

Abstract
Histidine-containing phosphotransfer proteins (HPts) take part in hormone signal transduction in higher plants. The overall pathway of this process is reminiscent of the two-component system initially identified in prokaryotes. HPts function in histidine-aspartate phosphorelays where they mediate the signal from sensory kinases (usually membrane proteins) to response regulators in the nucleus. Here we report the crystal structure of an HPt protein from Medicago truncatula (MtHPt1) determined at 1.45 Å resolution and refined to an R factor of 16.7% using low-temperature synchrotron-radiation X-ray diffraction data. There is one MtHPt1 molecule in the asymmetric unit of the crystal lattice with P212121 symmetry. The protein fold consists of six α-helices, four of which form a C-terminal helix bundle. The coiled-coil structure of the bundle is stabilized by a network of S-aromatic interactions involving highly conserved sulfur-containing residues. The structure reveals a solvent-exposed side chain of His79, which is the phosphorylation site, as demonstrated by autoradiography combined with site-directed mutation. It is surrounded by highly conserved residues present in all plant HPts. These residues form a putative docking interface for either the receiver domain of the sensory kinase, or for the response regulator. The biological activity of MtHPt1 was tested by autoradiography. It demonstrated phosphorylation by the intracellular kinase domain of the cytokinin receptor MtCRE1. Complex formation between MtHPt1 and the intracellular fragment of MtCRE1 was confirmed by thermophoresis, with a dissociation constant Kd of 14 μM.
Keywords: HPt, Histidine-containing phosphotransfer protein, histidine-aspartate phosphorelay, two-component relay system, phytohormone, cytokinin, hormone signal transduction, legume plants, MtCRE1, cytokinin receptor
Introduction
Phosphorylation is a ubiquitous regulation mechanism used by all living organisms. Eukaryotic pathways usually involve either tyrosine or serine/threonine phosphorylation by kinases. In prokaryotes, a two-component system consisting of a histidine-aspartate (His-Asp) phosphorelay is responsible for signal transduction initiated by various stimuli [1]. A very similar process operates in fungi and plants [2], despite the evolutionary divergence of these kingdoms. Beside their obvious biochemical differences, bacteria, fungi and plants have to face the same problem of adaptation to environmental conditions. This can explain why the two-component systems is absent only in animals.
The eukaryotic cascade is more complex and consists of three elements: (i) a hybrid sensory histidine kinase that is anchored in the plasma membrane, (ii) a histidine-containing phosphotransfer protein (HPt), and (iii) a response regulator (RR) (Fig. 1). The sensory kinase is comprised of three domains, with the following N- to C-terminal arrangement: an extracellular cyclases/histidine kinases-associated sensory (CHASE) domain, and of two intracellular parts, namely histidine kinase (HK) and receiver (REC) domains. The signal transduction pathway is triggered by ligand binding to the CHASE domain. As a result, the receptor gains its kinase activity and the HK domain autophosphorylates a His residue, which in turn phosphorylates an Asp residue in the receiver domain. Subsequently, the phosphate group is transferred to a His residue of the HPt protein and passed downstream to an Asp residue in the RR protein. The latter initiates the final response by binding to a specific DNA element or to a target protein.
Fig. 1.

Model of cytokinin signal transduction pathway. The cytokinin receptor has the following domains: CHASE, cyclases/histidine kinases-associated sensory extracellular domain; HK, histidine kinase domain; REC, receiver domain. The asterisk designates the intracellular portion of the sensory kinase receptor (consisting of HK and REC), in this paper referred to as MtCRE1′. Histidine-containing phosphotransfer proteins (HPts) are activated by the sensory kinase that perceives the signal, and subsequently translocate to the nucleus to transfer the phosphate group to an Asp residue of a response regulator (RR). Adapted with modifications from [6].
In higher plants, it can be environmental stress or a hormone signal that starts the cascade. Ethylene [3] and cytokinins [4–6] are hormones known to trigger this type of response pathways. In this study, we focus on an HPt element from the cytokinin transduction pathway. It mediates phosphate group transfer from cytokinin receptor CRE1 to one of the RR proteins (Fig. 1).
There are some structures of plant HPt proteins deposited in the Protein Data Bank (PDB): 1yvi [Center for Eukaryotic Structural Genomics, unpublished], 2q4f [7], or 1wn0 [8], however, all of them were solved at medium resolution, not higher than 2.0 Å. In the present study, we describe a high-resolution (1.45 Å) crystal structure of the first monomeric plant HPt protein, from Medicago truncatula (MtHPt1). It is also the only example of an HPt protein from a legume plant. The designation “1” in the name acronym indicates that MtHPt1 has the highest (69%) amino acid sequence identity (among the seven M. truncatula sequences known [9]) to the standard A. thaliana histidine-containing phosphotransfer protein AHP1. Importantly, we were able to demonstrate in two assays that the MtHPt1 protein used for the crystallization experiments has the biologically relevant structure. Firstly, the enzymatic phosphorylation of MtHPt1 by the intracellular fragment of the M. truncatula Cytokinin Receptor (MtCRE1′) was successfully performed, as confirmed by autoradiography with 32P-labeled phosphate. Secondly, a thermophoretic analysis of the MtCRE1′:MtHPt1 protein complex permitted the determination of the dissociation constant of the complex, Kd.
Results and discussion
Overall structure of MtHPt1
The protein crystallizes in the orthorhombic P212121 space group with one protein molecule in the asymmetric unit. It does not form any quaternary structure and is monomeric in solution, as confirmed by size exclusion chromatography. Also the PDBe PISA web server [10] predicts no multimeric assemblies of MtHPt1. The complete sequence of MtHPt1 is comprised of 153 amino acid residues and its molecular weight is 17.8 kDa. The Matthews coefficient (Vm) is 2.1 Å3/Da, which corresponds to 41.3% solvent content.
The N-terminal methionine (plus an SNA- cloning artifact) and four residues at the C terminus (-MELN) are missing in the electron density maps. The remaining residues of the model have excellent definition in electron density. The entire Glu2-Met149 main chain of the native protein and the majority of side chains could be traced without ambiguity. Eleven side chains were modeled in double conformation. The refined model includes 159 water molecules and no metal cations.
The fold of MtHPt1 is entirely α-helical and consists of six α-helices (Fig. 2A, B). Their lengths vary from 8 (α2) to 34 residues (α6). Four of the helices (α3, α4, α5 and α6) form an antiparallel coiled-coil four-helix bundle at the C terminus (Fig. 2C). In general, 116 (78%) out of the 148 amino acids that are present in the coordinate set, have α-helical conformation. Helix α3 is kinked and leans away from the protein core. As a result, the characteristic 3.613 α-helical hydrogen-bonding pattern is distorted.
Fig. 2.

Overall structure of MtHPt1. A Secondary structure elements (helices α1- α6) corresponding to the amino acid sequence. Five β and two γ turns are also labeled. B A ribbon diagram illustrating the six α-helices and the phosphorylation site His79. Four of the helices (α3-α6) form a helix bundle. C View down the axis of the four-helix bundle. The His79 side chain of the active site is highly exposed to the solvent.
All main-chain ϕ/ψ angles map to the most favored or allowed regions of the Ramachandran plot. There are five n+3 (β) turns. They involve residues 21–24, 35–38, 36–39, 38–41 and 66–69. The three overlapping turns map to the longest stretch of disordered residues, within loop 33–41. There are also two n+2 (γ) turns in the structure, between residues 69–71 and 89–91.
Sulfur-aromatic interactions in MtHPt1
The protein has an unusually high fraction (6.5%) of sulfur-containing residues (six Met and four Cys). None of the cysteine residues create a disulfide bond. Even though the side chains of Cys104 and Cys112 point toward each other, the distance between their S atoms (>4 Å) precludes such an interaction. Many of the S atoms, including the two mentioned above, interact with aromatic residues [11]. It is known that these interactions are strongest when the S-centroid distance is less than 4 Å with the sulfur atom lying on the normal to the aromatic ring plane [12]. They can stabilize ligand binding and participate in the enzymatic mechanism, as observed e.g. in NodS N-methyltransferase from B. japonicum in complex with S-adenosyl-L-homocysteine (SAH) [13]. In the core region of protein structures, however, the S-aromatic interactions are observed most frequently with the S-centroid line inclined to the aromatic ring at an angle of less than 60° and a distance of up to 6 Å [11, 12]. This is also the case of MtHPt1. Within helix α1 there is an interaction between Met17 and Tyr13 (Fig. 3A). There are also six S-aromatic interactions within the four-helix bundle (Fig. 3B), evidently contributing to the integrity of the bundle. The interaction between Phe81 and Cys97 links the helices α4 and α5 together. Residues Phe71, Phe100, Phe103, Cys104, Cys112 and Cys115 form a network of S-aromatic interactions that stabilize one end of the four-helix bundle. Phe100, Cys104 and Cys115 in this network are modeled with dual conformations but for each alternative an S-aromatic interaction is formed (Fig. 3B). The residues forming the S-aromatic interactions in MtHPt1 are highly conserved among homologous sequences (Fig. S1). In particular, the residues at positions corresponding to Phe71, Cys97, Phe100, Cys104, Cys112 and Cys115 in MtHPt1 are nearly universally conserved in the plant orthologs. Such a high degree of conservation suggests an indispensable role in the protein structure and function. It can be hypothesized that these interactions have evolved to stabilize the coiled-coil structure of this class of proteins.
Fig. 3.

Sulfur-aromatic interactions (dash lines) in MtHPt1. A Interaction between Met17 and Tyr13 within helix α1. B Six S-aromatic bonds in the hydrophobic core of the four-helix bundle. The network of these interactions is created by residues Phe71, Phe81, Cys97, Phe100, Phe103, Cys104, Cys112 and Cys115. Phe100, Cys104 and Cys115 are modeled with alternative conformations, but in each case an S-π bond is formed. The S-centroid distances to the aromatic rings are given in Å. The 2Fo-Fc electron density map is contoured at 1σ.
Comparison with other HPt proteins
HPt proteins are found in bacteria, fungi and plants. A phylogram of amino acid sequences from these kingdoms is shown in Fig. 4 (see Fig. S1 for full alignment). In this article, the numbering of residues and helices corresponds to MtHPt1. In addition to the obvious systematic division, it is also useful to divide the HPt proteins into enzymatically active and inactive orthologs. The active orthologs have a conserved histidine residue in a position corresponding to His79 in MtHPt1 that acts as the phosphorelay center. The identity of the enzymatically active residue can be deduced by analogy to the system studied in maize [14]. It has been additionally verified by site-directed mutagenesis (H79N) in the present work. Also, in a complex of S. cerevisiae osmosensing histidine protein kinase RR domain (SLN1-R1) and phosphorelay intermediate protein 1 (YPD1), the phosphotransfer reaction was captured in the crystal structure [15], with the BeF3− phosphate mimic observed next to this His residue. Proteins belonging to the second HPt group (inactive orthologs) have Asn instead of the active His residue. A. thaliana histidine-containing phosphotransfer protein 6 (AHP6) is an example of an inactive ortholog. According to the results of functional studies, that protein acts as an inhibitor of the cytokinin receptor [16]. Since AHP6 is unable to transfer the phosphate group, it functions to block the cytokinin signaling pathway. In the M. truncatula genome, two proteins are encoded that could potentially function in this inhibitory manner. In this paper we term them MtHPt6a and MtHPt6b.
Fig. 4.

A phylogram of amino acid sequences of HPt proteins [UniProt accession numbers in brackets]. Source organisms: E. coli (ArcB) [C6EH17]; A. thaliana (AHP1-6) [Q9ZNV9, Q9ZNV8, Q9SAZ5, Q9LU15, Q8L9T7, Q9SSC9]; M. truncatula (MtHPt1-6b) [B7FGU6, G7I2T8, A2Q3H8, G7IHZ7, G7I7G5, G7JGY0, G7II71]; O. sativa (OsHPT3) [Q6VAK4]; Z. mays (ZmHPT2) [Q9SLX1] and S. cerevisiae (YPD1) [Q07688]. Proteins with determined crystal structures are underlined. The scale bar at the bottom indicates the number of amino acid substitution per site.
Distant homologues of both the active and inactive HPt proteins show only a low level of overall sequence similarity in the neighborhood of the active site. However, when the set of sequences is limited to the active orthologs from plants, the number of conserved residues is much higher. The characteristic sequence signature in this case is 79HQ[LFQ]KGSS[SAT]S[IV]GAx[RK]V93, with the active histidine shown in bold.
From the structural point of view, HPt proteins from prokaryotes, fungi and plants differ widely. Fig. 5 shows a superposition of the HPt domain of the E. coli hybrid sensor ArcB, of S. cerevisiae YPD1, and of the plant MtHPt1 structure from this work. The HPt proteins from the lower organisms lack the α1 helix at the N terminus. In addition, in ArcB the α2 helix lies farther away from the protein core. The helices α3-α5, however, superpose very well, and the active histidine in α4 occupies nearly exactly the same position. More differences can be seen towards the C terminus. In YPD1, there is a long insert between helices α5 and α6, which is mostly disordered. Helix α6 is the longest helix in the MtHPt1 structure. Its extension follows the same general direction as in YPD1. In ArcB, this C-terminal helix is kinked towards the long axis of the molecule. It occupies part of the space used up by α1 in MtHPt1.
Fig. 5.

Structural alignment of HPt orthologs from three different kingdoms. MtHPt1 (plants), orange (this work); YDP1 (fungi), cyan (S. cerevisiae), and ArcB (bacteria), green (E. coli). The latter two proteins have been extracted from their complexes (PDB codes 1oxk and 1bdj, respectively). The well-superposed elements are shown in semi-transparent mode, the divergent elements are in solid color. Note the absence of helix α1 in the lower organisms and a long insert between helices α5 and α6 in YDP1. The active histidine (ball-and-stick model) occupies nearly the same position in all proteins. The lysine and glycine residues from the KGSS sequence motif that is conserved in all kingdoms are also shown to indicate the structural location of this motif in HPt proteins. These residues form part of the positively charged surface area (compare Fig. 6B). In general, the structural divergence increases with the distance from the active site. Numbering of the residues corresponds to MtHPt1.
Root-mean-square deviations (RMSD) and Q-scores for the structures of eukaryotic HPt proteins are shown in Table 2. The conserved residues in plant HPt sequences are located mainly in the helices forming the four-helix bundle and surrounding the His79 phosphorylation site. Their conservation suggests that they might be involved in binding an important partner protein, either the REC domain of the sensory kinase or the RR response regulator. The active His79, together with Lys82 and Gly83, are conserved throughout all active HPt orthologs. This lysine and histidine take part in the formation of the positive charge patch that surrounds the latter residue. Two subsequent serines, from the HXXKGSS motif, are present only in eukaryotic HPt proteins but their role has not been elucidated yet. The most important conclusion from this comparison is that the structural differences fade away in the geometrical vicinity of the active histidine.
Table 2.
Structural alignment of eukaryotic HPt proteins with the present MtHPt1 structure from M. truncatula.
| PDBa | RMSD of Cα atoms (Å) | Number of aligned residues | Sequence identity (%) | Number of secondary structure elements | Q-scoreb |
|---|---|---|---|---|---|
| 1oxk | 1.28 | 108 | 19 | 5 | 0.43 |
| 1yvi | 1.28 | 136 | 48 | 6 | 0.78 |
| 1wn0 | 1.29 | 136 | 50 | 6 | 0.77 |
The PDB accession codes correspond to the following HPt structures: 1oxk, Saccharomyces cerevisiae; 1yvi, Oryza sativa; 1wn0, Zea mays
Q-score represents the quality function of Cα alignment. It reduces the effect of RMSD - Nalgn (number of aligned residues) balance on the estimation of alignments (Nres1 and Nres2 stand for the number of residues in the aligned proteins, and the empirical parameter R0 is set to 3 Å): Q = (Nalgn·Nalgn)/[(1+(RMSD/R0)2)·Nres1·Nres2]
The active site His
The active His79 in MtHPt1 forms hydrogen bonds with two water molecules (Fig. 6). The protein crystalizes at pH 5.5 and in this milieu both nitrogen atoms of the His79 imidazole ring are protonated and the positive charge is delocalized over both N atoms. The water molecules can, therefore, only act as hydrogen-bond acceptors. They are also important markers for the orientation of the histidine ring.
Fig. 6.
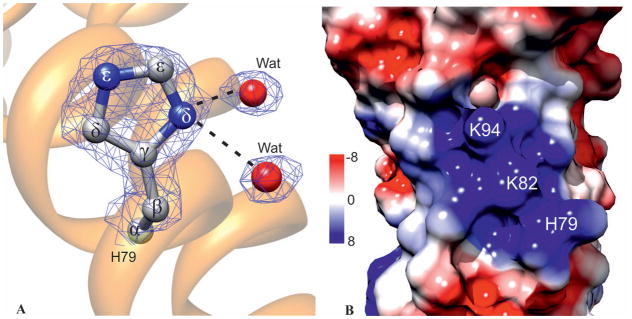
The active site His79. A His79 in ball-and-stick representation shown in 2Fo-Fc electron density map contoured at the 1.2σ level. Two water molecules form the only hydrogen bonds with the His79 side chain. B Electrostatic potential of the molecular surface of MtHPt1 in the vicinity of His79. Note the large patch of positive charge created by Lys82, Lys94 and the active His79. The electrostatic potential is color-coded according to the scale in kT units shown on the left.
The electrostatic potential of the surface reveals a strong positive patch in the vicinity of the active His79. The positive charge is created by His79 itself and by Lys82 and Lys94 (Fig. 6B). These three residues are part of the conserved HPt sequence motif and are likely to take part in binding of a phosphorelay partner.
Our attempts to crystalize MtHPt1 in the phosphorylated state have been unsuccessful, most likely because N-phosphorylated histidine is very labile [17–19], especially at the acidic pH (5.5) at which MtHPt1 normally crystallizes. Although it was possible to carefully increase the pH of crystallization to 7.0, the electron density for those crystals showed absence of a phosphate group (not shown).
In the PDB there are three crystal structures of proteins containing a phosphorylated histidine residue (HIP, PDB symbol for a modified His residue). None of them has any relation to HPt proteins. It is useful, however, to examine the modification scheme of the histidine side chain. In all three cases, the N atom proximal to the Cα atom (Nδ) is the phosphorylation site. Two of the structures (PDB codes 1nsq, 1nsp) were obtained upon crystallization at pH 8.0 [20]. Unfortunately it is not possible to inspect the corresponding electron density maps, as no structure factors were deposited with those structures. The third structure was determined for Discoidin II from D. discoideum at pH 7.5 (2vme) [21]. Although the phosphorylation is clearly only partial (negative peak in Fo-Fc map at −4.6σ), there is no doubt about the phosphate group location.
Biochemical properties
We have conducted an in vitro phosphorylation assay to confirm that the recombinant MtHPt1 protein is active as a phosphate acceptor. In this assay, recombinant MtHPt1 was phosphorylated only when MtCRE1′ was present (Fig. 7A, B). The intensity of the band on the autoradiography gel was the highest when the MtCRE1′:MtHPt1 molar ratio was 1:50. To test the specificity of the phosphorylation reaction, an H79N site-directed mutant of MtHPt1 was utilized in an analogous set of experiments. No phosphorylation of the mutated protein was observed. It is interesting to note that Asn substitutes for the active His residue in the biologically inactive HPt variants [16].
Fig. 7.

Biochemical properties of MtHPt1. A, B Autoradiography of enzymatic phosphorylation of MtHPt1 by MtCRE1′ with γ32P- labeled ATP as a phosphate donor. The H79N mutant of MtHPt1 was utilized as a negative control. Different ratios of the interacting proteins were used as indicated above the gels. The same gel was analyzed using autoradiography (B) and stained with Coomassie Brilliant Blue R-250 (A). C Results of Micro-Scale Thermophoresis. The experimental points and the fitted curve are shown. Concentration of MtHPt1 is plotted in nM. Fifteen measurements were taken at different MtCRE1′:MtHPt1 ratios. The change in fluorescence is the result of complex formation. The migration rate of the labeled protein (MtCRE1′) in a thermal gradient is slower upon binding to MtHPt1.
From a Micro-Scale Thermophoretic (MST) analysis of the MtCRE1′:MtHPt1 interaction the dissociation constant (Kd) was determined as 14 μM (Fig. 7C). This indicates that binding between MtHPt1 and the intracellular portion of the receptor is quite strong even in the absence of phosphorylation, as ATP was not present in the reaction mixture. The changes of MtCRE1′ mobility upon binding of MtHPt1 were very significant, which makes the thermophoresis method very reliable in this case.
Experimental procedures
Cloning, overexpression and purification of wild-type MtHPt1
The gene coding for MtHPt1 was found in M. truncatula genome transcript [22] by BLAST [23] using the homologous sequence of A. thaliana ahp1 (GenBank: AB015141) as a probe. There are several HPt sequences in the M. truncatula genome, as revealed by the recently completed genome sequencing project [9, 24]. The MtHPt1 protein has “Evidence at protein level” annotation in the UniProt Knowledgebase and is accessible under the code B7FGU6.
The MtHPt1 coding sequence was amplified by polymerase chain reaction (PCR) using M. truncatula (ecotype J5) cDNA from roots as template (Forward CACCATGGAGGTGGGTCAG and Reverse TCAGTTCAGTTCCATCATAGGGA PCR primers were used). The reaction product was cloned into the pET-TOPO-151D (Invitrogen) vector and the correctness of the insert was confirmed by sequencing. Small-scale overexpression tests in E. coli BL21 cells showed poor solubility of the protein. This obstacle was overcome by using the pMCSG9 vector (from the Midwest Center for Structural Genomics), which adds an N-terminal His6 plus Maltose Binding Protein (MBP) fusion tag. The pMCSG9-Mthpt1 construct was obtained by ligase-independent cloning [25] after PCR gene amplification with the pET-TOPO-151D plasmid as template. With the use of the pMCSG9-Mthpt1 construct, the solubility of the expressed protein was significantly improved.
Overexpression was carried out in BL21 Magic E. coli cells. The bacteria were cultured with shaking in LB media supplemented with 120 μg/ml ampicillin and 25 μg/ml kanamycin at 37°C until the OD600 reached 1.0. The temperature was lowered to 18°C and isopropyl-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM. The culture was grown for 18 h and then centrifuged at 4500 rpm for 10 min at 4°C. Cell pellet from 1l culture was resuspended in 30 ml of binding buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1 mM TCEP), flash-frozen in liquid nitrogen and stored at −80°C. The samples were thawed and the cells were disrupted by sonication using 4 min bursts with appropriate intervals for cooling. Cell debris was removed by centrifugation at 15 000 rpm for 30 min at 4°C. The supernatant was applied on a column packed with 6 ml of Ni Sepharose HP resin (GE Healthcare), connected to VacMan (Promega) and the chromatographic process was accelerated with a vacuum pump. After binding, the column was washed 4 times with 30 ml of binding buffer and the purified protein was eluted with 15 ml of elution buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 300 mM imidazole, 1 mM TCEP). The His6-MBP-tag was cleaved with TEV protease and the excess of imidazole was removed by dialysis (overnight, 4°C) simultaneously. The solution was mixed with Ni Sepharose HP resin to get rid of the tags and the His6-tagged TEV protease. The flow-through was collected, concentrated to 4 ml and applied on a HiLoad Superdex 200 16/60 column (GE Healthcare) equilibrated with a buffer composed of 50 mM Tris-HCl, pH 8.0, 200 mM NaCl and 1 mM TCEP. The size exclusion chromatography step yielded a homogenous, monomeric protein fraction of about 6 ml. The sample was concentrated to 6 mg/ml, as determined by the method of Bradford [26], and used for crystallization.
Generation of H79N site-directed mutant of MtHPt1
Site-directed mutagenesis of MtHPt1 which changed the active His79 residue to Asn was performed using the Polymerase Incomplete Primer Extension (PIPE) technique [27]. The pMCSG9-Mthpt1 construct was used as a template. The mutation (bold in underlined codon) was introduced using two primers: 5′-GCTCATGTTAACCAGTTTAAGGGTAG-3′ (coding strand) and 5′-CTGGTTAACATGAGCATCAACTTTTTTG-3′ (complementary strand). The correctness of the resulting vector was confirmed by sequencing. The mutated MtHPt1 protein was overexpressed and purified as described for the wild type protein above.
Crystallization
Initial screening for crystallization conditions was performed using a Robotic Sitting Drop Vapor Diffusion setup (Mosquito) with Crystal Screen HT, Index HT and Salt Rx reagents from Hampton Research. Manual optimization of the crystallization hits in hanging drops gave the following crystallization conditions: 100 mM Bis-Tris, pH 5.5, 25% PEG3350 and 200 mM ammonium acetate. The drop was mixed from 1 μl of the reservoir solution and 3 μl of protein solution. Crystals grew within 4 days at 18°C. After 2 months they were harvested with 0.1 mm nylon loops (Hampton Research), washed with cryo-protectant solution containing 20% (v/v) glycerol in the reservoir cocktail, and vitrified in liquid nitrogen for synchrotron-radiation data collection.
Data collection and processing
X-Ray diffraction data were collected at the Advanced Photon Source (ANL, Argonne, IL, USA) 22-BM beamline. A total of 200 diffraction images with 0.5° oscillation were indexed, integrated and scaled using XDS [28]. Details of data collection and processing are presented in Table 1.
Table 1.
Data-collection and refinement statistics.
| Data collection | |
|---|---|
| Wavelength (Å) | 1.0000 |
| Crystal-to-detector distance (mm) | 153 |
| Oscillation (°) | 0.5 |
| Temperature (K) | 100 |
| Space group | P212121 |
| Unit cell parameters | |
| a, b, c (Å) | 38.6, 44.9, 85.9 |
| Resolution (Å) | 31.0-1.45 (1.53-1.45)a |
| Total reflections | 98484 |
| Unique reflections | 25816 |
| Completeness (%) | 94.3 (73.7) |
| Multiplicity | 3.8 (2.5) |
| Rmerge (%) | 6.1 (52.4) |
| <I/σ(I)> | 14.0 (2.0) |
| Refinement | |
| No. of reflections, working set | 24814 |
| No. of reflections, test set | 1002 |
| No. of atoms (non H) | |
| protein | 1250 |
| solvent | 159 |
| Rwork/Rfree (%) | 16.7/19.0 |
| RMSD from ideal geometry | |
| bond lengths (Å) | 0.019 |
| Ramachandran statistics (%) | |
| most favored | 97.7 |
| allowed | 2.3 |
Values in parentheses correspond to the highest resolution shell.
Structure solution and refinement
The crystal structure of MtHPt1 was solved by molecular replacement using Phaser [29]. The crystal structure of a homologous protein from rice (Oryza sativa; PDB code 1yvi; Center for Eukaryotic Structural Genomics) was used as a search probe. Automatic model building was carried out with the on-line version of ARP/wARP [30]. Coot [31] was applied for manual fitting in electron density maps between model refinement and validation cycles carried out in phenix.refine [32]. An attempt to refine anisotropic atomic displacement parameters (ADPs) resulted in a significant increase of Rfree and, therefore, the isotropic model was accepted as final, with seven TLS [33] groups defined as suggested by the TLSMD server [34]. The final refinement statistics are listed in Table 1.
Overexpression and purification of the intracellular part of cytokinin receptor
Sequence analysis of the M. truncatula cytokinin receptor MtCRE1 (GenBank: 357521450) revealed that it contains an N-terminal part consisting of an extracellular cytokinin-binding domain and a transmembrane domain. The intracellular C-terminal part (MtCRE1′) comprised of residues Ile352 -- Ser1003 was amplified using PCR reaction with M. truncatula (ecotype J5) cDNA from roots as template. The reaction product was cloned into the pMCSG9 vector, and overexpression and purification was carried out using the same protocol as described for MtHPt1 (vide supra). The final purification step, size exclusion chromatography, yielded a homogenous fraction of dimeric MtCRE1′ (3 mg of protein from 1 l culture). The dimerization was additionally confirmed by dynamic light scattering, DLS (not shown).
In vitro phosphorylation
Recombinant MtCRE1′ was mixed with wild-type MtHPt1 or its H79N mutant at different molar ratios in a buffer containing 50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 10 mM MgCl2 and 1 mM TCEP in 20 μl aliquots. Reactions (at room temperature) were initiated by the addition of 1 μl of [γ-32P]ATP solution (1 μCi/μl and 5 mM final ATP). After 20 min, they were quenched by the addition of SDS-PAGE loading buffer, and immediately subjected to electrophoretic analysis using a Mini-Protean TGX system (Bio-Rad). The autoradiography was performed using an enhancing screen followed by radioisotope imaging with an FLA 5100 image analyzer (Fuji). The gel was stained with Coomassie Brilliant Blue R-250 and scanned using a regular desktop scanner.
Micro-scale thermophoresis
Micro-scale thermophoresis (MST) is a method developed for quantitative analysis of interactions between biomolecules [35]. It interprets the directed movement of particles in a temperature gradient. Changes in macromolecular shape, size or hydration shell can be observed using MST. One of the biomolecules of interest is labeled with a fluorescent marker and the experiment is performed in capillaries with different ratios of the interacting partners. An IR laser is pointed towards the sample (usually for 30 s) causing local temperature increase and thus the directed movement of particles in the temperature gradient. Relaxation to the initial state is also measured after the laser is switched off. With the use of this method, binding affinities and kinetics can be determined. MST is most useful when protein-protein interactions are evaluated, since the changes in molecular movement are substantial after binding of a large partner. To perform the MST experiment, MtCRE1′ was labeled with NT-647 dye using NanoTemper’s Protein Labeling Kit RED (L001, NanoTemper Technologies). The labeled MtCRE1′ was kept at a constant concentration of 20 nM, while the MtHPt1 solution (initial concentration 500 μM) was prepared as a dilution series of 15 concentrations. The analysis was carried out in a buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2 and 0.05% Tween. Each reaction mixture was incubated in 20 μl for 10 min followed by thermophoresis in hydrophilic MST-grade glass capillaries in a Monolith NT.115 apparatus.
Other software used
Molecular figures were created with UCSF Chimera [36]. Assignment of secondary structure elements was based on the DSSP [37] algorithm. Structural alignments based on Cα atoms were performed using PDBe Fold v2.51 (http://www.ebi.ac.uk/msd-srv/ssm) [38]. Sequence alignments and phylogenetic trees were calculated in ClustalW [39]. Phylograms and alignments were edited in TreeView [40] and GenDoc [41], respectively.
Conclusions and outlook
The present structural analysis of histidine-containing phosphotransfer protein from M. truncatula is characterized by the highest resolution achieved for a protein from this class. The MtHPt1 structure is similar to other plant HPt models. It is, however, the only structure of a plant HPt protein shown to be fully active in a biochemical assay.
One of the pending questions concerns the structural features of the HPt proteins related to their redundancy, i.e. numerous coding sequences found in a given genome [42]. It is not known why there are multiple HPt genes in plant genomes, for example, seven in M. truncatula or six in A. thaliana. Also, the problem of specificity of HPt proteins for sensory kinases of different types and for response regulators has not been solved yet. In this paper, it was demonstrated that MtHPt1 in the unphosphorylated state has a high affinity for the intracellular fragment (MtCRE1′) of the plasma-membrane-anchored MtCRE1 cytokinin receptor. Phosphorylation of MtHPt1, on the other hand, should have structural consequences, enabling the protein to migrate to the nucleus and to phosphorylate its target response regulator, RR.
An important aspect of MtHPt1 function is its ability to form complexes with protein partners in the signaling cascade. Our observation of almost strict conservation of the HPt residues that map on the protein surface of MtHPt1 in the vicinity of the solvent-exposed active His79 suggests that this structurally conserved area is in all probability the binding interface for protein-protein interactions. Using our experimental model of MtHPt1 and a homology-modeled structure of the MtCRE1 REC domain (not shown) we were able to build a convincing model of the REC:MtHPt1 complex (not shown) using a complex of osmosensing histidine protein kinase domain (SLN1-R1) and phosphorelay intermediate protein 1 (YPD1) from S. cerevisiae as a template (PDB code 2r25). It is more challenging to propose a hypothetical model for the MtHPt1:RR complex, mainly because there are no structures or even obvious templates for modeling this type of an RR response regulator. Work is in progress, however, with the utilization of both molecular modeling tools and crystallography, with the aim of arriving at reliable models for MtHPt1 interactions with its binding partners.
Supplementary Material
Alignment of known amino acid sequences of HPt proteins from plants (M. truncatula, A. thaliana, O. sativa, Z. mays), yeast (S. cerevisiae) and bacteria (E. coli). The level of conservation is expressed by the darkness of the lettering background. Residues forming sulfur-aromatic interactions in MtHPt1 are highlighted in bold red font. Source organisms are appended to the sequences. The following UniProt sequences have been used: MtHPt1 [B7FGU6], MtHPt2 [G7I2T8], MtHPt3 [A2Q3H8], MtHPt4 [G7IHZ7], MtHPt5 [G7I7G5], MtHPt6a [G7JGY0], MtHPt6b [G7II71], AHP1 [Q9ZNV9], AHP2 [Q9ZNV8], AHP3 [Q9SAZ5], AHP4 [Q9LU15], AHP5 [Q8L9T7], AHP6 [Q9SSC9], OsHPT3 [Q6VAK4], ZmHPT2 [Q9SLX1], YPD1 [Q07688], ArcB [C6EH17].
Acknowledgments
We are grateful to Dr. Stephen Blanke and Dr. Stefan Duhr (NanoTemper Technologies) for making the thermophoresis equipment available to us and to Prof. Jan Wrzesinski (IBCh, PAS, Poznan, Poland) for help with the autoradiography experiment. Financial support of the project was provided by the European Union within the European Regional Developmental Fund and by the Polish Ministry of Science and Higher Education (grant No. NN 301 003739). The pMCSG9 vector was acquired from the Midwest Center for Structural Genomics. X-Ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline of the APS/ANL. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. The Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, is supported by an NIH grant P41 RR001081.
Abbreviations
- ADP
atomic displacement parameter
- CHASE
cyclases/histidine kinases-associated sensory extracellular domain
- DLS
dynamic light scattering
- HK
histidine kinase domain of MtCRE1
- HPt
histidine-containing phosphotransfer protein
- IPTG
isopropyl-D-thiogalactopyranoside
- MBP
Maltose Binding Protein
- MST
Micro-Scale Thermophoresis
- MtCRE1
Medicago truncatula Cytokinin Receptor 1
- MtCRE1′
intracellular part of MtCRE1
- PDB
Protein Data Bank
- PIPE
Polymerase Incomplete Primer Extension
- REC
receiver domain of MtCRE1
- RMSD
root-mean-square deviation
- RR
response regulator
- SLN1-R1
S. cerevisiae osmosensing histidine protein kinase RR domain
- TCEP
tris(2-carboxyethyl)phosphine
- TEV
Tobacco Etch Virus
- YPD1
phosphorelay intermediate protein 1
References
- 1.West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci. 2001;26:369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 2.Schaller GMD, Gribskov M, Walker JC. In: Two-component signaling elements and histidyl-toaspartyl phosphorelays. Somerville C, Meyerowitz E, editors. The Arabidopsis Book, American Society of Plant Biologists; 2002. [Google Scholar]
- 3.Chang C, Stadler R. Ethylene hormone receptor action in Arabidopsis. Bioessays. 2001;23:619–627. doi: 10.1002/bies.1087. [DOI] [PubMed] [Google Scholar]
- 4.Schmulling T. CREam of cytokinin signalling: receptor identified. Trends Plant Sci. 2001;6:281–284. doi: 10.1016/s1360-1385(01)02011-8. [DOI] [PubMed] [Google Scholar]
- 5.Haberer G, Kieber JJ. Cytokinins. New insights into a classic phytohormone. Plant Physiol. 2002;128:354–362. doi: 10.1104/pp.010773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira FJ, Kieber JJ. Cytokinin signaling. Curr Opin Plant Biol. 2005;8:518–525. doi: 10.1016/j.pbi.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Levin EJ, Kondrashov DA, Wesenberg GE, Phillips GN., Jr Ensemble refinement of protein crystal structures: validation and application. Structure. 2007;15:1040–1052. doi: 10.1016/j.str.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugawara H, Kawano Y, Hatakeyama T, Yamaya T, Kamiya N, Sakakibara H. Crystal structure of the histidine-containing phosphotransfer protein ZmHP2 from maize. Protein Sci. 2005;14:202–208. doi: 10.1110/ps.041076905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young ND, Debelle F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–524. doi: 10.1038/nature10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Reid KSC, Lindley PF, Thornton JM. Sulphur-aromatic interactions in proteins. FEBS. 1985;190:209–213. [Google Scholar]
- 12.Ringer AL, Senenko A, Sherrill CD. Models of S/pi interactions in protein structures: comparison of the H2S benzene complex with PDB data. Protein Sci. 2007;16:2216–2223. doi: 10.1110/ps.073002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cakici O, Sikorski M, Stepkowski T, Bujacz G, Jaskolski M. Crystal structures of NodS N-methyltransferase from Bradyrhizobium japonicum in ligand-free form and as SAH complex. J Mol Biol. 2010;404:874–889. doi: 10.1016/j.jmb.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 14.Sakakibara H, Hayakawa A, Deji A, Gawronski SW, Sugiyama T. His-Asp phosphotransfer possibly involved in the nitrogen signal transduction mediated by cytokinin in maize: molecular cloning of cDNAs for two-component regulatory factors and demonstration of phosphotransfer activity in vitro. Plant Mol Biol. 1999;41:563–573. doi: 10.1023/a:1006391304881. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Copeland DM, Soares AS, West AH. Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J Mol Biol. 2008;375:1141–1151. doi: 10.1016/j.jmb.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahonen AP, Bishopp A, Higuchi M, Nieminen KM, Kinoshita K, Tormakangas K, Ikeda Y, Oka A, Kakimoto T, Helariutta Y. Cytokinin signaling and its inhibitor AHP6 regulate cell fate during vascular development. Science. 2006;311:94–98. doi: 10.1126/science.1118875. [DOI] [PubMed] [Google Scholar]
- 17.Puttick J, Baker EN, Delbaere LT. Histidine phosphorylation in biological systems. Biochim Biophys Acta. 2008;1784:100–105. doi: 10.1016/j.bbapap.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Quezada CM, Hamel DJ, Gradinaru C, Bilwes AM, Dahlquist FW, Crane BR, Simon MI. Structural and chemical requirements for histidine phosphorylation by the chemotaxis kinase CheA. J Biol Chem. 2005;280:30581–30585. doi: 10.1074/jbc.M505316200. [DOI] [PubMed] [Google Scholar]
- 19.Zu XL, Besant PG, Imhof A, Attwood PV. Mass spectrometric analysis of protein histidine phosphorylation. Amino Acids. 2007;32:347–357. doi: 10.1007/s00726-007-0493-4. [DOI] [PubMed] [Google Scholar]
- 20.Morera S, Chiadmi M, LeBras G, Lascu I, Janin J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry. 1995;34:11062–11070. [PubMed] [Google Scholar]
- 21.Aragao KS, Satre M, Imberty A, Varrot A. Structure determination of Discoidin II from Dictyostelium discoideum and carbohydrate binding properties of the lectin domain. Proteins. 2008;73:43–52. doi: 10.1002/prot.22038. [DOI] [PubMed] [Google Scholar]
- 22.Moskal W, Chan A, Cheung F, Xiao Y, Town CD. Plant Genomics. J Craig Venter Institute; 9704 Medical Center Dr, Rockville, MD 20850, USA: 2008. Direct Submission. [Google Scholar]
- 23.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branca A, Paape TD, Zhou P, Briskine R, Farmer AD, Mudge J, Bharti AK, Woodward JE, May GD, Gentzbittel L, et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc Natl Acad Sci U S A. 2011;108:E864–870. doi: 10.1073/pnas.1104032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim Y, Babnigg G, Jedrzejczak R, Eschenfeldt WH, Li H, Maltseva N, Hatzos-Skintges C, Gu M, Makowska-Grzyska M, Wu R, et al. High-throughput protein purification and quality assessment for crystallization. Methods. 2011;55:12–28. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol. 2009;498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 28.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 34.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 35.Duhr S, Braun D. Why molecules move along a temperature gradient. Proc Natl Acad Sci U S A. 2006;103:19678–19682. doi: 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 39.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 40.Page RDM. Tree View: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 41.Nicholas KBN, HB, Deerfield DW. Embnew News. 1997. Genedoc: Analysis and Visualization of Genetic Variation; p. 4. [Google Scholar]
- 42.Hutchison CE, Li J, Argueso C, Gonzalez M, Lee E, Lewis MW, Maxwell BB, Perdue TD, Schaller GE, Alonso JM, et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell. 2006;18:3073–3087. doi: 10.1105/tpc.106.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of known amino acid sequences of HPt proteins from plants (M. truncatula, A. thaliana, O. sativa, Z. mays), yeast (S. cerevisiae) and bacteria (E. coli). The level of conservation is expressed by the darkness of the lettering background. Residues forming sulfur-aromatic interactions in MtHPt1 are highlighted in bold red font. Source organisms are appended to the sequences. The following UniProt sequences have been used: MtHPt1 [B7FGU6], MtHPt2 [G7I2T8], MtHPt3 [A2Q3H8], MtHPt4 [G7IHZ7], MtHPt5 [G7I7G5], MtHPt6a [G7JGY0], MtHPt6b [G7II71], AHP1 [Q9ZNV9], AHP2 [Q9ZNV8], AHP3 [Q9SAZ5], AHP4 [Q9LU15], AHP5 [Q8L9T7], AHP6 [Q9SSC9], OsHPT3 [Q6VAK4], ZmHPT2 [Q9SLX1], YPD1 [Q07688], ArcB [C6EH17].