

Abstract
Genetic studies have clearly shown that primary headaches (migraine, tension-type headache and cluster headache) are multifactorial disorders characterized by a complex interaction between different genes and environmental factors. Genetic association studies have highlighted a potential role in the etiopathogenesis of these disorders for several genes related to vascular, neuronal and neuroendocrine functions. A potential role as a therapeutic target is now emerging for some of these genes. The main purpose of this review is to describe new advances in our knowledge regarding the role of MTHFR, KCNK18, TRPV1, TRPV3 and HCRTR genes in primary headache disorders. Involvement of these genes in primary headaches, as well as their potential role in the therapy of these disorders, will be discussed.
Keywords: Primary headaches, Genes, MHTFR, KCNK18, HCRTR1, HCRTR2
Introduction
Primary headache disorders, according to the International Classification of Headache Disorders 2nd edition (ICHD-II), include migraine, tension-type headache, cluster headache, and other primary headaches [1]. Primary headaches represent a common and major health problem worldwide and significantly impair patients’ quality of life [2,3]. These disorders may affect individuals from childhood and are most troublesome in the productive years of life, thus generating an economic burden for both society and healthcare systems [4,5]. Recently, Global Burden of Disease (GBD) studies have rated primary headaches among the top ten disorders causing significant disability [6].
In recent years, genetic studies have provided substantial evidence supporting the notion that primary headaches are complex, multifactorial disorders. Population, family and twin studies have shown that migraine, tension-type headache and cluster headache have a significant heritable component [7,8]. Different genetic factors may, therefore, be involved in the generation of a specific “headache threshold”. In rare primary headache subtypes, such as Familial Hemiplegic Migraine (FHM), single gene mutations co-segregate with disease phenotype [9-11]. In the more common forms of primary headaches, as in other complex diseases, the phenotype is thought to be caused by an interaction of multiple genetic variants, each of them having a small to medium effect, with different environmental factors.
Due to the complexity of these disorders, the isolation of different genetic factors involved in primary headaches has proven to be difficult. Genetic association studies have provided evidence that genes involved in vascular, neuronal and endocrine functions may have a significant role in primary headaches [12,13]. The purpose of this review is to highlight recent discoveries, in particular about methylenetetrahydrofolate reductase (MTHFR), potassium channel, subfamily K member 18 (KCNK18), transient related potential vanilloid type 1 (TRPV1), transient related potential vanilloid type 3 (TRPV3), hypocretin (orexin) receptor 1 (HCRTR1) and hypocretin (orexin) receptor 2 (HCRTR2) genes which are involved in different subtypes of primary headaches and that, in the near future, might be of relevance as novel therapeutic targets.
Review
In the last two decades, molecular genetic studies provided substantial evidence concerning the potential role of multiple genes in primary headaches. The majority of these studies evaluated genetic factors involved in migraine, while molecular genetics of cluster headache and tension-type headache has been little studied, so far.
Vascular genes and primary headaches
Migraine and cluster headache have long been considered vascular disorders. Even if the so-called “vascular theory” of migraine has been shown to be inadequate in explaining the complex symptoms of the disorder, both migraineurs and cluster headache patients show abnormalities in both cranial and extracranial vascular reactivity [14,15]. In addition, both disorders are characterized by a significant comorbidity with diseases such as stroke, myocardial infarction, hypertension and Raynaud’s phenomenon [16-18]. Several genes involved in vascular functions, such as endothelin-1 (ETA-1), angiotensin-converting enzyme (ACE), Neurogenic Locus Notch Homolog Protein 4 (NOTCH4) and methylenetetrahydrofolate reductase (MTHFR) genes have been studied in patients with primary headaches and some of these, such as the MTHFR gene, were significantly associated with migraine [19-24].
The MTHFR gene is located on chromosome 1p36.3; it consists of 11 exons and encodes for the methylenetetrahydrofolate reductase, a crucial enzyme involved in purine and thymidylate biosynthesis, methylation of DNA and amino acids, and neurotransmitters synthesis. The MTHFR enzyme catalyzes the reduction of 5, 10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a substrate needed for the conversion of homocysteine to methionine (Figure 1). This pathway is folate-dependent and a lack of dietary folate can produce an increase in homocysteine levels. The clinical consequences of increased homocysteine plasma concentrations include endothelial cells injury and alterations in coagulant properties of blood [25-27]. Furthermore, homocysteine derivatives act as NMDA receptor agonists and they may enhance glutamatergic neurotransmission, thereby increasing the spontaneous trigeminal cells firing and predisposing cortical neurons to hyperexcitability [28,29].
Figure 1.
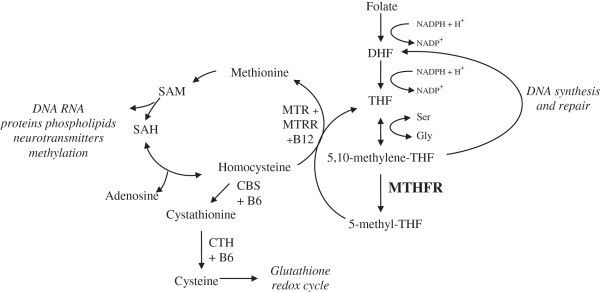
Production of homocysteine as part of the amino acid and purine biosynthesis pathway. DHF = dihydrofolate, THF = tetrahydrofolate, MTHFR = methylene-tetrahydrofolate-reductase, TS = thymidylate-synthase, MTR = methionine-synthase, MTRR o MSR = methionine-synthase-reductase.
Several genetic variants have been described in the MTHFR gene. Genetic research investigating the role of MTHFR in primary headaches has focused almost exclusively on two common polymorphisms, due to their functional activity. These are a cytosine (C) > thymine (T) change at position 677 in exon 4, that results in a substitution of an alanine into a valine amino acid (Ala222Val) in the catalytic domain, and an adenine (A) > cytosine (C) change occurring at position 1298 in exon 8, that changes a glutamate into an alanine amino acid (Glu429Ala). The C677T genetic variant allele produces a 35% reduction of MTHFR enzyme activity whereas the A1298C variant results in decreased MTHFR activity to a somewhat lesser degree [30]. These two variants have been extensively associated with the pathogenesis of several disorders, such as cardiovascular disease, cerebrovascular disease, and psychiatric disorders [31-35].
A large number of studies also provided evidence of an association between migraine and the C677T polymorphism in the MTHFR gene [22-24]. This association seems significant mainly in patients affected by migraine with aura (MA) while in patients affected by migraine without aura (MO) the results are conflicting. Two recent genetic meta-analysis provided clear evidence of a significant association between MA and the MTHFR gene: the carriage of the T allele was shown to be associated with an approximately two-fold increased risk [36,37].
More recently, in Norfolk Island Population, three MTHFR single nucleotide polymorphisms (SNPs) were associated with migraine. These three SNPs are located in intron 7 (rs6696752), in the 3′ untranslated region (rs4846048), and in exon 11 (rs2274976, non synonymous, producing a substitution of arginine to glutamine, R594Q) and have not previously been reported to show any genetic association with migraine. These findings reinforced the potential role of MTHFR in migraine susceptibility [37].
A recent case–control study examined the association between MTHFR polymorphisms and cluster headache in a group of 147 cases and 599 Caucasians controls. This study found no evidence of association between genotypes of the MTHFR 677C>T polymorphism and cluster headache overall. However, subgroup analyses suggested that carriers of the MTHFR 677 T allele may have an increased risk for chronic cluster headache, suggesting a need for additional studies in order to evaluate a possible role of MHTFR as modifier gene in the disorder [38]. At present, no study examined the potential association between tension-type headache and MTHFR polymorphisms.
Hyperomocysteinemia has been reported in patients with migraine [39]. Folic acid, vitamin B6 and vitamin B12 supplementation has been found to be effective in reducing the occurrence of migraine attacks [40]. Therefore, a recent pharmacogenetic study evaluated the effects of different MTHFR and 5-methyltetrahydrofolate-homocysteine methyltransferase reductase (MTRR) genotypes on the occurrence of migraine in a double-blinded placebo-controlled trial of daily vitamin B supplementation. Patients carrying the C allele of the MTHFR C677T variant showed a higher reduction in homocysteine levels, severity of pain and migraine disability, when compared with those with the T allele. MTRR catalyzes the remethylation of homocysteine to methionine, and, similarly, the A allele carriers of the MTRR A66G variants showed a higher degree of reduction in homocysteine levels, severity of pain and percentage of severe migraine disability, when compared with those carrying the GG genotypes [41]. This pivotal study suggests that both MTHFR and MTRR gene variants may influence the response to treatment with vitamin B in migraineurs. Conversely, genetic data concerning the role of vascular genes in tension-type headache are still scarce. A recent meta-analysis investigated the genetic role of the endothelin type A receptor (EDNRA) – one of the two receptors of the potent vasoconstrictor ETA-1 – in migraineurs and in patients with tension-type headache [42]. This meta-analysis included 440 migraineurs, 222 patients with tension-type headaches and 1323 controls from three previous studies reporting conflicting results about EDNRA -231G>A polymorphism. It found a significant difference in the frequency of AA genotype between migraine subjects and healthy controls. However, no differences were found in the distribution of the EDNRA -231G>A SNP between patients with tension-type headaches and controls.
Neuronal genes and primary headaches
Several clinical and experimental data support the concept of abnormal cortical excitability as the pivotal physiological disturbance in migraine [43,44]. Mutations in genes that code for ion channels or pumps (CACNA1A, ATP1A2, and SCN1A) have been described in FHM, strongly supporting the hypothesis that migraine may be classified as a “cerebral ionopathy” [45]. CaV 2.1 (CACNA1A) calcium channels are located in the presynaptic terminal of both excitatory and inhibitory neurons, NaV 1.1 (SCN1A) sodium channels are expressed in inhibitory interneurons while Na+/K+ATPase (ATP1A2) is located at the surface of glial cells (astrocytes). Knock-in mouse models carrying such mutations showed an increased susceptibility to cortical spreading depression, the likely underlying mechanism of migraine aura [46,47]. Finally, an interesting comorbidity between migraine and epilepsy has been described, further supporting a role for ion homeostasis genes in migraine pathophysiology [48,49]. However, until few years ago, no ion channel gene involvement has been described in the common form of migraine or in other primary headaches disorders.
In 2010, a frameshift mutation in the KCNK18 gene which segregates perfectly with typical MA in a large, multigenerational pedigree was reported [50]. This gene codes for TWIK-related spinal cord potassium channel (TRESK), a member of the two-pore domain (K2P) potassium channel family. Functional characterization of the F139WfsX24 mutation demonstrated that it causes a complete loss of TRESK function and that the mutant subunit suppresses the wild-type channel function through a dominant-negative effect.
The KCNK18 gene is located on chromosome 10; it encodes a protein containing 4 transmembrane domains (TMDs), and two pore-forming domains. The extracellular domain located between TMD1 and TMD2 contains a conserved cysteine residue that may form a disulfide bridge to aid channel dimerization, and a conserved N-linked glycosylation site, important for surface expression of the channel [51]. In humans, the family of KP2 channels includes 15 related channels but TRESK is unique in having a large intracellular regulatory domain located between TMD2 and TMD3. TRESK is abundantly expressed in the dorsal root ganglion (DRG), and in other sensory ganglia such as the trigeminal ganglion (TG). TRESK was also found in human autonomic nervous system ganglia, such as the stellate ganglion and paravertebral sympathetic chain [50,52].
The TRESK is an outwardly rectifying K+ current channel that contributes to the resting potential and is the most important background potassium channel in DRG. TRESK is activated in a complex manner by intracellular calcium signalling. The calcium/calmodulin-dependent protein phosphatase, calcineurin, activates TRESK function [53,54]. Calcineurin also regulates nuclear factor of activated T cells (NFATs), transcription factors that regulate inducible expression of many cytokines. In addition, recent studies have identified volatile anesthetics as highly potent TRESK agonists, interacting directly with the channel. On the contrary, cyclosporin A and tacrolimus, two potent immunosuppressants that specifically inhibit the calcineurin activation of NFATs, mimic a TRESK loss of function by keeping the channel insensitive to increases in intracellular Ca2+.
The physiological functions of TRESK have been mainly investigated in knock-out (KO) mice. The KO mice show no gross anatomical or behavioral phenotype. The gene ablation has minimal effect on the resting membrane potential. However, DRG neurons from TRESK KO mice displayed a lower threshold for activation, reduced action potential duration, and slightly higher amplitudes of after-hyperpolarization, suggesting that DRG neurons from KO mice were more excitable than wild-type DRG neurons [55]. In addition, due to the coupling of TRESK to the histamine H1 receptor, the channel may reduce neuronal excitability in inflammatory conditions when histamine or other inflammatory modulators are released into the surrounding tissue. Taken together, these data suggest an important role of TRESK in both acute and chronic pain conditions [56].
After the identification of KCNK18 gene mutation in a Canadian MA pedigree, a large cohort of unrelated MA patients and healthy controls was screened for gene mutation. Several missense variants (R10G, A34V, C110R, S231P and A233V) were found. These variants either had no apparent functional effect, or they caused a reduction in channel activity. The A34V was identified in a single Australian migraine proband for which family samples were not available, but it was not detected in controls. By contrast, the R10G, C110R, and S231P variants were found in both migraineurs and controls. The authors concluded that the presence of a single non-functional variant in the KCNC18 gene is probably not sufficient to determine whether an individual develops migraine [57].
In a recent study, we examined the presence of KCNK18 gene mutations in a large data set of Italian migraine patients (both with MA and MO) and healthy controls. We confirmed the presence of KCNK18 gene mutations in MA and also found gene mutations in MO patients [58]. Some of these gene variants have not previously been described. However, the functional relevance of these mutations still need further investigations. Finally, KCNK18 gene involvement in tension-type headache or cluster headache has not been investigated yet.
The TRESK K2P channel is a novel and interesting component of the migraine pathogenesis pathway and represents an excellent opportunity for development of antimigraine therapy, given its highly selective expression pattern in neuronal structures, which is known to be important in disease pathogenesis. Its highly specific expression pattern in TG, DRG and parasympathetic neurons and the presumed role in abating neuronal excitability under inflammatory conditions make it an excellent target for development of new migraine therapeutics [59,60]. Specific agonists could upregulate TRESK activity and may have a potential in both acute and preventive migraine therapy, as well as in other pain disorders.
Recently, the transient receptor potential (TRP) channels gained increased interest for their potential involvement in primary headaches [61,62]. There are at least 30 members of the mammalian TRP family, which are coded by several, different genes. TRP channels are distributed in many peripheral tissues as well as central and peripheral nervous system. Several TRP family members, including the TRPV1 (Transient Related Potential Vanilloid Type 1), TRPV2 (Transient Related Potential Vanilloid Type 2), TRPV3 (Transient Related Potential Vanilloid Type 3) and TRPV4 (Transient Related Potential Vanilloid Type 4), TRPM8 (Transient Related Potential Metastatin Type 8) channels, are polymodal sensors expressed in the sensory neurons of dorsal root ganglia (DRG) and trigeminal ganglia (TG) [63]. In particular, TRPV1 receptor is highly co-expressed with calcitonin-gene related peptide (CGRP) a potent vasodilator with an important role in migraine. TRPV1 is also co-expressed with other pain signalling molecules such as substance P, P2X3 purinergic receptors and other markers of nociceptive C and Aδ fibers [64]. It has been proposed to play a crucial role as mediators of neuropathic pain and have been proposed to play a role in migraineous allodynia and sensitization phenomena [65].
Using a genetic association strategy, in 2012 Carreno et al. found a significant association between SNPs within the TRPV1 and TRPV3 genes and migraine in the Spanish population [66]. Interestingly, TRPV1 and TRPV3 are located in close proximity on the 17p13 chromosomal region and they share a high sequence homology. In the meanwhile, two genome-wide association studies (GWAS) found evidence of a significant association between genetic markers in or near the TRPM8 gene and migraine [67,68]. TRPM8 gene is expressed by a different TRPV1-negative neuronal subpopulation in DRG and TG. These preliminary data significantly support a role for TRP channels in the pathogenesis of primary headaches.
TRP channels have been among the most aggressively pursued drug targets over the past few years and several studies suggested these channels as potential therapeutic targets in migraine. Both peripheral and central nerve terminals at the spinal cord can be targeted to induce pain relief by TRPV1 agonists. In particular, the analgesic effects of the TRPV1 antagonist SB-705498 on trigeminovascular sensitization and neurotransmission have been studied in an animal model of neurovascular head pain [69,70]. Recently, a phase II clinical trial using SB-705498 has been conducted for the acute treatment of migraine attacks but results are pending (ClinicalTrials.gov).
Neuroendocrine genes and primary headaches
A large number of endocrine abnormalities has been described in patients with primary headaches [71,72]. The hypothalamus, with its paramount control of the endocrine system, as well as its widespread connections with both central and autonomic nervous system, exerts a pivotal role in the pathogenesis of both migraine and cluster headache [73]. Therefore, genes that code for proteins involved in endocrine functions are candidate genes for primary headache disorders. Polymorphisms in genes that code for estrogen and progesterone receptors have been intensively studied in migraineurs, with contrasting results [74,75].
In 1998, two research groups independently discovered a new hypothalamic peptidergic system. The former group named the peptides “hypocretins”, because of their hypothalamic location and structural similarity to the incretin family of hormones [76]. The latter group named the peptides “orexins”, due to the appetite-enhancing properties when administered centrally to rats [77].
Subsequent studies revealed complex and interesting neurobiological effects of these peptides, with particular relevance to the pathophysiology of primary headaches [78].
The hypocretins (Hcrt-1 and Hcrt-2), also called orexins, are peptides derived by proteolytic cleavage from the same 130 amino acid precursor peptide (prepro-hypocretin). A single gene located on chromosome 17q21 in humans is responsible for encoding prepro-hypocretin. The human prepro-hypocretin gene consists of 2 exons and 1 intron. The hypocretins bind to 2 G-protein coupled receptors, termed HCRTR1 and HCRTR2. The HCRTR1 gene in humans is located on chromosome1p33 whereas the HCRTR2 gene is located on chromosome 6p11. Both genes consist of 7 exons and 6 introns. Hcrt-1 has equal affinity for both HCRTR1 and HCRTR2, with Hcrt-2 demonstrating a 10-fold higher affinity for HCRTR2 than HCRTR1. Activation of both receptors results in elevated levels of the intracellular Ca2+ concentrations, and this in turn results in the enhancement of the Gq-mediated stimulation of phospholipase C. Hypocretin immunoreactive cell bodies have been observed mainly in the hypothalamus [79]. Hypocretin-containing neurons have widespread projections throughout the CNS with particularly dense excitatory projections to monoaminergic and serotonergic brainstem centers [80]. The hypocretin system influences a wide range of physiological processes in mammals, such as feeding, arousal, rewards, and drug addiction [81,82]. Recently, a number of studies in experimental animals showed that hypocretins are involved in pain modulation within the CNS, and suggested an important role for these peptides in primary headaches [83].
In 2004, our research group investigated the possible involvement of the hypocretin transmission in cluster headache. We selected several DNA polymorphisms of the three genes that constitute the hypocretin system and, using a case–control strategy, we evaluated possible allelic and genotypic differences in a group of 109 CH patients and 211 controls. Genetic analysis revealed that both allelic and genotypic frequencies of the G1246A polymorphism in the HCRTR2 gene were significantly different between CH patients and controls [84]. Subjects homozygous for the G allele, in comparison with the remaining genotypes, were 5-fold more likely to develop the disease. This association was confirmed in a large group of CH patients and controls from Germany [85]. On the contrary, Baumber et al. found no association between CH and the HCRTR2 gene in a cohort of 259 patients of Danish, Swedish, and British origin [86]. To resolve this issue, we performed a genetic meta-analysis of the previous studies (593 cases and 599 controls) and a haplotype analysis: both these studies confirmed the presence of a significant association between the HCRTR2 gene and CH [87]. At present, the possible involvement of the hypocretin system in migraine has been scarcely investigated. Studies in CH patients prompted two independent research groups to evaluate the association of the G1246A polymorphisms in the HCRTR2 gene with migraine. Both studies found no association between this polymorphism and migraine or its clinical subtypes [88,89]. Recently, we performed a genetic case–control study to investigate whether genetic variants in the HCRTR1 gene could modify the occurrence and the clinical features of migraine. Using a case–control strategy we genotyped 384 migraine patients and 259 controls for three SNPs in the HCRTR1 gene. Genotypic and allelic frequencies of the rs2271933 non-synonymous polymorphism were different between migraineurs and controls [90]. The carriage of the A allele was associated with an increased migraine risk. This study supports the hypothesis that the HCRTR1 gene could represent a genetic susceptibility factor for migraine and suggests that the hypocretin system may have a role also in the pathophysiology of migraine. Unfortunately, no data regarding involvement of HCRTR genes in tension-type headache are currently available.
The growing knowledge concerning the role of hypocretins/orexins in different neurological conditions has generated considerable interest in developing small-molecule hypocretin receptor antagonists as a novel therapeutic strategy. Hypocretin antagonists, especially those that block Hcrtr1 or both Hcrtr1 and Hcrtr2 receptors, have been studied mainly as new drugs for sleep disorders. In experimental animals hypocretin/orexin antagonists (almorexant, suvorexant) clearly promote sleep, and clinical results are encouraging [91-94]. Considering the high frequency of sleep disorders occurring in patients with migraine and CH, these drugs offer a new perspective in the treatment of these disorders. Finally, a pivotal role for these peptides in drug reward and drug seeking has been established and their potential role as anti-relapse medication in drug addiction is currently under investigation in experimental animals [95].
Conclusions
The main goal of genetic studies is to unravel molecular pathways underlying primary headache disorders, in order to discover new therapeutic targets. Recent studies have highlighted a potential role for new genes, like MTHFR, KCNK18, TRPV1, TRPV3, and new neurotransmission systems, like the hypocretin system, both in migraine and cluster headache. Additional experimental and clinical studies are needed to better elucidate the involvement of these new genes in primary headaches and to evaluate new therapeutic strategies.
Competing interest
The authors declare no competing interest regarding this manuscript.
Authors’ contribution
IR and ER planned the review and wrote the manuscript, with input from other authors. KP reviewed the manuscript. AG and AV performed bibliographic research and draw the pictures. PDM, SG, PS and LP supervised the project. All authors read and approved the final manuscript.
Contributor Information
Innocenzo Rainero, Email: innocenzo.rainero@unito.it.
Elisa Rubino, Email: elisa.rubino@unito.it.
Koen Paemeleire, Email: koen.paemeleire@uzgent.be.
Annalisa Gai, Email: annalisa.gai5@gmail.com.
Alessandro Vacca, Email: aletorinese@hotmail.it.
Paola De Martino, Email: paola.demartino@unito.it.
Salvatore Gentile, Email: sgentile@cittadellasalute.to.it.
Paola Sarchielli, Email: paola.sarchielli@gmail.com.
Lorenzo Pinessi, Email: lorenzo.pinessi@unito.it.
Acknowledgements
This Review Article will be presented at the XXVII National Congress of the Italian Society for the Study of Headaches – 26–28 September 2013.
References
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders: 2nd edn. Cephalalgia. 2004;14(Suppl 1):9–160. doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- Lantéri-Minet M, Duru G, Mudge M, Cottrell S. Quality of life impairment, disability and economic burden associated with chronic daily headache, focusing on chronic migraine with or without medication overuse: a systematic review. Cephalalgia. 2011;14(7):837–850. doi: 10.1177/0333102411398400. [DOI] [PubMed] [Google Scholar]
- Raggi A, Giovannetti AM, Quintas R, D’Amico D, Cieza A, Sabariego C, Bickenbach JE, Leonardi M. A systematic review of the psychosocial difficulties relevant to patients with migraine. J Headache Pain. 2012;14(8):595–606. doi: 10.1007/s10194-012-0482-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradalier A, Auray JP, El Hasnaoui A, Alzahouri K, Dartigues JF, Duru G, Henry P, Lantéri-Minet M, Lucas C, Chazot G, Gaudin AF. Economic impact of migraine and other episodic headaches in France: data from the GRIM2000 study. PharmacoEconomics. 2004;14(15):985–999. doi: 10.2165/00019053-200422150-00003. [DOI] [PubMed] [Google Scholar]
- Bloudek LM, Stokes M, Buse DC, Wilcox TK, Lipton RB, Goadsby PJ, Varon SF, Blumenfeld AM, Katsarava Z, Pascual J, Lanteri-Minet M, Cortelli P, Martelletti P. Cost of healthcare for patients with migraine in five European countries: results from the International Burden of Migraine Study (IBMS) J Headache Pain. 2012;14(5):361–378. doi: 10.1007/s10194-012-0460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M. et al. Years lived with disability (YLD) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systemati c analysis for the Global Burden of Disease Study 2010. Lancet. 2012;14(9859):2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MB. Genetics in primary headaches. J Headache Pain. 2007;14(3):190–195. doi: 10.1007/s10194-007-0389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson DA, Larsson B, Waldenlind E, Pedersen NL. Shared rearing environment in migraine: results from twins reared apart and twins reared together. Headache. 2003;14(3):235–244. doi: 10.1046/j.1526-4610.2003.03047.x. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN. et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;14:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Marconi R, Silvestri L. et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;14:192–196. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G. et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;14:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- Colson NJ, Lea RA, Quinlan S, Griffiths LR. The role of vascular and hormonal genes in migraine susceptibility. Mol Genet Metab. 2006;14:107–113. doi: 10.1016/j.ymgme.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Maher BH, Griffiths LR. Identification of molecular genetic factors that influence migraine. Mol Genet Genomics. 2011;14(6):433–446. doi: 10.1007/s00438-011-0622-3. [DOI] [PubMed] [Google Scholar]
- Vanmolkot FH, Van Bortel LM, de Hoon JN. Altered arterial function in migraine of recent onset. Neurology. 2007;14(19):1563–1570. doi: 10.1212/01.wnl.0000260964.28393.ed. [DOI] [PubMed] [Google Scholar]
- Barriga FJ, Cuadrado ML, Bueno A, Barón M, Dobato JL, Vela L, Pareja JA. Clus ter headache: orbital hemodynamic changes during Valsalva maneuver. Headache. 2006;14(2):298–305. doi: 10.1111/j.1526-4610.2006.00287.x. [DOI] [PubMed] [Google Scholar]
- Kurth T. The association of migraine with ischemic stroke. Curr Neurol Neurosci Rep. 2010;14(2):133–139. doi: 10.1007/s11910-010-0098-2. [DOI] [PubMed] [Google Scholar]
- Kurth T, Gaziano JM, Cook NR, Logroscino G, Diener HC, Buring JE. Migraine and risk of cardiovascular disease in women. JAMA. 2006;14(3):283–291. doi: 10.1001/jama.296.3.283. [DOI] [PubMed] [Google Scholar]
- Sacco S, Ricci S, Carolei A. Migraine and vascular diseases: a review of the evidence and potential implications for management. Cephalalgia. 2012;14(10):785–795. doi: 10.1177/0333102412451361. [DOI] [PubMed] [Google Scholar]
- Tzourio C, El Amrani M, Poirier O, Nicaud V, Bousser MG, Alpérovitch A. Association between migraine and endothelin type A receptor (ETA −231 A/G) gene polymorphism. Neurology. 2001;14(10):1273–1277. doi: 10.1212/WNL.56.10.1273. [DOI] [PubMed] [Google Scholar]
- Tronvik E, Stovner LJ, Schrader H, Bovim G. Involvement of the renin-angiotensin system in migraine. J Hypertens. 2006;14:139–143. doi: 10.1097/01.hjh.0000220419.86149.11. [DOI] [PubMed] [Google Scholar]
- Rubino E, Fenoglio P, Gallone S, Govone F, Vacca A, De Martino P, Giobbe ML, Boschi S, Pinessi L, Gentile S, Rainero I. Genetic variants in the NOTCH4 gene influence the clinical features of migraine. J Headache Pain. 2013;14(1):28. doi: 10.1186/1129-2377-14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher AI, Terwindt GM, Verschuren WM, Kruit MC, Blom HJ, Kowa H, Frants RR, van den Maagdenberg AM, van Buchem M, Ferrari MD, Launer LJ. Migraine and MTHFR C677T genotype in a population-based sample. Ann Neurol. 2006;14(2):372–375. doi: 10.1002/ana.20755. [DOI] [PubMed] [Google Scholar]
- Liu A, Menon S, Colson NJ, Quinlan S, Cox H, Peterson M, Tiang T, Haupt LM, Lea RA, Griffiths LR. Analysis of the MTHFR C677T variant with migraine phenotypes. BMC Res Notes. 2010;14:213. doi: 10.1186/1756-0500-3-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaan Z, Gaysina D, Cohen-Woods S, Craddock N, Jones L, Korszun A, Owen M, Mente A, McGuffin P, Farmer A. Methylenetetrahydrofolate reductase gene variant (MTHFR C677T) and migraine: a case control study and meta-analysis. BMC Neurol. 2011;14:66. doi: 10.1186/1471-2377-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann Clin Lab Sci. 2009;14(3):219–232. [PubMed] [Google Scholar]
- Dionisio N, Jardín I, Salido GM, Rosado JA. Homocysteine, intracellular signaling and thrombotic disorders. Curr Med Chem. 2010;14(27):3109–3119. doi: 10.2174/092986710791959783. [DOI] [PubMed] [Google Scholar]
- Boldyrev A, Bryushkova E, Mashkina A, Vladychenskaya E. Why is Homocysteine toxic for the nervous and immune systems? Curr Aging Sci. 2012. [Epub ahead of print] [DOI] [PubMed]
- Zieminska E, Lazarewicz JW. Excitotoxic neuronal injury in chronic homocysteine neurotoxicity studied in vitro: the role of NMDA and group I metabotropic glutamate receptors. Acta Neurobiol Exp (Wars) 2006;14(4):301–309. doi: 10.55782/ane-2006-1619. [DOI] [PubMed] [Google Scholar]
- Yeganeh F, Nikbakht F, Bahmanpour S, Rastegar K, Namavar R. Neuroprotective effects of NMDA and Group I metabotropic glutamate receptor antagonists against neurodegeneration induced by homocysteine in rat hippocampus: in vivo study. J Mol Neurosci. 2013. [Epub ahead of print] [DOI] [PubMed]
- Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, Rozen R. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;14(1):111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- Trabetti E. Homocysteine, MTHFR gene polymorphisms, and cardio-cerebrovascular risk. J Appl Genet. 2008;14(3):267–282. doi: 10.1007/BF03195624. [DOI] [PubMed] [Google Scholar]
- Pizza V, Bisogno A, Lamaida E, Agresta A, Bandieramonte G, Volpe A, Galasso R, Galasso L, Caputo M, Tecce MF, Capasso A. Migraine and coronary artery disease: an open study on the genetic polymorphism of the 5, 10 methylenetetrahydrofolate (MTHFR) and angiotensin I-converting enzyme (ACE) genes. Cent Nerv Syst Agents Med Chem. 2010;14(2):91–96. doi: 10.2174/187152410791196404. [DOI] [PubMed] [Google Scholar]
- Sazci A, Ergul E, Tuncer N, Akpinar G, Kara I. Methylenetetrahydrofolate reductase gene polymorphisms are associated with ischemic and hemorrhagic stroke: dual effect of MTHFR polymorphisms C677T and A1298C. Brain Res Bull. 2006;14(1–3):45–50. doi: 10.1016/j.brainresbull.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Almeida OP, McCaul K, Hankey GJ, Norman P, Jamrozik K, Flicker L. Homocysteine and depression in later life. Arch Gen Psychiatry. 2008;14(11):1286–1294. doi: 10.1001/archpsyc.65.11.1286. [DOI] [PubMed] [Google Scholar]
- Peerbooms OL, van Os J, Drukker M, Kenis G, Hoogveld L, de Hert M, Delespaul P, van Winkel R, Rutten BP. MTHFR in Psychiatry Group. Meta-analysis of MTHFR gene variants in schizophrenia, bipolar disorder and unipolar depressive disorder: evidence for a common genetic vulnerability? Brain Behav Immun. 2011;14(8):1530–1543. doi: 10.1016/j.bbi.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Rubino E, Ferrero M, Rainero I, Binello E, Vaula G, Pinessi L. Association of the C677T polymorphism in the MTHFR gene with migraine: a meta-analysis. Cephalalgia. 2009;14:818–825. doi: 10.1111/j.1468-2982.2007.01400.x. [DOI] [PubMed] [Google Scholar]
- Schürks M, Rist PM, Kurth T. MTHFR 677C>T and ACE D/I polymorphisms in migraine: a systematic review and meta-analysis. Headache. 2010;14(4):588–599. doi: 10.1111/j.1526-4610.2009.01570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürks M, Neumann FA, Kessler C, Diener HC, Kroemer HK, Kurth T, Völzke H, Rosskopf D. MTHFR 677C>T polymorphism and cluster headache. Headache. 2010;14(2):201–207. doi: 10.1111/j.1526-4610.2010.01780.x. [DOI] [PubMed] [Google Scholar]
- Moschiano F, D’Amico D, Usai S, Grazzi L, Di Stefano M, Ciusani E, Erba N, Bussone G. Homocysteine plasma levels in patients with migraine with aura. Neurol Sci. 2008;14(Suppl 1):S173–S175. doi: 10.1007/s10072-008-0917-2. [DOI] [PubMed] [Google Scholar]
- Lea R, Colson N, Quinlan S, Macmillan J, Griffiths L. The effects of vitamin supplementation and MTHFR (C677T) genotype on homocysteine-lowering and migraine disability. Pharmacogenet Genomics. 2009;14(6):422–428. doi: 10.1097/FPC.0b013e32832af5a3. [DOI] [PubMed] [Google Scholar]
- Menon S, Lea RA, Roy B, Hanna M, Wee S, Haupt LM, Oliver C, Griffiths LR. Genotypes of the MTHFR C677T and MTRR A66G genes act independently to reduce migraine disability in response to vitamin supplementation. Pharmacogenet Genomics. 2012;14(10):741–749. doi: 10.1097/FPC.0b013e3283576b6b. [DOI] [PubMed] [Google Scholar]
- Miao J, Wang F, Fang Y. Association of 231G>A polymorphism of endothelin type A receptor gene with migraine: a meta-analysis. J Neurol Sci. 2012;14(1–2):232–235. doi: 10.1016/j.jns.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Haigh S, Karanovic O, Wilkinson F, Wilkins A. Cortical hyperexcitability in migraine and aversion to patterns. Cephalalgia. 2012;14(3):236–240. doi: 10.1177/0333102411433301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Pierelli F, Schoenen J. Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia. 2007;14(12):1427–1439. doi: 10.1111/j.1468-2982.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- Ferrari MD, van der Maagdenberg AM, Frants RR, Goadsby PJ. In: Molecular Neurology. Waxman SG, editor. Amsterdam: Elsevier; 2007. Migraine as a cerebral ionopathy with impaired central sensory processing; pp. 439–461. [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci. 2011;14(15):5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo L, Gherardini L, Barone V, De Fusco M, Pietrobon D, Pizzorusso T, Casari G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2012;14(6):e1002129. doi: 10.1371/journal.pgen.1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchin MM, Londero RG, Lima JE, Bigal ME. Migraine and epilepsy: a focus on overlapping clinical, pathophysiological, molecular, and therapeutic aspects. Curr Pain Headache Rep. 2010;14(4):276–283. doi: 10.1007/s11916-010-0121-y. [DOI] [PubMed] [Google Scholar]
- Striano P, Belcastro V, Verrotti A, Parisi P. “Comorbidity” between epilepsy and headache/migraine: the other side of the same coin! J Headache Pain. 2011;14(5):577–578. doi: 10.1007/s10194-011-0371-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenière RG, Cader MZ, Poulin JF, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafrenière F, McLaughlan S, Dubé MP, Marcinkiewicz MM, Ramagopalan S, Ansorge O, Brais B, Sequeiros J, Pereira-Monteiro JM, Griffiths LR, Tucker SJ, Ebers G, Rouleau GA. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med. 2010;14(10):1157–1160. doi: 10.1038/nm.2216. [DOI] [PubMed] [Google Scholar]
- Egenberger B, Polleichtner G, Wischmeyer E, Döring F. N-linked glycos ylation determines cell surface expression of two-pore-domain K+ channel TRESK. Biochem Biophys Res Commun. 2010;14(2):1262–1267. doi: 10.1016/j.bbrc.2009.12.056. [DOI] [PubMed] [Google Scholar]
- Kang D, Mariash E, Kim D. Functional expression of TRESK-2, a new member of the tandem-pore K+ channel family. J Biol Chem. 2004;14(27):28063–28070. doi: 10.1074/jbc.M402940200. [DOI] [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Structural delineation of the calcineurin–NFAT inte raction and its parallels to PP1 targeting interactions. J Mol Biol. 2004;14(5):1659–1674. doi: 10.1016/j.jmb.2004.07.068. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Braun G, Czirják G. TRESK: the lone ranger of two-pore domain potassium channels. Mol Cell Endocrinol. 2012;14(1–2):75–81. doi: 10.1016/j.mce.2011.11.009. [DOI] [PubMed] [Google Scholar]
- Dobler T, Springauf A, Tovornik S, Weber M, Schmitt A, Sedlmeier R, Wischmeyer E, Döring F. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol. 2007;14(Pt 3):867–879. doi: 10.1113/jphysiol.2007.145649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DY, Yu BW, Fan QW. Roles of TRESK, a novel two-pore domain K+ channel, in pain pathway and general anesthesia. Neurosci Bull. 2008;14(3):166–172. doi: 10.1007/s12264-008-0225-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres-Enguix I, Shang L, Stansfeld PJ, Morahan JM, Sansom MS, Lafrenière RG, Roy B, Griffiths LR, Rouleau GA, Ebers GC, Cader ZM, Tucker SJ. Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci Rep. 2012;14:237–240. doi: 10.1038/srep00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainero I, Rubino E, Fenoglio P, Gallone S, Zavarise P, Carli D, Boschi S, Gai A, Pinessi L, Dalla Volta G. Investigation of KCNK18 (TRESK) Genetic Variants in Migraine with and without Aura. In: Abstracts of the 65th AAN Annual Meeting, San Diego. Neurology. 2013;14(Meeting Abstracts 1):S55.003. [Google Scholar]
- Marsh B, Acosta C, Djouhri L, Lawson SN. Leak K+ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci. 2012;14(3):375–386. doi: 10.1016/j.mcn.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X. TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain. 2011;14:30. doi: 10.1186/1744-8069-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R, De Cesaris F, Pedretti P, Geppetti P. TRPS and migraine. The Open Drug Discovery Journal. 2010;14:55–63. doi: 10.2174/1877381801002030055. [DOI] [Google Scholar]
- Oxford GS, Hurley JH. The role of TRP channels in migraine. The Open Pain Journal. 2013;14(Suppl 1):37–49. [Google Scholar]
- Vennekens R, Menigoz A, Nilius B. TRPs in the Brain. Rev Physiol Biochem Pharmacol. 2012;14:27–64. doi: 10.1007/112_2012_8. [DOI] [PubMed] [Google Scholar]
- Bae YC, Oh JM, Hwang SJ, Shigenaga Y, Valtschanoff JG. Expression of vanilloid receptor TRPV1 in the rat trigeminal sensory nuclei. J Comp Neurol. 2004;14(1):62–71. doi: 10.1002/cne.20272. [DOI] [PubMed] [Google Scholar]
- Meents JE, Neeb L, Reuter U. TRPV1 in migraine pathophysiology. Trends Mol Med. 2010;14(4):153–159. doi: 10.1016/j.molmed.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Carreño O, Corominas R, Fernández-Morales J, Camiña M, Sobrido MJ, Fernández-Fernández JM, Pozo-Rosich P, Cormand B, Macaya A. SNP variants within the vanilloid TRPV1 and TRPV3 receptor genes are associated with migraine in the Spanish population. Am J Med Genet B Neuropsychiatr Genet. 2012;14(1):94–103. doi: 10.1002/ajmg.b.32007. [DOI] [PubMed] [Google Scholar]
- Chasman DI, Schürks M, Anttila V, de Vries B, Schminke U, Launer LJ, Terwindt GM, van den Maagdenberg AM, Fendrich K, Völzke H, Ernst F, Griffiths LR, Buring JE, Kallela M, Freilinger T, Kubisch C, Ridker PM, Palotie A, Ferrari MD, Hoffmann W, Zee RY, Kurth T. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;14(7):695–698. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilinger T, Anttila V, de Vries B, Malik R, Kallela M, Terwindt GM, Pozo-Rosich P, Winsvold B, Nyholt DR, van Oosterhout WP, Artto V, Todt U, Hämäläinen E, Fernández-Morales J, Louter MA, Kaunisto MA, Schoenen J, Raitakari O, Lehtimäki T, Vila-Pueyo M, Göbel H, Wichmann E, Sintas C, Uitterlinden AG, Hofman A, Rivadeneira F, Heinze A, Tronvik E, van Duijn CM, Kaprio J. et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;14(7):777–782. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GA, Davis JB, Appleby JM, Chizh BA, Hoskin KL, Zagami AS. The effects of the TRPV1 receptor antagonist SB-705498 on trigeminovascular sensitisation and neurotransmission. Naunyn Schmiedebergs Arch Pharmacol. 2009;14(4):311–325. doi: 10.1007/s00210-009-0437-5. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cruz F, Geppetti P. RPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med. 2006;14(11):545–554. doi: 10.1016/j.molmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Silberstein SD. The role of sex hormones in headache. Neurology. 1992;14(Suppl 2):37–42. [PubMed] [Google Scholar]
- Nappi RE, Nappi G. Neuroendocrine aspects of migraine in women. Gynecol Endocrinol. 2012;14(Suppl 1):37–41. doi: 10.3109/09513590.2012.651931. [DOI] [PubMed] [Google Scholar]
- Alstadhaug KB. Migraine and the hypothalamus. Cephalalgia. 2009;14(8):809–817. doi: 10.1111/j.1468-2982.2008.01814.x. [DOI] [PubMed] [Google Scholar]
- Schürks M, Rist PM, Kurth T. Sex hormone receptor gene polymorphisms and migrain e: a systematic review and meta-analysis. Cephalalgia. 2010;14:1306–1328. doi: 10.1177/0333102410364155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi G, Pradhan S, Mittal B. Role of the oestrogen receptor (ESR1 PvuII and ESR1 325 C->G) and progesterone receptor (PROGINS) polymorphisms in genetic susceptibility to migraine in a North Indian population. Cephalalgia. 2010;14(3):311–320. doi: 10.1111/j.1468-2982.2009.01967.x. [DOI] [PubMed] [Google Scholar]
- De Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;14(1):322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;14(4):573–585. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Rainero I, De Martino P, Pinessi L. Hypocretins and primary headaches: neurobiology and clinical implications. Expert Rev Neurother. 2008;14(3):409–416. doi: 10.1586/14737175.8.3.409. [DOI] [PubMed] [Google Scholar]
- Nishino S. The hypothalamic peptidergic system, hypocretin/orexin and vigilance control. Neuropeptides. 2007;14(3):117–133. doi: 10.1016/j.npep.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Mori T, Ito S, Kuwaki T, Yanagisawa M, Sakurai T, Sawaguchi T. Monoaminergic neuronal changes in orexin deficient mice. Neuropharmacology. 2012;14(4–5):826–832. doi: 10.1016/j.neuropharm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Girault EM, Yi CX, Fliers E, Kalsbeek A. Orexins, feeding, and energy balance. Prog Brain Res. 2012;14:47–64. doi: 10.1016/B978-0-444-59489-1.00005-7. [DOI] [PubMed] [Google Scholar]
- Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G. Multiple roles for orexin/hypocretin in addiction. Prog Brain Res. 2012;14:79–121. doi: 10.1016/B978-0-444-59489-1.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Saito O, Shono K, Aoe T, Chiba T. Anti-mechanical allodynic effect of intrathecal and intracerebroventricular injection of orexin-A in the rat neuropathic pain model. Neurosci Lett. 2003;14(3):183–186. doi: 10.1016/S0304-3940(03)00716-X. [DOI] [PubMed] [Google Scholar]
- Rainero I, Gallone S, Valfrè W, Ferrero M, Angilella G, Rivoiro C, Rubino E, De Martino P, Savi L, Ferrone M, Pinessi L. A polymorphism of the hypocretin receptor 2 gene is associated with cluster headache. Neurology. 2004;14(7):1286–1288. doi: 10.1212/01.WNL.0000142424.65251.DB. [DOI] [PubMed] [Google Scholar]
- Schürks M, Kurth T, Geissler I, Tessmann G, Diener HC, Rosskopf D. Cluster heada che is associated with the G1246A polymorphism in the hypocretin receptor 2 gene. Neurology. 2006;14(12):1917–1919. doi: 10.1212/01.wnl.0000215852.35329.34. [DOI] [PubMed] [Google Scholar]
- Baumber L, Sjöstrand C, Leone M, Harty H, Bussone G, Hillert J, Trembath RC, MB R e. A genome-wide scan and HCRTR2 candidate gene analysis in a European cluster headache cohort. Neurology. 2006;14(12):1888–1893. doi: 10.1212/01.wnl.0000219765.95038.d7. [DOI] [PubMed] [Google Scholar]
- Rainero I, Rubino E, Valfrè W, Gallone S, De Martino P, Zampella E, Pinessi L. Association between the G1246A polymorphism of the hypocretin receptor 2 gene and cluster headache: a meta-analysis. J Headache Pain. 2007;14(3):152–156. doi: 10.1007/s10194-007-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinessi L, Binello E, De Martino P, Gallone S, Gentile S, Rainero I, Rivoiro C, Rubino E, Savi L, Valfrè W, Vaula G. The 1246G–>A polymorphism of the HCRTR2 gene is not associated with migraine. Cephalalgia. 2007;14(8):945–949. doi: 10.1111/j.1468-2982.2007.01347.x. [DOI] [PubMed] [Google Scholar]
- Schürks M, Limmroth V, Geissler I, Tessmann G, Savidou I, Engelbergs J, Kurth T, Di ener HC, Rosskopf D. Association between migraine and the G1246A polymorphism in the hypocretin receptor 2 gene. Headache. 2007;14(8):1195–1199. doi: 10.1111/j.1526-4610.2007.00863.x. [DOI] [PubMed] [Google Scholar]
- Rainero I, Rubino E, Gallone S, Fenoglio P, Picci LR, Giobbe L, Ostacoli L, Pinessi L. Evidence for an association between migraine and the hypocretin receptor 1 gene. J Headache Pain. 2011;14(2):193–199. doi: 10.1007/s10194-011-0314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inutsuka A, Yamanaka A. The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front Endocrinol (Lausanne) 2013;14:18. doi: 10.3389/fendo.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang GM, Dürst T, Bürki H, Imobersteg S, Abramowski D, Schuepbach E, Hoyer D, F endt M, Gee CE. The dual orexin receptor antagonist almorexant induces sleep and decreases orexin- induced locomotion by blocking orexin 2 receptors. Sleep. 2012;14(12):1625–1635. doi: 10.5665/sleep.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kennedy WP, Wilbraham D, Lewis N, Calder N, Li X, Ma J, Yee KL, Ermlich S, Mangin E, Lines C, Rosen L, Chodakewitz J, Murphy GM. Effects of suvorexant, an orexin receptor antagonist, on sleep parameters as measured by polysomnography in healthy men. Sleep. 2013;14(2):259–267. doi: 10.5665/sleep.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Sakurai T. Orexin (hypocretin) receptor agonists and antagonists for treatment of sleep disorders. Rationale for development and current status. CNS Drugs. 2013;14(2):83–90. doi: 10.1007/s40263-012-0036-8. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ghee SM, Chan C, Lin L, Cameron MD, Kenny PJ, See RE. Orexin-1 receptor mediation of cocaine seeking in male and female rats. J Pharmacol Exp Ther. 2012;14(3):801–809. doi: 10.1124/jpet.111.187567. [DOI] [PMC free article] [PubMed] [Google Scholar]