration of morphine in the rat. Neuropharmacology 30: 1219–1223. [DOI] [PubMed] [Google Scholar]
- 17. Kang B-N, Jeong K-S, Park S-J, Kim S-H, Kim T-H, et al. (2001) Regulation of apoptosis by somatostatin and substance P in peritoneal macrophages. Regulatory peptides 101: 43–49. [DOI] [PubMed] [Google Scholar]
- 18. Lallemend F, Lefebvre P, Hans G, Rigo J-m, Van de Water T, et al. (2003) Substance P protects spiral ganglion neurons from apoptosis via PKC-Ca2+-MAPK/ERK pathways. Journal of neurochemistry 87: 508–521. [DOI] [PubMed] [Google Scholar]
- 19. Gerfen CR, McGinty J, Young WS (1991) Dopamine differentially regulates dynorphin, substance P, and enkephalin expression in striatal neurons: in situ hybridization histochemical analysis. The Journal of neuroscience 11: 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jessell T, Tsunoo A, Kanazawa I, Otsuka M (1979) Substance P: depletion in the dorsal horn of rat spinal cord after section of the peripheral processes of primary sensory neurons. Brain research 168: 247–259. [DOI] [PubMed] [Google Scholar]
- 21. Verge V, Richardson P, Wiesenfeld-Hallin Z, Hokfelt T (1995) Differential influence of nerve growth factor on neuropeptide expression in vivo: a novel role in peptide suppression in adult sensory neurons. The Journal of neuroscience 15: 2081–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Theriault E, Otsuka M, Jessell T (1979) Capsaicin-evoked release of substance P from primary sensory neurons. Brain research 170: 209. [DOI] [PubMed] [Google Scholar]
- 23. Marguet D, Baggio L, Kobayashi T, Bernard A-M, Pierres M, et al. (2000) Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proceedings of the National Academy of Sciences 97: 6874–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tinoco AD, Kim YG, Tagore DM, Wiwczar J, Lane WS, et al. (2011) A peptidomics strategy to elucidate the proteolytic pathways that inactivate Peptide hormones. Biochemistry 50: 2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Le Greves P, Nyberg F, Terenius L, Hökfelt T (1985) Calcitonin gene-related peptide is a potent inhibitor of substance P degradation. European journal of pharmacology 115: 309–311. [DOI] [PubMed] [Google Scholar]
- 26. Lee CM, Sandberg BE, Hanley MR, Iversen LL (1981) Purification and Characterisation of a Membrane-Bound Substance-P-Degrading Enzyme from Human Brain. European Journal of Biochemistry 114: 315–327. [DOI] [PubMed] [Google Scholar]
- 27. Blumberg S, Teichberg V, Charli J, Hersh L, McKelvy J (1980) Cleavage of substance P to an N-terminal tetrapeptide and a C-terminal heptapeptide by a post-proline cleaving enzyme from bovine brain. Brain research 192: 477–486. [DOI] [PubMed] [Google Scholar]
- 28. Bunnett N, Turner A, Hryszko J, Kobayashi R, Walsh J (1988) Isolation of endopeptidase-24.11 (EC 3.4. 24.11, “phalinase” from the pig stomach. Hydrolysis of substance P, gastrin-releasing peptide 10,[Leu5] enkephalin, and [Met5] enkephalin. Gastroenterology 95: 952. [DOI] [PubMed] [Google Scholar]
- 29. Diekmann O, Tschesche H (1994) Degradation of kinins, angiotensins and substance P by polymorphonuclear matrix metalloproteinases MMP 8 and MMP 9. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas/Sociedade Brasileira de Biofisica [et al] 27: 1865. [PubMed] [Google Scholar]
- 30. Heymann E, Mentlein R (1978) Liver dipeptidyl aminopeptidase IV hydrolyzes substance P. FEBS letters. 91: 360. [DOI] [PubMed] [Google Scholar]
- 31. Karlsson K, Nyberg F (1998) Purification of substance P endopeptidase (SPE) activity in human spinal cord and subsequent comparative studies with SPE in cerebrospinal fluid and with chymotrypsin. J Mol Recognit 11: 266–269. [DOI] [PubMed] [Google Scholar]
- 32. Karlsson K, Nyberg F (2000) Purification of Substance P endopeptidase activity in the rat ventral tegemental area with the Äkta-Purifier chromatographic system. Journal of Chromatography A 893: 107–113. [DOI] [PubMed] [Google Scholar]
- 33. Yokosawa H, Endo S, Ogura Y, Ishii S-i (1983) A new feature of angiotensin-converting enzyme in the brain: hydrolysis of substance P. Biochemical and biophysical research communications. 116: 735–742. [DOI] [PubMed] [Google Scholar]
- 34. Zimmer A, Zimmer AM, Baffi J, Usdin T, Reynolds K, et al. (1998) Hypoalgesia in mice with a targeted deletion of the tachykinin 1 gene. Proceedings of the National Academy of Sciences 95: 2630–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu B, Gerard NP, Kolakowski Jr LF, Bozza M, Zurakowski D, et al. (1995) Neutral endopeptidase modulation of septic shock. The Journal of experimental medicine 181: 2271–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim YG, Lone AM, Nolte WM, Saghatelian A (2012) Peptidomics approach to elucidate the proteolytic regulation of bioactive peptides. Proc Natl Acad Sci U S A 109: 8523–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tinoco AD, Tagore DM, Saghatelian A (2010) Expanding the dipeptidyl peptidase 4-regulated peptidome via an optimized peptidomics platform. Journal of the American Chemical Society 132: 3819–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40: D343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ducret A, Van Oostveen I, Eng JK, Yates JR, Aebersold R (1998) High throughput protein characterization by automated reverse-phase chromatography electrospray tandem mass spectrometry. Protein Science 7: 706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karlsson K, Eriksson U, Andrén P, Nyberg F (1997) Purification and characterization of substance P endopeptidase activities in the rat spinal cord. Preparative biochemistry & biotechnology 27: 59–78. [DOI] [PubMed] [Google Scholar]
- 41. Nyberg F, Le Greves P, Sundqvist C, Terenius L (1984) Characterization of substance P(1–7) and (1–8) generating enzyme in human cerebrospinal fluid. Biochem Biophys Res Commun 125: 244–250. [DOI] [PubMed] [Google Scholar]
- 42. Brou C, Logeat F, Gupta N, Bessia C, LeBail O, et al. (2000) A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Molecular cell 5: 207–216. [DOI] [PubMed] [Google Scholar]
- 43. Sekar V, Hageman JH (1979) Specificity of the serine protease inhibitor, phenylmethylsulfonyl fluoride. Biochemical and biophysical research communications 89: 474–478. [DOI] [PubMed] [Google Scholar]
- 44. Siviter RJ, Nachman RJ, Dani MP, Keen JN, Shirras AD, et al. (2002) Peptidyl dipeptidases (Ance and Acer) of Drosophila melanogaster: major differences in the substrate specificity of two homologs of human angiotensin I-converting enzyme. Peptides 23: 2025–2034. [DOI] [PubMed] [Google Scholar]
- 45. Bertenshaw GP, Turk BE, Hubbard SJ, Matters GL, Bylander JE, et al. (2001) Marked differences between metalloproteases meprin A and B in substrate and peptide bond specificity. Journal of Biological Chemistry 276: 13248–13255. [DOI] [PubMed] [Google Scholar]
- 46. Skidgel RA, Engelbrecht S, Johnson AR, Erdös EG (1984) Hydrolysis of substance P and neurotensin by converting enzyme and neutral endopeptidase. Peptides 5: 769–776. [DOI] [PubMed] [Google Scholar]
- 47. Johnson GD, Stevenson T, Ahn K (1999) Hydrolysis of Peptide Hormones by Endothelin-converting Enzyme-1 A COMPARISON WITH NEPRILYSIN. Journal of Biological Chemistry 274: 4053–4058. [DOI] [PubMed] [Google Scholar]
- 48. Chow KM, Gakh O, Payne I, Juliano MA, Juliano L, et al. (2009) Mammalian Pitrilysin: Substrate Specificity and Mitochondrial Targeting†. Biochemistry 48: 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, et al.. (2010) Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5. [DOI] [PMC free article] [PubMed]
- 50. Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176. [DOI] [PubMed] [Google Scholar]
- 51. Scheer M, Grote A, Chang A, Schomburg I, Munaretto C, et al. (2011) BRENDA, the enzyme information system in 2011. NUCLEIC ACIDS RESEARCH 39: D670–D676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takayama J, Takaoka M, Yamamoto S, Nohara A, Ohkita M, et al. (2008) Actinonin, a meprin inhibitor, protects ischemic acute kidney injury in male but not in female rats. European journal of pharmacology 581: 157–163. [DOI] [PubMed] [Google Scholar]
- 53. Oefner C, D'Arcy A, Hennig M, Winkler FK, Dale GE (2000) Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. Journal of molecular biology 296: 341–349. [DOI] [PubMed] [Google Scholar]
- 54. Kukkola PJ, Savage P, Sakane Y, Berry JC, Bilci NA, et al. (1995) Differential structure-activity relationships of phosphoramidon analogues for inhibition of three metalloproteases: endothelin-converting enzyme, neutral endopeptidase, and angiotensin-converting enzyme. Journal of cardiovascular pharmacology 26: S65. [PubMed] [Google Scholar]
- 55. Grobelny D, Poncz L, Galardy RE (1992) Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry 31: 7152–7154. [DOI] [PubMed] [Google Scholar]
- 56. Whittaker M, Floyd CD, Brown P, Gearing AJH (1999) Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chemical Reviews 99: 2735–2776. [DOI] [PubMed] [Google Scholar]
- 57. Turk B (2006) Targeting proteases: successes, failures and future prospects. Nature reviews Drug discovery 5: 785–799. [DOI] [PubMed] [Google Scholar]
- 58. Kato T, Nakano T, Kojima K, Nagatsu T, Sakakibara S (1980) Changes in prolyl endopeptidase during maturation of rat brain and hydrolysis of substance P by the purified enzyme. Journal of neurochemistry 35: 527–535. [DOI] [PubMed] [Google Scholar]
- 59. Matsas R, Fulcher IS, Kenny AJ, Turner AJ (1983) Substance P and [Leu] enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proceedings of the National Academy of Sciences 80: 3111–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nolte WM, Tagore DM, Lane WS, Saghatelian A (2009) Peptidomics of prolyl endopeptidase in the central nervous system. Biochemistry 48: 11971–11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sandberg BEB, Iversen LL (1982) Substance P. Journal of medicinal chemistry. 25: 1009–1015. [DOI] [PubMed] [Google Scholar]
- 62. Andren PE, Caprioli RM (1995) In vivo metabolism of substance P in rat striatum utilizing microdialysis/liquid chromatography/micro-electrospray mass spectrometry. Journal of Mass Spectrometry 30: 817–824. [Google Scholar]
- 63. Keshishian H, Addona T, Burgess M, Mani DR, Shi X, et al. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics 8: 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beaudry F, Vachon P (2006) Determination of substance P in rat spinal cord by high-performance liquid chromatography electrospray quadrupole ion trap mass spectrometry. Biomedical Chromatography 20: 1344–1350. [DOI] [PubMed] [Google Scholar]
- 65. Klohs J, Baeva N, Steinbrink J, Bourayou R, Boettcher C, et al. (2009) In vivo near-infrared fluorescence imaging of matrix metalloproteinase activity after cerebral ischemia. Journal of Cerebral Blood Flow & Metabolism 29: 1284–1292. [DOI] [PubMed] [Google Scholar]
- 66. Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF (2004) Activity-based probes for the proteomic profiling of metalloproteases. Proceedings of the National Academy of Sciences of the United States of America 101: 10000–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kastin AJ (2006) Handbook of biologically active peptides. Amsterdam; Boston: Academic Press. xliii, 1595 p., 1540 p. of col. plates p.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow chart outlining the strategy used at each step to identify or eliminate a candidate SP-degrading enzyme. Ultimately this approach identified a GM6001-sensitive pathway in the spinal cord to validate the hypothesis that proteolysis regulates SP levels.
(PNG)
ECE2 is not responsible for SP-degrading activity in lysates. Fractionation of spinal cord lysate by successive solvation in the detergent deoxycholate coupled with an LCMS-based assay for SP 1–9 production shows that ECE2 abundance does not correlate with the activity of interest.
(PNG)
Existence of a proteolytic activity in spinal cord lysates that converts SP 1–9 to SP1–7. SP1–7 is produced when SP 1–9 is incubated in spinal cord lysate indicating the presence of a SP1–9 to SP1–7 activity.
(PNG)