

Abstract
EGR1 transcription factor orchestrates a plethora of signaling cascades involved in cellular homeostasis and its down-regulation has been implicated in the development of prostate cancer. Herein, using a battery of biophysical tools, we show that the binding of EGR1 to DNA is tightly regulated by solution pH. Importantly, the binding affinity undergoes an enhancement of more than an order of magnitude with increasing pH from 5 to 8, implying that the deprotonation of an ionizable residue accounts for such behavior. This ionizable residue is identified as H382 by virtue of the fact that its substitution to non-ionizable residues abolishes pH-dependence of the binding of EGR1 to DNA. Notably, H382 inserts into the major groove of DNA and stabilizes the EGR1-DNA interaction via both hydrogen bonding and van der Waals contacts. Remarkably, H382 is predominantly conserved across other members of EGR1 family, implying that histidine protonation-deprotonation may serve as a molecular switch for modulating protein-DNA interactions central to this family of transcription factors. Collectively, our findings uncover an unexpected but a key step in the molecular recognition of EGR1 family of transcription factors and suggest that they may act as sensors of pH within the intracellular environment.
Keywords: Protein-DNA thermodynamics, Protein dynamics, Zinc fingers, Histidine protonation, Intracellular pH
INTRODUCTION
EGR1 transcription factor, also known as Zif268, bolsters the classical TA-DB modular architecture, where the TA is the N-terminal transactivation domain and DB is the C-terminal DNA-binding domain. Such a modular design exquisitely befits the role of EGR1 in coupling extracellular stimuli such as hormones, neurotransmitters and growth factors to changes in gene expression responsible for a myriad of cellular activities ranging from cell growth and proliferation to apoptosis and oncogenic transformation [1–4]. Importantly, cellular expression of EGR1 is down-regulated in glioblastoma, lymphoma, and cancers of the lung and breast [5–8], implying that EGR1 plays a tumor suppressive role in various cancers. This view is further supported by the observation that tumor suppressors such as PTEN, p53 and TGFβ are direct targets of EGR1 [9–11]. Paradoxically, expression of EGR1 is up-regulated in prostate tumors [12–16], implying that the role of EGR1 is tissue-dependent and that it likely serves as a double-edged sword depending on the biological context.
Regardless of the complexity of physiological actions of EGR1, it primarily exerts its effects by virtue of its ability to bind to the promoters of target genes containing the GCGTGGGCG consensus motif, referred to hereinafter as Zif268 response element (ZRE), in a sequence-dependent manner. The EGR1-DNA interaction is driven by the binding of DB domain as a monomer to the major groove within the ZRE duplex [17]. This mode of DNA-binding is somewhat unusual in that transcription factors usually recognize their promoter elements either as homodimers or heterodimers. However, the DB domain of EGR1 is comprised of three tandem copies of C2H2-type zinc fingers, designated herein ZFI, ZFII and ZFIII, which come together in space to assemble into an arc-shaped architecture that snugly fits into the major groove of DNA (Figure 1a). Importantly, each zinc finger within the DB domain contains an α-helix and an antiparallel double-stranded (β1-β2) β-sheet that together sandwich a Zn2+ divalent ion, the latter being coordinated in a tetrahedral arrangement by two histidine residues and two cysteine residues. Remarkably, the EGR1-DNA interaction is driven by the binding of each zinc finger to one of the three subsites, each subsite being comprised of a trinucleotide sequence, within the 9-bp GCGTGGGCG consensus motif (Figure 1b). The three zinc fingers within the DB domain thus act as a cooperative unit and bind to their cognate DNA in a manner akin to the cooperativity observed between monomeric units of dimeric transcription factors. In particular, at each of the three subsites within the ZRE duplex occupied by one of the three zinc fingers, the protein-DNA contacts are largely afforded by the α-helix, which fits into the major groove of DNA, and β2-strand, which contacts the DNA phosphate backbone (Figure 1a). Notably, β1-strand appears to provide a scaffolding role and makes no discernable contacts with DNA.
Figure 1.

Protein and DNA analysis. (a) Structural model of the DB domain (residues 336–420) of human EGR1 in complex with ZRE duplex containing the GCGTGGGCG consensus sequence. Note that the DB domain is comprised of three tandem C2H2-type zinc fingers, designated herein ZFI (green), ZFII (blue) and ZFIII (magenta). Each zinc finger is comprised of an α-helix and a double-stranded (β1-β2) antiparallel β-sheet that together sandwich a Zn2+ divalent ion indicated by a sphere (cyan). In each zinc finger, the Zn2+ ion is coordinated by two histidine residues and two cysteine residues in a tetrahedral arrangement. The DNA backbone is shown in yellow and the bases are colored gray for clarity. Note also that each zinc finger recognizes a triplet of bases within the 9-bp GCGTGGGCG consensus sequence. The sidechain moieties of amino acid residues E354 (ZFI), H382 (ZFII) and E410 (ZFIII) within the DB domain that contact DNA are shown in red. (b) ZRE duplex containing the GCGTGGGCG tripartite consensus motif. The consensus nucleotides within this motif are capitalized whilst the flanking nucleotides are shown in small letters. The three sub-sites within the consensus motif are marked for clarity and accommodate ZFI (right site), ZFII (middle site) and ZFIII (left site) within the DB domain. The numbering of various nucleotides with respect to the central nucleotide of the middle site (which is arbitrarily assigned zero) are indicated.
Of Particular note is the observation that the binding of DB domain of EGR1 appears to be strongly governed by numerous van der Waals contacts in addition to an extensive network of intermolecular hydrogen bonding and ion pairing [17]. Ironically, detailed examination of the atomic structure of the DB domain of EGR1 in complex with the ZRE duplex shows that an histidine residue (H382), located within the first turn of α-helix (αII) of ZFII but not involved in coordinating the zinc ligand, protrudes deep into the major groove at the protein-DNA interface (Figure 1a). It should be noted that the imidazole ring of H382 is coplanar with G0 and stacks against the pyrimidine ring of T-1 within the ZRE duplex. Importantly, the H382-G0 interaction appears to be stabilized via a two-prong mechanism: firstly, the coplanar alignment of the imidazole ring of H382 and the purine ring of G0, the central guanine of the middle trinucleotide subsite that accommodates ZFII within the DB domain, facilitates the formation of an hydrogen bond between the Hε2 atom of H382 and N7 atom of G0; and secondly, stacking of the imidazole ring of H382 against the pyrimidine ring of T-1 promotes van der Waals contacts between the protein and DNA. Given that pKa values of histidine residues located within the binding and catalytic centers of proteins are frequently perturbed [18, 19], we wondered whether protonation-deprotonation of H382 may be involved in modulating EGR1-DNA interaction in response to changes in solution pH. Importantly, H382 located within ZFII is replaced by a glutamate residue in ZFI (E354) and ZFIII (E410) at the structurally-equivalent positions. Such lack of conservation of H382 in ZFI and ZFIII implicates a unique role of ZFII in dictating the binding of EGR1 to DNA.
In an attempt to test our hypothesis that the protonation-deprotonation of H382 may be involved in modulating EGR1-DNA interactions, we analyzed the pH-dependence of the binding of DB domain of EGR1 to a 15-mer dsDNA oligo containing the ZRE motif using various biophysical tools. Our study shows that the binding of EGR1 to DNA is tightly regulated by solution pH by virtue of the ability of H382 to undergo protonation-deprotonation equilibrium. Remarkably, H382 is predominantly conserved across other members of EGR1 family, implying that histidine protonation-deprotonation may serve as a molecular switch for modulating protein-DNA interactions central to this family of transcription factors. Collectively, our findings uncover an unexpected but a key step in the molecular recognition of EGR1 family of transcription factors and suggest that they may act as sensors of pH within the intracellular environment.
RESULTS and DISCUSSION
Protonation-deprotonation of H382 modulates the binding of EGR1 to DNA
In an attempt to test our hypothesis that H382 within EGR1 may be subject to protonation-deprotonation equilibrium, we measured the effect of varying solution pH ranging from 5 to 8 on the binding of ZRE duplex to wildtype DB (DB_WT) domain of EGR1 using ITC (Figure 2 and Table 1). Our data show that the binding of DB_WT domain to DNA is strongly pH-dependent. Thus, while the DB-WT domain binds to DNA with an affinity of close to 2μM at pH 5, the binding increases by more than an order of magnitude to around 150nM at pH 8 (Table 1). This finding strongly implies that the protonation of an ionizable residue with a pKa close to neutral pH most likely accounts for such enhancement in the binding of DB domain of EGR1 to DNA. To test our hypothesis that H382 serves as the site for such protonation-deprotonation, we next introduced and measured the effect of the binding of DB domain of EGR1 containing the H382A substitution (DB_H382A) to DNA. We anticipate that the H382A substitution should be expected to remove the contribution of the ionizable imidazole moiety of H382 and thereby eliminate the pH-dependence of the binding of DB domain to DNA. Consistent with this rationale, our comparative analysis reveals that while the binding of DB_WT domain to DNA monotonically increases as a function of pH, the binding of DB_H382A domain displays no dependence on solution pH (Figure 3).
Figure 2.
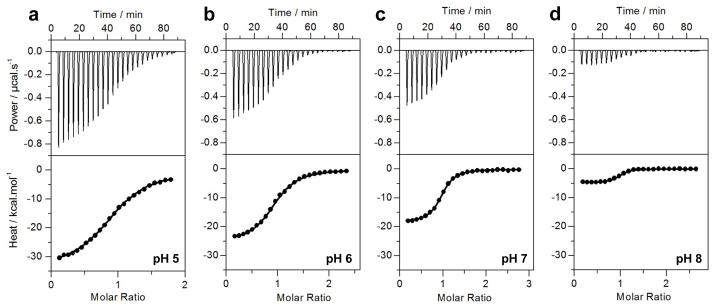
Representative ITC isotherms at 25°C for the binding of ZRE duplex to the DB_WT domain of EGR1 at pH 5 (a), pH 6 (b), pH 7 (c) and pH 8 (d). The upper panels show raw ITC data expressed as change in thermal power with respect to time over the period of titration. In the lower panels, change in molar heat is expressed as a function of molar ratio of ZRE duplex to DB domain. The solid lines in the lower panels show the fit of data to a one-site model, as embodied in Eq [1], using Microcal Origin software.
Table 1.
Thermodynamic parameters for the binding of ZRE duplex to wildtype DB domain (DB_WT) of EGR1 at various pH
| Kd/nM | ΔH/kcal.mol−1 | TΔS/kcal.mol−1 | ΔG/kcal.mol−1 | |
|---|---|---|---|---|
| pH 5.0 | 1962 ± 485 | −33.43 ± 0.73 | −25.63 ± 0.58 | −7.80 ± 0.15 |
| pH 5.5 | 2045 ± 363 | −31.01 ± 0.68 | −23.23 ± 0.57 | −7.77 ± 0.11 |
| pH 6.0 | 806 ± 77 | −25.26 ± 0.30 | −16.94 ± 0.25 | −8.32 ± 0.06 |
| pH 6.5 | 311 ± 30 | −22.49 ± 0.65 | −13.60 ± 0.70 | −8.89 ± 0.06 |
| pH 7.0 | 236 ± 27 | −18.71 ± 0.46 | −9.67 ± 0.39 | −9.05 ± 0.07 |
| pH 7.5 | 186 ± 16 | −14.34 ± 0.31 | −5.15 ± 0.25 | −9.19 ± 0.05 |
| pH 8.0 | 149 ± 15 | −4.53 ± 0.34 | +4.80 ± 0.28 | −9.32 ± 0.06 |
The binding stoichiometries to the fits agreed to within ±10%. Errors were calculated from at least three independent measurements. All errors are given to one standard deviation.
Figure 3.

Dependence of [1/Kd] on pH for the binding of ZRE duplex to DB_WT (black), DB_H382A (red), DB-H382K (green), DB_H382R (blue), DB_H382E (magenta), DB_HH (brown) domains of EGR1 at 25°C. The error bars were calculated from at least three independent measurements to one standard deviation.
To provide further support for our hypothesis, we also introduced and measured the effect of the binding of DB domain of EGR1 containing the H382K (DB_H382K) and H382R (DB_H382R) substitutions to DNA. Notably, these substitutions were introduced to mimic the effect of a protonated histidine containing a net positive charge at H382. As expected, the binding of neither DB_H382K nor DB_H382R domain to DNA exhibited pH-dependence (Figure 3). However, both DB_H382K and DB_H382R domains bound to DNA with affinities similar to those observed for the binding of DB_WT domain at pH 5 in lieu of its enhanced binding at pH 8 (Tables 1 and 2). We believe that this is most likely due to the fact that while H382K and H382R may carry a net positive charge in a manner akin to protonated H382, their non-aromatic sidechain moieties do not structurally resemble the imidazole ring of H382 and therefore unlikely to faithfully substitute its role in its ability to engage in close intermolecular contacts with the DNA. Given that the H382 residue located within the ZFII of DB domain of EGR1 is replaced by a glutamate residue at the structurally-equivalent positions within ZFI (E354) and ZFIII (E410) (Figure 1a), we also wondered how H382E substitution might influence the binding of DB domain to DNA. Toward this goal, we introduced and measured the effect of the binding of DB domain of EGR1 containing the H382E substitution (DB_H382E) to DNA. Consistent with our hypothesis that H382 is responsible for the pH-dependent binding of DB domain to DNA, our data reveal that the binding of DB_H382E domain to DNA is independent of solution pH (Figure 3). Prompted by this promising observation, we next wondered how E354H and E410H substitutions may affect the binding of DB domain of EGR1 to DNA. To accomplish this goal, we introduced and measured the effect of the binding of DB domain of EGR1 containing the E354H/E410H double-substitution (DB_HH) to DNA. Remarkably, the binding of DB_HH domain to DNA displays pH-dependence in a manner that is even stronger than that observed for the DB_WT domain (Figure 3). Thus, while the binding of DB_WT domain to DNA undergoes an eight-fold increase in affinity as solution pH is raised from a value of 5 to 7, the DB_HH domain experiences close to 30-fold enhancement over the same pH range (Table 1 and 3). Taken together, these observations unequivocally demonstrate that the protonation-deprotonation of H382 accounts for the binding of EGR1 to DNA in a pH-dependent manner.
Table 2.
Thermodynamic parameters for the binding of ZRE duplex to wildtype (DB_WT) and various mutant constructs of the DB domain of EGR1 at pH 7.0
| Kd/nM | ΔH/kcal.mol−1 | TΔS/kcal.mol−1 | ΔG/kcal.mol−1 | |
|---|---|---|---|---|
| DB_WT | 236 ± 27 | −18.71 ± 0.46 | −9.67 ± 0.39 | −9.05 ± 0.07 |
| DB_H382A | 812 ± 137 | −14.34 ± 0.69 | −6.02 ± 0.58 | −8.32 ± 0.10 |
| DB_H382K | 1759 ± 160 | −5.20 ± 0.37 | +2.66 ± 0.31 | −7.86 ± 0.05 |
| DB_H382R | 1901 ± 235 | −6.98 ± 0.22 | +0.84 ± 0.14 | −7.81 ± 0.07 |
| DB_H382E | 1492 ± 237 | −2.34 ± 0.14 | +5.62 ± 0.23 | −7.96 ± 0.10 |
| DB_HH | 192 ± 18 | −5.43 ± 0.20 | +3.75 ± 0.26 | −9.17 ± 0.06 |
The binding stoichiometries to the fits agreed to within ±10%. Errors were calculated from three independent measurements. All errors are given to one standard deviation.
Table 3.
Thermodynamic parameters for the binding of ZRE duplex to the double-mutant DB domain containing the E354H/E410H substitutions (DB_HH) of EGR1 at various pH
| Kd/nM | ΔH/kcal.mol−1 | TΔS/kcal.mol−1 | ΔG/kcal.mol−1 | |
|---|---|---|---|---|
| pH 5.0 | 5456 ± 1079 | −21.39 ± 0.61 | −14.20 ± 0.50 | −7.19 ± 0.12 |
| pH 5.5 | 3860 ± 645 | −20.39 ± 0.40 | −12.99 ± 0.29 | −7.40 ± 0.10 |
| pH 6.0 | 3736 ± 1076 | −14.87 ± 0.57 | −7.45 ± 0.74 | −7.42 ± 0.17 |
| pH 6.5 | 2177 ± 696 | −9.37 ± 0.33 | −1.63 ± 0.14 | −7.75 ± 0.19 |
| pH 7.0 | 192 ± 18 | −5.57 ± 0.20 | +3.75 ± 0.14 | −9.17 ± 0.06 |
The binding stoichiometries to the fits agreed to within ±10%. Errors were calculated from at least three independent measurements. All errors are given to one standard deviation.
It is also noteworthy that the enthalpic change associated with the binding of the DB_WT domain of EGR1 to ZRE duplex was observed to be independent of ionization enthalpy of reaction buffers such as phosphate and Tris. This implies that the protonation-deprotonation of H382 in EGR1 is solely dependent upon solution pH and that it is not coupled to DNA-binding. Given that the protonation of H382 hampers the binding of EGR1 to DNA, the lack of such proton-coupled equilibrium to DNA-binding would indeed be thermodynamically unfavorable and thus highly undesirable. In short, our data strongly suggest that the solution pH is likely to play a key regulatory role in fine tuning the binding of EGR1 to DNA under physiological conditions within the living cell.
Binding of EGR1 to DNA is enthalpy-entropy compensated
In addition to the demonstration that the solution pH modulates the binding of EGR1 to DNA, our data also shed light into the underlying thermodynamics governing this key protein-DNA interaction (Figure 4 and Table 1). Interestingly, while enthalpy drives the EGR1-DNA interaction accompanied by opposing entropic forces under physiological pH range, the enthalpic contributions appear to monotonically decrease with increasing pH ranging from 5 to 8 (Figure 4a). Noting that enthalpic contributions most likely result from favorable intermolecular hydrogen bonding, ion pairing and van der Waals forces, the most straightforward interpretation of this finding is that increasing pH disrupts or mitigates the effect of such intermolecular forces on protein-DNA interactions. On the other hand, the entropic contributions to the free energy become more favorable with increasing pH (Figure 4b). Such loss of opposing entropic change most probably results from the change in the interaction of water molecules with protein and DNA with increasing solution pH. It is noteworthy that the release of hydrogen bonded and trapped water within the crevices and cavities in protein and DNA is a major contributor to the favorable entropic change upon association. Accordingly, the changes in solution pH would directly affect the equilibrium between bulk water and trapped water and thereby the entropic contributions to the overall free energy. Notably, the loss of enthalpic contributions is more or less compensated by an equal but opposite favorable increase in entropic contributions such that there is little or no net gain in the overall free energy (Figures 4a and 4b). This reciprocal relationship between enthalpy and entropy lies in the enthalpy-entropy compensation phenomenon [20–24]. Indeed, as shown in Figure 4c, the binding of EGR1 to DNA as a function of pH exquisitely follows such enthalpy-entropy compensation phenomenon.
Figure 4.

Dependence of thermodynamics on pH for the binding of ZRE duplex to DB_WT domain of EGR1 at 25°C. (b) ΔH-pH plot. (b) TΔS-pH plot. (c) TΔS-ΔH plot. The error bars were calculated from at least three independent measurements to one standard deviation.
Neutral pH has little or negligible effect on the structure of the DB domain of EGR1
Given that the binding of EGR1 to DNA is tightly regulated by pH, we next analyzed the extent to which pH may also affect the secondary structure and stability of DB_WT domain using far-UV CD (Figure 5). Our analysis reveals that the far-UV CD spectral features of DB_WT domain are characterized by a positive band centered around 195nm and a negative band centered around 208nm with a shoulder at 225nm (Figure 5a). These observations are consistent with the αβ-fold of the DB domain of EGR1. Importantly, while increasing solution pH from 5 to 6 appears to enhance the spectral intensity of DB_WT domain in the 200–240nm region, there is little or negligible effect on the spectral intensity as the pH is further raised from 6 to 8. This implies that while pH below 6 significantly compromises the structural integrity of the DB_WT domain, its secondary structure undergoes little or no change in the pH range from 6 to 8. Next, to test how changes in secondary structure affect the stability of the DB domain, we probed the dependence of mean ellipticity observed at a wavelength of 222nm, [θ222], as a function of pH over the temperature range 20–100°C using far-UV CD (Figure 5b). In striking contrast to our secondary structural analysis above, our thermal scans suggest that the DB_WT domain displays a melting temperature (Tm) of around 55°C under all pH conditions from 5 to 8. This finding argues that while acidic pH may destabilize the secondary structure of the DB_WT domain, such loss of structure does not necessarily translate into lower thermal stability. Collectively, our far-UV CD analysis shows that while solution pH in the range from 6 to 8 significantly enhances the binding affinity of DB domain of EGR1 toward its cognate DNA by virtue of the ability of H382 to undergo protonation-deprotonation, it has little or negligible effect on its secondary structure and thermal stability. However, it should be borne in mind that far-UV CD is a bulk technique that probes the overall global average structure in lieu of providing information into specific regions or residues which may be under structural fluctuation in a transient manner. Accordingly, our far-UV CD analysis presented above may have overlooked the effect of solution pH on the structure of DB domain at atomic level.
Figure 5.
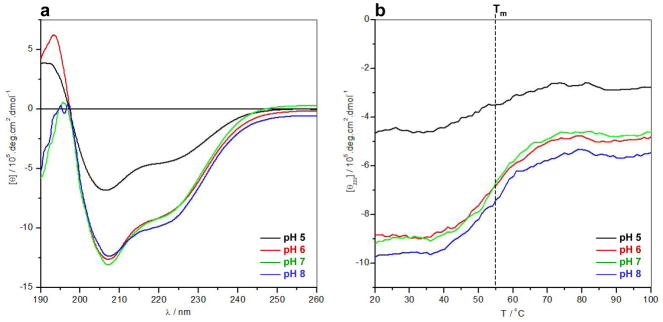
Far-UV CD analysis of DB_WT domain of EGR1 at pH 5 (black), pH 6 (red), pH 7 (green) and pH 8 (blue). (a) Representative far-UV spectra as a function of pH over the wavelength (λ) range 190–260nm at 25°C. (b) Representative melting curves as a function of pH over the temperature (T) range 20–100°C expressed in terms of the mean ellipticity observed at a wavelength of 222nm, [θ222]. Note that the vertical dashed line indicates the melting transition (Tm) of the curves.
Protonation of H382 compromises thermodynamic contacts at protein-DNA interface
In order to rationalize the effect of protonation of H382 on electrostatics at the protein-DNA interface, we generated electrostatic surface potential maps of the DB domain of EGR1 containing H382 in the unprotonated and protonated state in complex with the ZRE duplex (Figure 6). Our data lend interesting insights into how such protonation transforms electrostatic polarization of protein surface at H382 so as to render it thermodynamically less favorable for coming into contact with DNA. In the unprotonated state, H382 occupies what appears to be a largely apolar surface destined to engage in close van der Waals contacts with DNA by virtue of the ability of the imidazole ring of H382 to stack against the pyrimidine ring of T-1 (Figure 6a). Such stacking also results in the coplanar alignment of the imidazole ring of H382 and the purine ring of G0 and thereby facilitates the formation of an hydrogen bond between the Hε2 atom of H382 and N7 atom of G0. Importantly, upon protonation of Nδ1 atom of H382, the local surface becomes positively charged (Figure 6b). Such a scenario would compromise the ability of H382 to engage in intermolecular hydrogen bonding and van der Waals contacts with DNA and thereby eliminate an important thermodynamic component contributing to the free energy driving EGR1-DNA interaction. Accordingly, the unfavorable interactions of the protonated state compared to unprotonated state of H382 would weaken protein-DNA contacts in agreement with our demonstration that increasing pH enhances EGR1-DNA interactions. In short, the aforementioned electrostatic surface potential maps of the DB domain of EGR1 argue strongly that the protonation of H382 would result in the loss of favorable thermodynamic factors that would facilitate the two molecular surfaces to come in close proximity to attain a tight molecular fit.
Figure 6.

Electrostatic surface potential maps of the structural models of the DB_WT domain of EGR1 containing H382 in unprotonated (a) and protonated (b) forms in complex with the ZRE duplex. Note that in the unprotonated form, H382 is protonated only at Nε2 atom within the imidazole ring, while it is protonated at both Nε2 and Nδ1 atoms in the protonated form. The blue and red colors respectively denote the density of positive and negative charges, while the apolar and polar surfaces are indicated by white/gray color on the molecular surfaces. In the expanded views, the location of H382 is clearly marked on the molecular surfaces with the parenthesis indicating the overall charge on this residue under unprotonated (0) and protonated (+) forms. The ZRE duplex is displayed as a “stick” model and colored green for clarity.
Protonation of H382 mitigates structural stability and alters protein-DNA dynamics
Our analysis presented above suggests strongly that the protonation of H382 serves as a molecular switch in modulating the EGR1-DNA interactions. To probe the effect of such protonation on protein stability and dynamics at atomic level, we next conducted MD simulations on the DB domain of EGR1 containing H382 in the unprotonated and protonated state in complex with the ZRE duplex (Figure 7). As shown in Figure 7a, the MD trajectories reveal that while the unprotonated state reaches structural equilibrium after about 20ns with an overall root mean square deviation (RMSD) of ~1.5Å, its structural stability is somewhat compromised upon protonation with an RMSD greater than 2.0Å. This observation suggests that the protonation of H382 most likely destabilizes protein-DNA contacts in agreement with our analysis presented above. While the overall global changes in protein dynamics between protonated and unprotonated forms of DB domain may not appear to be very drastic, a close inspection of how protonation affects the dynamics of αII helix (harboring the H382 residue) within the ZFII of DB domain is telling. Thus, while the average RMSD per residue for αII helix is close to 0.2Å within the unprotonated form of DB domain, it appears to hover around 1.5Å within the protonated form (Figure 7b). This salient observation suggests strongly that while αII helix is highly ordered in the unprotonated form of DB domain, protonation of H382 results in substantial disorder. In agreement with our thermodynamic data presented above, we believe that such order—disorder transition of αII helix upon protonation of H382 severely compromises the protein-DNA contacts resulting in the loss of high-affinity binding.
Figure 7.
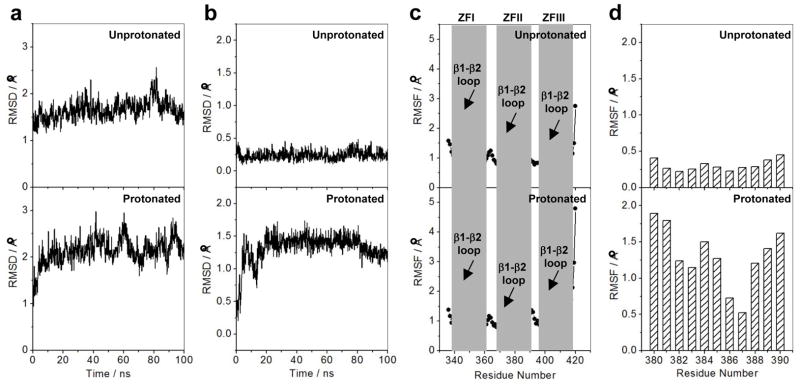
Effect of protonation on the dynamics of DB domain of EGR1 as determined from MD analysis on the structural models of the DB_WT domain of EGR1 containing H382 in unprotonated and protonated forms in complex with the ZRE duplex. Note that in the unprotonated form, H382 is protonated only at Nε2 atom within the imidazole ring, while it is protonated at both Nε2 and Nδ1 atoms in the protonated form. (a) RMSD of backbone atoms (N, Cα and C) within each simulated structure relative to the initial modeled structure of DB domain as a function of simulation time in unprotonated (top panel) and protonated (bottom panel) forms. (b) RMSD per residue within each simulated structure relative to the initial modeled structure for the αII helix (residues 380–390) located within the ZFII of DB domain as a function of simulation time in unprotonated (top panel) and protonated (bottom panel) forms. (c) RMSF of backbone atoms (N, Cα and C) averaged over the entire course of corresponding MD trajectory of DB domain as a function of residue number in unprotonated (top panel) and protonated (bottom panel) forms. Note that the vertical gray boxes denote the boundaries of residues encompassing ZFI, ZFII and ZFIII within the DB domain. The position of β1-β2 loop within each zinc finger is also indicated. (d) RMSF per residue averaged over the entire course of corresponding MD trajectory for the αII helix (residues 380–390) located within the ZFII of DB domain as a function of residue number in unprotonated (top panel) and protonated (bottom panel) forms.
An alternative means to assess mobility and stability of macromolecular complexes is through an assessment of the root mean square fluctuation (RMSF) of specific atoms over the course of MD simulation. In Figure 7c, we provide such analysis for the backbone atoms of each residue within the DB domain. The RMSF analysis reveals that while a majority of residues within the DB domain appear to be well-ordered in both the unprotonated and the protonated state, there are subtle differences within the loop regions. Thus, for example, residues within the β1-β2 loop of ZFI display similar mobilities between the protonated and unprotonated state. In sharp contrast, the mobility of residues within the β1-β2 loop of ZFII is markedly greater in the unprotonated state compared to those in the corresponding loop in the protonated state, while the opposite trend is observed for the residues within the β1-β2 loop of ZFIII. Additionally, residues located within the C-terminal of αIII helix of ZFIII appear to display much greater mobility than the corresponding residues in ZFI and ZFII. This implies that that protonation of H382 located within ZFII not only affects protein dynamics locally but also globally that stretch across all three zinc fingers. Accordingly, this finding argues that the three zinc fingers most likely bind to DNA in a cooperative manner and that such allosteric communication is most likely transmitted through protein dynamics in lieu of structure changes. Of particular note is the observation that the residues 380–390 spanning the αII helix display substantially higher fluctuations in the protonated form relative to the unprotonated form of DB domain (Figure 7d). This further corroborates the notion that the protonation of H382 results in order-disorder transition of αII helix.
Protonation of H382 affects structural integrity of αII helix
In an attempt to further shed light into how protonation of H382 affects the dynamics of αII helix located within ZFII of DB domain, we next analyzed how MD simulations affect its secondary structural features (Figure 8). Notably, protonation of H382 results in a dramatic loss of the propensity of residues 380–390 spanning αII helix to adopt α-helical conformation, particularly those located within the N- and C-terminal regions (Figure 8a). Equally importantly, the loss of such helicity within αII helix exquisitely correlates with changes in the backbone torsion angles φ and ψ (Figure 8b). Thus, while residues spanning αII helix within the unprotonated form respectively adopt φ and ψ torsion angles around −65° and −40° (characteristic of an ideal α helix on the Ramachandran plot), the φ and ψ angles in the protonated form respectively take up values of around −75° and −25°. This observation suggests that upon protonation of H382, the αII helix not only becomes more dynamic but may also resemble some features of 310 helix that typically occupies the bottom left quadrant of the Ramachandran plot with φ and ψ values of around −50° and −25°. This notion gains further credibility in light of our analysis showing that while intramolecular hydrogen bonding network within the αII helix is dominated between residues i and (i+4)—characteristic of an ideal α helix—in the unprotonated form of DB domain, protonation of H382 not only perturbs this network but also favors the formation of hydrogen bonds between residues i and (i+3) that feature heavily in a 310 helix (Figure 8c).
Figure 8.
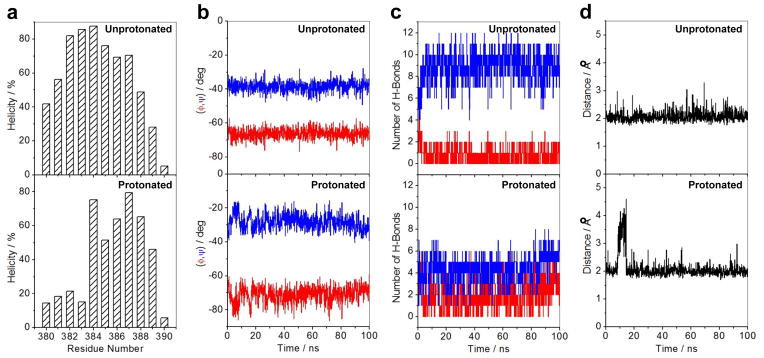
Effect of protonation on the structural stability of αII helix (residues 380–390) located within the ZFII of DB domain of EGR1 as determined from MD analysis on the structural models of the DB_WT domain of EGR1 containing H382 in unprotonated and protonated forms in complex with the ZRE duplex. Note that in the unprotonated form, H382 is protonated only at Nε2 atom within the imidazole ring, while it is protonated at both Nε2 and Nδ1 atoms in the protonated form. (a) Helicity as a function of residue number within the αII helix of ZFII in unprotonated (top panel) and protonated (bottom panel) forms of DB domain. Note that helicity is defined as the percentage of time each residue samples the α-helical conformation over the course of the simulation. (b) Dependence of backbone torsion angles φ (red) and ψ (blue) within the αII helix of ZFII in unprotonated (top panel) and protonated (bottom panel) forms of DB domain. (c) Variation of number of intramolecular backbone hydrogen bonds (H-bonds) between residue i and (i+3) characteristic of an 310-helical conformation (red) as well as between residue i and (i+4) characteristic of an α-helical conformation (blue) observed within the αII helix of ZFII in unprotonated (top panel) and protonated (bottom panel) forms of DB domain. (d) Distance between Hε2 atom (the hydrogen atom directly attached to Nε2) within the imidazole ring of H382 and N7 atom within the guanine base of G0 as a function of time in unprotonated (top panel) and protonated (bottom panel) forms of DB domain. Note that G0, according to the nomenclature shown in Figure 1b, is the central guanine of the middle trinucleotide subsite that accommodates ZFII within the DB domain.
Our thermodynamic data presented above suggest that the protonation of Nδ1 atom of H382 likely results in the disruption of an hydrogen bond between Hε2 atom of H382 and N7 atom of G0. To test this hypothesis using our MD simulations, we also plotted the distance between Hε2 atom and the N7 atom as a function of simulation time (Figure 8d). Our analysis reveals that while this distance remains constant within the unprotonated state of the DB domain at around 2Å throughout the 100-ns simulation cycle, it shows a fluctuation of greater than 4Å after about 10ns in the protonated state. This finding thus supports the notion that the hydrogen bond between Hε2 atom of H382 and N7 atom of G0 is less stable in the protonated state of the DB domain of EGR1. In sum, our MD simulations strongly argue that the protonation of H382 results in marked changes associated with protein dynamics and, in particular, results in the order-disorder transition of αII helix. It is important to note here that while our far-UV CD analysis presented above suggests that the solution pH has little or negligible effect on the secondary structure and thermal stability of DB domain, our MD simulations have provided key insights into how protonation of H382 alters protein structure and stability at atomic level.
pH-dependent binding to DNA appears to be a hallmark of all members of the EGR1 and the related KLF family
In an attempt to analyze the extent to which modulation of DNA-binding through protonation of H382 in EGR1 may also be shared by other members, we generated amino acid sequence alignment of the DB domains of all known members of the human EGR family and the related KLF family (Figure 9). It should be noted that the DB domains of all members of the EGR and KLF families are characterized by the presence of three tandem copies of C2H2-type zinc fingers, designated herein ZFI, ZFII and ZFIII, which are all expected to come together in space to assemble into an arc-shaped architecture so as to snugly fit into the major groove of DNA in a manner akin to the binding of EGR1 (Figure 1a). Importantly, our analysis reveals that the DB domains of all four members of EGR family (EGR1-EGR4) are remarkably well-conserved and display close to 80% sequence identity. However, the EGR family shares only around 35% sequence identity with the DB domains of KLF family members (KLF1-KLF17). Accordingly, these differences at amino acid sequence level must define the precise mechanism and differential specificity of recognition of DNA promoter elements by the EGR/KLF family members.
Figure 9.

Amino acid sequence alignment of DB domains of all known members of the human EGR1 family (EGR1-EGR4) and the related human KLF family (KLF1-KLF17). Absolutely conserved residues are shown in red, non-conserved residues at structurally-equivalent positions occupied by E354, H382 and E410 in EGR1 are colored blue, while all other residues are depicted in black. Each member is denoted by its acronym in the left column with the corresponding UniProt code provided in the right column for access to complete proteomic details on each member. The numerals denote the amino acid boundaries of DB domains for each member. The cysteine and histidine residues within each of the three C2H2-type zinc fingers of DB domains, denoted ZFI, ZFII and ZFIII, that coordinate the Zn2+ divalent ion in a tetrahedral arrangement are marked by asterisks. The vertical arrows indicate the position of E354/H382/E410 in EGR1 and their structural-equivalents in other DB domains.
Notably, while H382 within ZFII of EGR1 is fully conserved in all other EGR members, it is replaced by a glutamate at the structurally-equivalent position within ZFII of all members of KLF family. Strikingly, while the glutamate residues located at the structurally-equivalent position to H382 within ZFI (E354) and ZFIII (E410) of EGR1 are fully conserved within other members of EGR family, they are replaced by an histidine residue within all KLF members. Simply put, while the H382 within ZFII is not conserved in the KLF family members, the latter have evolved to acquire a structurally-equivalent histidine within ZFI and ZFII. Accordingly, this salient observation implies that these conserved histidine residues are likely to be subject to protonation-deprotonation in response to changes in solution pH. On the basis of this argument, we anticipate that the binding of all members of EGR and KLF families to DNA must be tightly regulated by solution pH. Importantly, our analysis also predicts that unlike the members of EGR family, the KLF members contain not one but two potential sites of protonation. Accordingly, the solution pH may play an even more intricate role in modulating the binding of the members of KLF family to DNA. This notion is supported by our thermodynamic data indicating that the binding of the DB_HH domain of EGR1 to DNA shows much stronger dependence on solution pH than the DB_WT domain (Tables 1 and 3).
CONCLUSIONS
The role of EGR1 in regulating a myriad of cellular activities ranging from cell growth and proliferation to apoptosis and oncogenic transformation is well-documented [1–4]. Our demonstration here that solution pH is likely to modulate the binding of EGR1 to DNA, and therefore by extension its transcriptional activity, adds another dimension to the functional complexity of this key player in cellular signaling. Tellingly, changes in intracellular pH regulate a plethora of cellular processes such as metabolic homeostasis and apoptosis [25]. Moreover, it is well-documented that ionizable residues within proteins sense such changes and activate a variety of proton pumps and ion transporters that in turn mediate extracellular transport of protons and anions to regulate intracellular pH [26–28]. Accordingly, it is tempting to speculate that changes in intracellular pH may also tightly regulate the transcriptional activity of EGR1 through modulating the ionization state of H382 located at the protein-DNA interface. Importantly, protonation-deprotonation of H382 would have important consequences on the contributions of intermolecular hydrogen bonding and van der Waals forces to the free energy available to drive this key protein-DNA interaction. It is noteworthy that the protonation-deprotonation of H382 may not necessarily require large changes but rather may be mediated by small changes in intracellular pH. In particular, under pathological states such as metabolic acidosis or alkalosis, the transcriptional activity of EGR1 is likely to be substantially altered. This would likely have serious consequences on cellular signaling cascades that rely on EGR1 for coupling extracellular information in the form of hormones, neurotransmitters and growth factors to changes in gene expression of specific target proteins.
In short, our study demonstrates that the solution pH tightly modulates the binding of EGR1 transcription factor to DNA by virtue of the ability of H382 to serve as a protonation-deprotonation site. Given that H382 is conserved in the structurally-equivalent positions within the C2H2-type zinc fingers of other members of EGR family as well as the related KLF family, our findings have important implications on the physiological function of these proteins. Our study invokes the notion that this group of transcription factors may also act as sensors of intracellular pH and thus bears important consequences for a paradigm shift of their molecular action.
EXPERIMENTAL PROCEDURES
Protein preparation
The wildtype DB (DB_WT) domain (residues 331–430) of human EGR1 (UniProt# P18146) was cloned into pET30 bacterial expression vector with an N-terminal His-tag using Novagen LIC technology (Novagen, Madison, WI, USA). The single-mutants of the DB domain of EGR1 containing H382A (DB_H382A), H382K (DB_H382K), H382R (DB_H382R) and H382E (DB_H382E) substitutions as well as the double-mutant containing the E354H/E410H (DB_HH) substitutions were generated through de novo DNA synthesis courtesy of GenScript Corporation (GenScript, Piscataway, NJ, USA) and subsequently cloned into pET30 bacterial expression as described for the DB_WT domain. Additionally, a tryptophan residue was added at both the N- and C-termini of the wildtype and mutant constructs to aid in protein characterization upon purification due to the fact that the DB domain of EGR1 does not contain a native tryptophan. All recombinant proteins were subsequently expressed in Escherichia coli BL21*(DE3) bacterial strain (Invitrogen, Carlsbad, CA, USA) and purified on a Ni-NTA affinity column as described previously for other systems from our laboratory [29–31]. Briefly, bacterial cells were grown at 20°C in Terrific Broth supplemented with 50μM ZnCl2 to an optical density of greater than unity at 600nm prior to induction with 0.5mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The bacterial culture was further grown overnight at 20°C and the cells were subsequently harvested and disrupted using a BeadBeater (Biospec, Bartlesville, OK, USA). After separation of cell debris using high-speed centrifugation, the cell lysate was loaded onto a Ni-NTA column and washed extensively with 20mM imidazole to remove non-specific binding of bacterial proteins to the column. The recombinant proteins were subsequently eluted with 200mM imidazole and dialyzed against an appropriate buffer to remove excess imidazole. The proteins were further passed through a Hiload Superdex 200 size-exclusion chromatography (SEC) column coupled in-line with GE Akta FPLC system (GE Healthcare, Milwaukee, WI, USA). This final step led to purification of recombinant proteins to apparent homogeneity as judged by SDS-PAGE analysis. Final yields were typically between 5–10mg protein of apparent homogeneity per liter of bacterial culture. Protein concentration was determined by the fluorescence-based Quant-It assay (Invitrogen, Carlsbad, CA, USA) and spectrophotometrically using an extinction coefficient of 12,865 M−1cm−1 calculated using the online software ProtParam at ExPasy Server. Results from both methods were in a good agreement.
DNA synthesis
15-mer DNA oligos containing the ZRE consensus site (GCGTGGGCG) were commercially obtained from Sigma Genosys (The Woodlands, TX, USA). The complete nucleotide sequence of the sense and antisense oligos constituting the ZRE duplex is presented in Figure 1b. Oligo concentrations were determined spectrophotometrically on the basis of their extinction co-efficients derived from their nucleotide sequences using the online software OligoAnalyzer 3.1 (Integrated DNA Technologies). Equimolar amounts of sense and antisense oligos were mixed together and heated at 95°C for 10min and then allowed to cool to room temperature to obtain double-stranded DNA (dsDNA) annealed oligos (ZRE duplex).
ITC measurements
Isothermal titration calorimetry (ITC) experiments were performed on a TA Nano-ITC instrument (New Castle, DE, USA). Briefly, wildtype and mutant DB domains of EGR1 and the ZRE dsDNA oligos were either dialyzed in 50mM Sodium acetate (for measurements conducted below pH 6) or 50mM Sodium phosphate (for measurements conducted at or above pH 6) containing 100mM NaCl and 5mM β-mercaptoethanol at a specified pH. All experiments were initiated by injecting 25 × 10μl aliquots of 100μM of ZRE duplex from the syringe into the calorimetric cell containing 0.95ml of 10–20μM of DB domain solution at 25°C. The change in thermal power as a function of each injection was automatically recorded using the integrated NanoAnalyze software. The raw data were further integrated to yield binding isotherms of heat release per injection as a function of molar ratio of ZRE duplex to DB domain. The heats of mixing and dilution were subtracted from the heat of binding per injection by carrying out a control experiment in which the same buffer in the calorimetric cell was titrated against the ZRE duplex in an identical manner. Control experiments with scrambled dsDNA oligos generated similar thermal power to that obtained for the buffer alone, implying that there was no non-specific binding of DB domain to non-cognate DNA sequences. To determine the binding constant (Kd) and the binding enthalpy (ΔH), the binding isotherms were iteratively fit to the following built-in function by non-linear least squares regression analysis using the integrated NanoAnalyze software:
| [1] |
where q(i) is the heat release (kcal/mol) for the ith injection, n is the binding stoichiometry, V is the effective volume of protein solution in the calorimetric cell (0.95ml), P is the concentration of each DB domain in the calorimetric cell (μM) and L is the concentration of ZRE duplex added (μM). It should be noted that Eq[1] is derived using the law of mass action assuming one-site binding model [32]. The free energy change (ΔG) upon DNA binding was calculated from the relationship:
| [2] |
where R is the universal molar gas constant (1.99 cal/mol/K) and T is the absolute temperature (298 K). The entropic contribution (TΔSobs) to the free energy of binding was calculated from the relationship:
| [3] |
Circular dichroism
Circular dichroism (CD) measurements were conducted on a Jasco J-815 spectropolarimeter (Jasco, Easton, MD, USA) thermostatically controlled at 25°C. Briefly, the wildtype and mutant DB domains of EGR1 were prepared in 50mM Sodium phosphate at a specified pH ranging from 5 to 8. Experiments were conducted on 10μM of protein and data were collected using a quartz cuvette with a 2-mm pathlength in the 190–260nm wavelength range. All data were recorded with a slit bandwidth of 2nm at a scan rate of 10nm/min and normalized against reference spectra to remove the background contribution of buffer. Each spectral data set represents an average of four scans acquired at 0.1nm intervals. Data were converted to mean ellipticity, [θ], as a function of wavelength (λ) of electromagnetic radiation using the equation:
| [4] |
where Δθ is the observed ellipticity in mdeg, c is the protein concentration in μM and l is the cuvette pathlength in cm. For temperature scans of wildtype and mutant DB domains of EGR1 to generate melting curves, the spectral intensity at a wavelength of 222nm was monitored in the temperature range 20–100°C at a scan rate of 1°C/min. It is noteworthy that the introduction of various single- and double-mutations did not lead to any substantial changes in the structure or stability of the DB domain.
Molecular modeling
Molecular modeling (MM) was employed to build a structural model of the DB domain of EGR1 in complex with the 15-mer ZRE duplex. Briefly, the structural model was built in two stages: first, the double-helical B-DNA conformation of the ZRE duplex was obtained on the basis of de novo modeling using 3D-DART [33]. Next, the crystal structure of DB domain of EGR1 in complex with a dsDNA oligo containing the ZRE consensus motif (PDB# 1ZAA), but with varying flanking sequences, and the de novo model of ZRE duplex were used as templates in a multi-template alignment fashion to calculate the overall structural model of the protein-DNA complex using the MODELLER software based on homology modeling [34]. A total of 100 structural models were calculated and the structure with the lowest energy, as judged by the MODELLER Objective Function, was selected for further analysis. The structural model was rendered using RIBBONS [35] and the electrostatic surface potentials maps were generated using MOLMOL [36].
Molecular dynamics
Molecular dynamics (MD) simulations on the structural models of the DB domain of EGR1 in unprotonated and protonated forms with respect to H382 in complex with the ZRE duplex were performed with the GROMACS software [37, 38] using the integrated AMBER99SB-ILDN force field [39, 40]. Briefly, the modeled structure of the DB domain of EGR1 was subjected to GROMACS and pre-protonated either at only Nε2 atom (unprotonated form) or protonated at both Nε2 and Nδ1 atoms (protonated form) within the imidazole ring of H382. Next, each structure was centered within a cubic box, hydrated using the extended simple point charge (SPC/E) water model [41, 42], and the ionic strength of solution was set to 100mM with NaCl. The hydrated structures were energy-minimized with the steepest descent algorithm prior to equilibration under the NPT ensemble conditions, wherein the number of atoms (N), pressure (P) and temperature (T) within the system were respectively kept constant at ~50000, 1 bar and 300 K. The Particle-Mesh Ewald (PME) method was employed to compute long-range electrostatic interactions with a 10Å cut-off [43] and the Linear Constraint Solver (LINCS) algorithm to restrain bond lengths [44]. All MD simulations were performed under periodic boundary conditions (PBC) using the leap-frog integrator with a time step of 2fs. For the final MD production runs, data were collected every 100ps over a time scale of 100ns. All simulations were run on a Linux workstation using parallel processors at the High Performance Computing facility within the Center for Computational Science of the University of Miami.
Acknowledgments
The authors remain deeply indebted to Mansoor Ahmed for many critical discussions and helpful suggestions. We thank the Sylvia Daunert Group for the use of its Jasco J-815 spectropolarimeter. This work was supported by funds from the National Institutes of Health (Grant# R01-GM083897) and the USylvester Braman Family Breast Cancer Institute to AF. CBM is a recipient of a postdoctoral fellowship from the National Institutes of Health (Award# T32-CA119929).
ABBREVIATIONS
- CD
Circular dichroism
- DB
DNA-binding (domain)
- EGR
Early growth response
- EGR1
Early growth response (protein) 1
- ITC
Isothermal titration calorimetry
- KLF
Krueppel-like factor
- LIC
Ligation-independent cloning
- MD
Molecular dynamics
- MM
Molecular modeling
- SEC
Size-exclusion chromatography
- RMSD
Root mean square deviation
- RMSF
Root mean square fluctuation
- TA
Transactivation (domain)
- ZF
Zinc finger
- ZRE
Zif268 (EGR1) response element
References
- 1.Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 2.Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed MM. Regulation of radiation-induced apoptosis by early growth response-1 gene in solid tumors. Curr Cancer Drug Targets. 2004;4:43–52. doi: 10.2174/1568009043481704. [DOI] [PubMed] [Google Scholar]
- 4.Adamson ED, Mercola D. Egr1 transcription factor: multiple roles in prostate tumor cell growth and survival. Tumour Biol. 2002;23:93–102. doi: 10.1159/000059711. [DOI] [PubMed] [Google Scholar]
- 5.Levin WJ, Press MF, Gaynor RB, Sukhatme VP, Boone TC, Reissmann PT, Figlin RA, Holmes EC, Souza LM, Slamon DJ. Expression patterns of immediate early transcription factors in human non-small cell lung cancer. The Lung Cancer Study Group. Oncogene. 1995;11:1261–1269. [PubMed] [Google Scholar]
- 6.Joslin JM, Fernald AA, Tennant TR, Davis EM, Kogan SC, Anastasi J, Crispino JD, Le Beau MM. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood. 2007;110:719–726. doi: 10.1182/blood-2007-01-068809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. 1997;72:102–109. doi: 10.1002/(sici)1097-0215(19970703)72:1<102::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 8.Calogero A, Arcella A, De Gregorio G, Porcellini A, Mercola D, Liu C, Lombari V, Zani M, Giannini G, Gagliardi FM, et al. The early growth response gene EGR-1 behaves as a suppressor gene that is down-regulated independent of ARF/Mdm2 but not p53 alterations in fresh human gliomas. Clin Cancer Res. 2001;7:2788–2796. [PubMed] [Google Scholar]
- 9.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol. 2001;3:1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 10.Krones-Herzig A, Mittal S, Yule K, Liang H, English C, Urcis R, Soni T, Adamson ED, Mercola D. Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 2005;65:5133–5143. doi: 10.1158/0008-5472.CAN-04-3742. [DOI] [PubMed] [Google Scholar]
- 11.Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. doi: 10.1038/sj.cgt.7700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thigpen AE, Cala KM, Guileyardo JM, Molberg KH, McConnell JD, Russell DW. Increased expression of early growth response-1 messenger ribonucleic acid in prostatic adenocarcinoma. J Urol. 1996;155:975–981. [PubMed] [Google Scholar]
- 13.Eid MA, Kumar MV, Iczkowski KA, Bostwick DG, Tindall DJ. Expression of early growth response genes in human prostate cancer. Cancer Res. 1998;58:2461–2468. [PubMed] [Google Scholar]
- 14.Baron V, De Gregorio G, Krones-Herzig A, Virolle T, Calogero A, Urcis R, Mercola D. Inhibition of Egr-1 expression reverses transformation of prostate cancer cells in vitro and in vivo. Oncogene. 2003;22:4194–4204. doi: 10.1038/sj.onc.1206560. [DOI] [PubMed] [Google Scholar]
- 15.Baron V, Duss S, Rhim J, Mercola D. Antisense to the early growth response-1 gene (Egr-1) inhibits prostate tumor development in TRAMP mice. Ann N Y Acad Sci. 2003;1002:197–216. doi: 10.1196/annals.1281.024. [DOI] [PubMed] [Google Scholar]
- 16.Virolle T, Krones-Herzig A, Baron V, De Gregorio G, Adamson ED, Mercola D. Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J Biol Chem. 2003;278:11802–11810. doi: 10.1074/jbc.M210279200. [DOI] [PubMed] [Google Scholar]
- 17.Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 18.Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 19.Rostkowski M, Olsson MH, Sondergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol. 2011;11:6. doi: 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumry R, Rajender S. Enthalpy-entropy compensation phenomena in water solutions of proteins and small molecules: a ubiquitous property of water. Biopolymers. 1970;9:1125–1227. doi: 10.1002/bip.1970.360091002. [DOI] [PubMed] [Google Scholar]
- 21.Eftink MR, Anusiem AC, Biltonen RL. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: general thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry. 1983;22:3884–3896. doi: 10.1021/bi00285a025. [DOI] [PubMed] [Google Scholar]
- 22.Cooper A, Johnson CM, Lakey JH, Nollmann M. Heat does not come in different colours: entropy-enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions. Biophys Chem. 2001;93:215–230. doi: 10.1016/s0301-4622(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 23.Sharp K. Entropy-enthalpy compensation: fact or artifact? Protein Sci. 2001;10:661–667. doi: 10.1110/ps.37801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starikov EB, Norden B. Enthalpy-entropy compensation: a phantom or something useful? J Phys Chem B. 2007;111:14431–14435. doi: 10.1021/jp075784i. [DOI] [PubMed] [Google Scholar]
- 25.Boron WF. Regulation of intracellular pH. Adv Physiol Educ. 2004;28:160–179. doi: 10.1152/advan.00045.2004. [DOI] [PubMed] [Google Scholar]
- 26.Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K, Fisher RJ, Fliegel L, Durum SK. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol. 2001;21:7545–7557. doi: 10.1128/MCB.21.22.7545-7557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khaled AR, Reynolds DA, Young HA, Thompson CB, Muegge K, Durum SK. Interleukin-3 withdrawal induces an early increase in mitochondrial membrane potential unrelated to the Bcl-2 family. Roles of intracellular pH, ADP transport, and F(0)F(1)-ATPase. J Biol Chem. 2001;276:6453–6462. doi: 10.1074/jbc.M006391200. [DOI] [PubMed] [Google Scholar]
- 28.Puceat M, Roche S, Vassort G. Src family tyrosine kinase regulates intracellular pH in cardiomyocytes. J Cell Biol. 1998;141:1637–1646. doi: 10.1083/jcb.141.7.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deegan BJ, Seldeen KL, McDonald CB, Bhat V, Farooq A. Binding of the ERalpha nuclear receptor to DNA is coupled to proton uptake. Biochemistry. 2010;49:5978–5988. doi: 10.1021/bi1004359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deegan BJ, Bhat V, Seldeen KL, McDonald CB, Farooq A. Genetic variations within the ERE motif modulate plasticity and energetics of binding of DNA to the ERalpha nuclear receptor. Arch Biochem Biophys. 2011;507:262–270. doi: 10.1016/j.abb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhat V, Kurouski D, Olenick MB, McDonald CB, Mikles DC, Deegan BJ, Seldeen KL, Lednev IK, Farooq A. Acidic pH promotes oligomerization and membrane insertion of the BclXL apoptotic repressor. Arch Biochem Biophys. 2012;528:32–44. doi: 10.1016/j.abb.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 33.van Dijk M, Bonvin AM. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 2009;37:W235–239. doi: 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative Protein Structure Modeling of Genes and Genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 35.Carson M. Ribbons 2.0. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 36.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 38.Hess B. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 39.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toukan K, Rahman A. Molecular-dynamics study of atomic motions in water. Physical Review B. 1985;31:2643–2648. doi: 10.1103/physrevb.31.2643. [DOI] [PubMed] [Google Scholar]
- 42.Berendsen HJC, Grigera JR, Straatsma TP. The Missing Term in Effective Pair Potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 43.Darden TA, York D, Pedersen L. Particle mesh Ewald: An N. log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 44.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]