

Abstract
Hepatocellular carcinoma and cholangiocarcinoma are primary liver cancers, both represent a growing challenge for clinicians due to their increasing morbidity and mortality.
In the last few years a number of in vivo models of hepatocellular carcinoma and cholangiocarcinoma have been developed. The study of these models is providing a significant contribution in unveiling the pathophysiology of primary liver malignancies. They are also fundamental tools to evaluate newly designed molecules to be tested as new potential therapeutic agents in a pre-clinical set. Technical aspects of each model are critical steps, and they should always be considered in order to appropriately interpret the findings of a study or its planning.
The purpose of this review is to describe the technical and experimental features of the most significant rodent models, highlighting similarities or differences between the corresponding human diseases. The first part is dedicated to the discussion of models of hepatocellular carcinoma, developed using toxic agents, or through dietary or genetic manipulations. In the second we will address models of cholangiocarcinoma developed in rats or mice by toxin administration, genetic manipulation and/or bile duct incannulation or surgery. Xenograft or syngenic models are also proposed.
Keywords: Animal models, Chemotoxic agents, Cholangiocarcinoma, Hepatocellular carcinoma, Xenograft models
1. Introduction
Hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) are primary liver cancers, both represent a growing challenge for clinicians due to their increasing morbidity and mortality.
HCC is the sixth most common cancer in the world, with 630,000 new cases diagnosed each year [1]. The clinical history of approximately 80% of HCC patients progresses from fibrosis, to cirrhosis and finally to cancer [2,3]. The three main causes of HCC are HBV and HCV infections and alcohol-induced liver injury. Less frequent causes are some autoimmune and metabolic diseases (starting from non-alcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH)). An additional rarer cause of liver carcinogenesis, especially in African and Asian Countries, is represented by aflatoxin B1 (AFB) [4]. The mechanisms by which these aetiologic factors may induce HCC involve a wide range of pathways and molecules, currently under study.
CCA arises as a malignant transformation of cholangiocytes, the epithelial cells lining the intra- and extrahepatic biliary epithelium. CCA is an aggressive disease, with increasing incidence in Western countries [5]; currently approximately 6000 new cases of CCA are diagnosed in the United States each year [6]. Diagnosis is often made when the disease is already in its late stages. The therapeutic options (medical or surgical) are limited, which results in a poor prognosis. The vast majority of the patients die within a few months from diagnosis [5,7].
The pathophysiology of CCA is poorly understood. The known definite or probable risk factors [such as Primary Sclerosing Cholangitis, liver fluke infections, hepatolithiasis or chronic hepatitis C, cirrhosis and toxins) share the common feature of inducing chronic cholestasis and biliary and/or liver inflammation [5,7]. Thus, the development of animal models for better understanding the aetiology of these deadly cancers is essential. Over the last years a broad number of in vivo models of HCC and CCA have been developed. The studyof these models are providing a significant contribution to unveiling the pathophysiology of primary liver malignancies. These models are also fundamental tools to evaluate newly designed molecules to be tested as new potential therapeutic agents in a pre-clinical set.
Because of the short lifespan and breeding capacity, rodents are widely employed for cancer research. Rats (Rattus norvegicus) or mice (Mus musculus) have also been favourite models for studying both HCC and CCA development. Mice are widely used to define the role of genetic modification through the use of knock or transgenic models, also because these models are easier to be handle.
The purpose of this review is to describe the technical and experimental features of the most significant rodent models, highlighting similarities or differences between the corresponding human diseases. For clarity, animal models were given specific names, in order to facilitate interpretation by readers.
2. Experimental models of HCC
2.1. Chemotoxic agents
Several chemicals damage the liver and induce progression and development of tumours (Table 1). Based on current literature, there are two types of carcinogenic compounds: (i) genotoxic agents that directly induce tumour formation and (ii) promoting agents that enhance tumour formation when in association with genotoxic agents [8]. The treatment with a tumour-promoting agents facilitates the clonal expansion of the preneoplastic cells, therefore enhancing both tumour development and its aggressiveness. The main advantage of chemically induced models is the similarity with the injury–fibrosis–malignancy cycle seen in humans.
Table 1.
Synopsis of the main experimental features of rodent models of HCC.
| Genotoxic agent | Promoting agent | Species | Tumour development (time) | Features | Metastatic foci | References |
|---|---|---|---|---|---|---|
| DEN | – | Mouse/rat | 100 weeks | Pure tumours, no fibrogenesis | No | [13–21] |
| DEN | PB | Mouse/rat | 12–40 weeks | Aggressive Tumours | Yes | [22,23] |
| DEN | PH | Rat | 4–8 weeks | Poorly reproducible | No | [24–27] |
| Peroxisome proliferators | – | Mouse | 50–100 weeks | Strain specific, mutations not known in humans | Yes | [28–33] |
| Aflatoxin | – | Mouse/rat | 50 weeks | Yes | [34–36] | |
| CCl4 | – | Mouse | 100 weeks | Inflammation and fibrosis | Yes | [37–41] |
| TAA | – | Mouse/rat | 50–70 weeks | Inflammation | No | [49] |
| Choline deficient diet | – (Ethionine) | Mouse/rat | 50 weeks (30–40 weeks) | Steatohepatitis | No | [42–47] |
| HBx transgenic | – (DEN) | Mouse | 80–100 weeks (30–50 weeks) | HBV related | No | [62,63] |
| Core, A, E transgenic mice | – (DEN) | Mouse | 60 weeks (30 weeks) | HCV related | No | [64,65] |
| P-TEN | – | Mouse | 40 weeks | Tumour, proliferation | No | [74,75] |
| TGF-b transgenic mice | – | Mouse | 30 weeks | Tumour, inflammation, fibrosis | No | [66] |
| NEMO | – | Mouse | 48 weeks | Tumour, inflammation, steatohepatitis | No | [68–70] |
| TAK 1 | – | Mouse | 48 weeks | Tumour, inflammation, fibrosis | No | [71] |
DEN = N-nitrosodiethylamine; CCl4 = carbon tetrachloride; TAA = thioacetamide; TGF = transforming growth factor.
2.1.1. The “N-nitrosodiethylamine” model
This model of HCC is developed by administering N-nitrosodiethylamine (DEN) to mice [9,10]. The carcinogenetic activity of DEN is exerted in two different ways: (i) by alkylating DNA structures thus causing DNA damage and subsequent cell degeneration and (ii) by inducing reactive oxygen species (ROS) formation through the activation of the cytochrome P450 in hepatocytes [11,12]. The DEN model has specific characteristics: (i) dose dependency; (ii) timing of the administration; (iii) sex-, age- and mice strain-related efficacy; and (iv) possible association with the simultaneous administration of promoting agents (Table 1) [13–17]. Administration of DEN, in a single dose to 15-day-old mice, leads to tumour development in 80% of cases, while a 100% success rate in tumour formation is obtained with long term DEN administration [18,19].
Among the promoting agents, phenobarbital (PB) needs to be taken into consideration. The effects of PB promotion on DEN-initiated mice also vary considerably depending upon strain, sex and age of the mice. Timing of initiation with DEN is a critical determinant: when adult male B6C3F1 mice are initiated with DEN between 6 and 10 weeks of age followed by exposure to PB in drinking water for 36 weeks, PB serves as a tumour promoting agent [20,21].
Another “two-step” hepatocarcinogenesis model is known as the Solt-Farber protocol [22]. In this model, initiation with a hepato-carcinogenic compound (DEN) is followed by partial hepatectomy (PH) [23].
The main limit of the DEN model is the long duration of the experiments, the average time being 50 weeks for HCC development. Specifically, in the different chemotoxic models, a single dose of DEN is simple and reproducible: although the incidence of tumour development is less than 100%, the single dose administration exposes the animals to a reduced external effect and the mechanism is more similar to a pathophysiological progression. The long-term protocols have the advantage of inducing tumour formation in a higher percentage of cases, however, the model is influenced by the multiple DEN injections. Promoting agents such as PB may induce a higher rate of carcinogenesis, but the characteristics of the tumour are slightly modified in addition to a significantly reduced reproducibility of the model itself [24,25]. Finally, the PH-method is based on a difficult surgical technique and, therefore, is an operator-dependent feature and less reproducible.
2.1.2. The “peroxisome proliferators” model
The peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that bind to fatty acid-derived ligands and activate the transcription of genes that regulate lipid metabolism [26,27]. PPARs ligand activates peroxisomal oxidase and induces ROS formation, thus promoting HCC-development [28,29]. This experimental model has specific characteristics such as the trabecular histological pattern, metastasis in 20–40% of cases and possible induction of gene mutations [30]. However, caution should be used in the extrapolation to the human disease, since the PPs induced hepatocarcinogenesis might be a species-specific process and PP models do not have much in common with human HCCs from a genetic point of view.
2.1.3. The “aflatoxin” model
A limited number of studies employed aflatoxin exposure to both mice and rats to study HCC formation. The hepatotoxin AFB is mainly produced by certain fungi of the Aspergillus genus, such as Aspergillus flavum, and exerts carcinogenic activity. In China and Western Africa, the combined high prevalence of AFB and HBV contributes to high rates of HCC [31]. Carcinogenic activity of AFB is strictly related to the induction of chromosomal aberrations, chromosomal strand breaks, DNA-adducts generation, micronuclei and uncontrolled DNA synthesis [32]. This model has been used in both mice and rats [33]. HCC development in 7-day-old mice, injected with 6 mg/kg of AFB, is obtained after 52 weeks with a success rate of almost 100% [34]. Experimental models involving AFB administration are useful to evaluate the mechanisms involved in AFB-induced hepatocarcinogenesis, yet limited to the specific cases in which the mechanisms of AFB-induced HCC need to be elucidated.
2.1.4. The “carbon tetrachloride” model
An important chemotoxin, when administered to mice or rats, is carbon tetrachloride (CCl4) [35]. The hepatotoxicity of CCl4 is mainly exerted in two different levels: first, CCl4 induction of cytochrome P450 and the consequent increased formation of ROS, and [36,37] induction of inflammatory response by Kupffer cells through production of cytokines, chemokines and other proinflammatory factors [38]. The repeated cycles of injury, inflammation and repair lead to fibrosis and eventually HCC. Several studies have mostly used CCl4 in association with other agents such as alcohol: weekly injections of CCl4 and alcohol administration through drinking water lead to HCC after 104 weeks in mice [35,38,39]. Other studies used CCl4 administration in rats leading to a 30% efficacy in HCC formation after 30 weeks [40].
2.2. Diet-induced HCC models
Studies have shown that HCC development can be achieved by the administration of a choline deficient diet (CDD). This diet was originally developed to induce steatohepatitis, fibrosis and cirrhosis in mice and rats [41,42]. More recently, it has been observed that mice subjected to CDD develop HCC after 50–52 weeks [41]. Similarly, rats on a CDD develop tumours in a significant percentage of cases. The main mechanisms related to HCC development in CDD-treated animals are related to the stimulation of oval cells, leading to an increased oxidative stress, DNA damage and genetic mutations or modifications.
The effects of CDD have been evaluated in association with the administration of chemotoxic compounds such as DEN or CCl4 [43]. Ethionine supplementation to CDD enhances oval cell stimulation increasing carcinogenetic potential [44,45]. Similarly, combining the CDD and DEN models induces HCC faster than CDD alone, while maintaining the specific features of the diet-induced liver injury, namely steatosis and inflammation [43]. In a similar fashion, the CDD has been employed in association with CCl4 or alcohol, resulting in increased number and size of liver tumours [43]. A small variation of the CDD is represented by the choline-deficient and iron-supplemented l-amino acid-defined (CDAA) diet that mimics the same effect of the CDD in a shorter time frame (Fig. 1) [42,46].
Fig. 1.

Representative images of macroscopic (top) and microscopic H&E staining (bottom) appearance of CDAA (choline-deficient and iron-supplemented l-amino acid-defined) + CCl4 (carbon tetrachloride)-induced HCC (hepatocellular carcinoma) nodules. Large nodules are visible on the surface of the mice livers after 6 month of CDAA diet associated with low dose chronic injection of CCl4 (0.2 mg/kg of body weight, once a week).
From: De Minicis et al., unpublished observations (2011).
2.3. The “TAA” model
An additional model used in the study of HCC is thioacetamide (TAA) administration, TAA is a hepatotoxin that can be administered either in drinking water (0.02–0.05%) or by intraperitoneal (IP) injections. Several studies have shown that repeated administration of TAA leads to fibrosis in mice over a period of 10–15 weeks. The main carcinogenetic effect of TAA is related to oxidative stress formation. Increased levels of ROS in the liver progressively lead to DNA damage and HCC development [47].
2.4. Xenograft models
Xenograft tumours grow rapidly, as a consequence of cancer cell replication, collagen deposition and neo-angiogenesis. The major advantages of this model are the rapid induction and easy surveillance of tumour growth, with direct nodule measurement over time [48]. In xenograft models, tumours are induced by injecting human cancer cells in immune deficient mice, such as athymic (nude) or severe combined immune deficient (SCID) mice [49]. The main xenograft models are: (i) the ectopic model, in which human cancer cells are directly injected subcutaneously in the hind flanks of mice and (ii) the orthotopic model, in which tumour cells are injected directly into the mouse liver. The orthotopic model allows a better understanding of the metastatic spread of the tumour [50].
Concerning the ectopic model, different cell lines are often used for chemotherapeutic drug screening with common chemotherapeutic agents. However, significant differences in tumour growth inhibition are present in literature [51,52].
An interesting and more reproducible setup consists of orthotopic implantations of HCC cells in fibrotic livers [53]. Using a fibrotic liver model, the authors demonstrated the faster development of tumours and their higher capacity to metastasize and form satellite nodules [54].
In summary, the main advantage of the present model is related to the short time span occurring between injection and tumour development. However, the pathophysiological processes associated with tumour development are completely related to the model and do not resemble the main changes observed in humans. Thus, the xenograft model is commonly used and is important for the study of drug reactions and tumour characteristics, but cannot be used to mimic human tumour development [55].
An additional method used for the study of cancer is the “hollow fibre assay (HFA)” [56]. In this model, tumour cell lines are inoculated into hollow (1 mm internal diameter) polyvinylidene fluoride fibres that are heat-sealed and cut at 2 cm intervals [57]. After 24–48 h of culture in vitro, multiple fibres may be implanted in athymic mice, subcutaneously or intraperitoneally. The main advantage of this method, in comparison to the other xenograft models is represented by the possibility of testing multiple cells lines in a single mouse [58].
2.5. Genetically modified models
Genetically modified mouse models (GMMs) have the ability to mimic pathophysiological and molecular features of HCC [59]. This approach represents the best tool to test the effects of oncogenes in the presence or absence of carcinogenic agents. GMMs may be further improved by using cDNA constructs containing a promoter able to target a specific cell type; this condition may allow the generation of tissue-specific expression of special genes [60]. Mice with albumin promoter are often used in this field.
Rather than constitutive tissue-specific deleted expression of genes, an alternative model could be represented by the induction of specific genes, the so-called transgenic mice. This approach allows the study of the role of several oncogenes in tumour maintenance. Several transgenic mice models are found in the literature on HCC (Table 2). Of these it is important to consider the transgenic mice models expressing viral genes for hepatitis.
Table 2.
Synopsis of the main experimental features of rodent models of HCC.
| Model | Promoting agent | Orthotopic | Genetic | Toxic | Abdominal surgery | Inflammation | References |
|---|---|---|---|---|---|---|---|
| DEN | √ | √ | √ | [13–21] | |||
| DEN | PB | √ | √ | √ | [22,23] | ||
| DEN | PH | √ | √ | √ | [24–27] | ||
| Peroxisome proliferators | √ | √ | √ | [28–33] | |||
| Aflatoxin | √ | √ | [34–36] | ||||
| CCl4 | √ | √ | √ | [37–41] | |||
| TAA | √ | √ | √ | [49] | |||
| Choline deficient diet | √ | √ | √ | [42–45] | |||
| Choline deficient diet | Ethionine | √ | √ | √ | [46,47] | ||
| HBx transgenic | (DEN) | √ | (√) | [62,63] | |||
| Core, A, E transgenic mice | (DEN) | √ | √ | (√) | [64,65] | ||
| P-TEN | √ | √ | √ | [74,75] | |||
| TGF-b transgenic mice | √ | √ | [66] | ||||
| NEMO | √ | √ | √ | [68–70] | |||
| TAK 1 | √ | √ | √ | [71] |
DEN = N-nitrosodiethylamine; CCl4 = carbon tetrachloride; TAA = thioacetamide; TGF = transforming growth factor.
Among the viral models, most of the HBV-related transgenic animals express the HBx genes, showing HCC development after 52–104 weeks [61–63]. In HCV-models transgenic mice, expressing core E1 and E2 structural proteins, develop HCC after 60 weeks [64]. The addition of DEN injections accelerated HCC development to only 32 weeks [65].
Other mouse models of HCC have been generated from transgenic mice expressing oncogenes [66], such as c-Myc, β-catenin, or from mice with mutation/deletion of several genes: PDGF, TGFβ1, NEMO, TAK1, alpha-1 antitrypsin and PTEN (tumour suppressor gene that regulates the PKB/akt pathway) [67–71].
Among these models, an important contribution to cancer research has been the PTEN-deficient mice [19,72–74]. Liver-specific PTEN-deficient mice develop HCC after 40–44 weeks, in addition to hepatic steatosis, inflammation and fibrosis [75].
3. Experimental models of CCA
3.1. Rat models of CCA
3.1.1. The “syngenic” model
The syngenic model of CCA was proposed by Sirica et al. [76] and consists of the intra-hepatic implantation of cells from a rat-derived CCA cell line (BDEneu) into Fisher 344 rats. This approach yielded tumour formation in 100% of the injected animals, with a high level of consistency of tumoural mass after 20–22 days from the inoculation (Table 3). The course of tumour development showed an exponential trend, being greater at 25–26 days than at 15–16 days after cell inoculation. Significant increases in bilirubin serum levels were observed. Intrahepatic growth of the tumours was also paralleled by a concomitant development of peritoneal metastases and by a progressive reduction of body weight.
Table 3.
Synopsis of the experimental protocols and outcomes of rodent models of CCA.
| Model | Rodent background | Protocol | Time for tumour development | Yield | Metastases | References |
|---|---|---|---|---|---|---|
| Syngenic | Fisher rat | 4 × 106 BDEneu cells, resuspended in 0.1 ml Hanks' balanced salt solution, are injected in the left hepatic duct. of adult Fischer 344 rats. Such an approach yielded the 100% of tumour formation in the injected animals, with a high level of consistency in tumoral mass after 20–22 days from the inoculation, yet independently from the number of culture passages of the cell line | 17 days | 100% after 20–22 days from inoculation | Peritoneala | [76,78,79] |
| TAA | Sprague-Dawley, Fisher, Zucker rat | 0.03% TAA in drinking water, at a standard dose of 0.03% for 24 weeks | 16–24 weeks | 100% | Lung | [80,81–88] |
| 9th week: foci of cholangiocyte proliferation and dysplasia | ||||||
| 12th week: cancer microfoci | ||||||
| 16th week: visible CCA tumours | ||||||
| 24th week: consistent CCA tumours in treated animalsb | ||||||
| Smad4-Pten knock out | Smad4Co/Co PtenCo/Co Al/b-Cre mouse | Cross-breeding of Smad4Co and/or PtenCo mice with Alb-Cre mice: generation of Smad4Co/Co PtenCo/Co Alb-Cre mice | 24–28 weeks | 4–7 month old mice: consistent CCA tumours of age | Not reported | [91] |
| 2–3 month old mice: hyperplastic foci of the biliary epithelium | ||||||
| Time-dependent progression to dysplasia and cancer in situ | ||||||
| p53 knock out-CCl4 | p53−/− C57B16 mouse | p53+/− mice bred to produce p53 +/+, +/− and −/− | 29 weeks (p53−/−) | 54% (p53−/−) | Not reported | [98] |
| CCl4 administration (10 μL/g body weight, i.p.) starts at the age of 6 months. | 53 weeks(p53+/−) | 18%(p53+/−) | ||||
| Schedule: three injections per week for 4 months. Follow up to 53 weeks | ||||||
| Xenograft | Nude mouse | Subcutaneous implant of cancer cell lines of human origin | 3–11 weeks | 100% | – | [105–113] |
| Rapid tumour growth, as a consequence of cancer cell replication, collagen deposition and neoangiogenesis | ||||||
| Detectable changes in tumour size from week 2 | ||||||
| DEN-LMBDL | Balb/c mouse | Young adult mice subjected to two separate weekly IP injections of DEN | 28 weeks | Not indicated | Not reported | [129] |
| After 2 weeks: LMBDL | ||||||
| After 1 week: DEN feeding (oral gavage, 25 mg/kg body weight, once a week) | ||||||
| 8th week: cyst formation | ||||||
| 12th week: biliary hyperplasia | ||||||
| 16th week: cholangiomas and adenomas | ||||||
| 28th week: CCA |
TAA = thioacetamide; DEN = N-nitrosodiethylamine; LMBDL = left median bile duct ligation.
Implanted in the liver via the inoculation of the left hepatic lobe.
Mansuroglu et al. demonstrated consistent development of CCA nodules in the 100% of the animals in 18 week 0.05% TAA treated rats.
The authors also proposed a slightly different model, in which BDEneu cells were implanted in the liver after having subjected the animal to common bile duct ligation (BDL). After 21 days, tumour growth was found to be significantly greater than that observed in animals not subjected to BDL. Extra-hepatic, peritoneal tumoural nodules were found in animals injected with BDEneu cells and subjected to BDL, but not in animals injected with cells and sham operated.
The current model has the advantage of employing cells that show biological features similar to the ones observed in human disease, such as TRAIL expression, COX-2 over expression and ERK1/2 hyper-phosphorylation [7,76–78]. In addition, and in accordance with human CCA, the model is associated with biliary obstruction, by which tumour development is further increased, and with progressive body weight loss.
From an experimental point of view, the model has two advantages: (i) tumour nodules develop consistently and (ii) within a short period of time (Table 3). These features make this model suitable for testing novel therapeutic molecules in pre-clinical studies.
Consistently, using this model, sorafenib was shown to reduce CCA growth. Sorafenib treatment produced a significant reduction in tumoural liver invasion, with complete regression in 22% of the treated animals [79].
More recently, BDEneu cell implanted rats were treated with JP1584, a small-molecule second mitochondria-derived activator of caspase (smac) mimetic [78], which resulted in a significant reduction in peritoneal metastatization, as compared to vehicle treated ones [78].
The limits of this model reside in the absence of de novo CCA development and the implantation of malignant cells in the absence of chronic biliary/liver injury, which differs from human disease. From an experimental point of view, the model requires abdominal manipulation and left bile duct incannulation, thus possibly altering the cytokine milieu within the liver and limiting its extensive employment in larger numbers. This model has been developed in rats, but probably has limited applications for pathophysiological studies in transgenic animals.
3.1.2. The “thioacetamide” model
Administration of TAA in rodents is a commonly used model for the induction of liver fibrosis and cirrhosis [47]. Over two decades ago, however, it was observed that oral feeding of rats with TAA caused biliary dysplasia and CCA [80,81]. Since then, the TAA rat model of CCA has been the one most studied and employed. TAA is given in drinking water, at a standard dose of 0.03%; this, in time, induces progressive weight loss, liver injury and fibrosis [81–84]. By the 9th week, foci of cholangiocyte proliferation and dysplasia can be detected and, by the 12th week, microfoci of cancerous cells develop [81,83,84]. Whitish, visible CCA tumours are observed from the 16th week of treatment, with the incidence of larger and invasive tumours increasing progressively to 100% of the animals by the 24th week (Fig. 2A) [81,83–86]. This CCA developmental path was independent from the rat strain (Table 3) [81,83–86]. Animal mortality is virtually null [81,83–86], with some experiments carried on for up to 40 weeks [87]. At 24 weeks, lung metastases can be detected (Fig. 2A) [86]; intra-hepatic CAA nodules persist even after TAA discontinuation at least for a period of observation of 8 weeks [86].
Fig. 2.
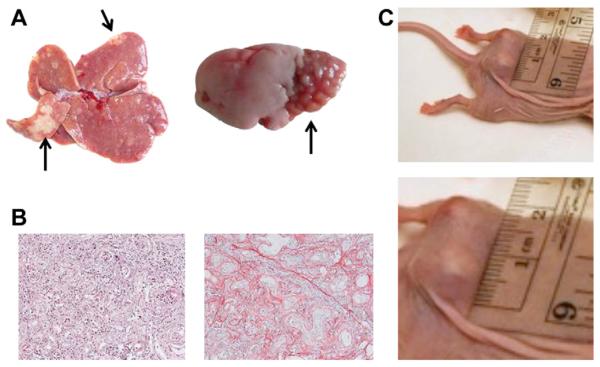
(A) Representative image of macroscopic appearance of TAA (thioacetamide)-induced IH-CCA (intrahepatic-cholangiocarcinoma) nodules. White-yellowish large nodules/arrows are consistently visible on the surface of the liver of treated animals after 24 weeks of 0.03% TAA administration to rats (left). Representative image of lung metastases due to TAA-induced IH-CCA. Bottom segments of left lung from a 24 week 0.03% TAA administration show clear evidence of nodules, metastases from IH-CCA (right). (From: Marzioni and Nilsson, unpublished observations (2011)). (B) Representative image of H&E staining of a TAA-induced IH-CCA nodule. Tumour is composed of deranged and irregular, duct forming tissue, together with a dense inflammatory infiltrate (original magnification 20x, left). Representative image of Sirius-Red staining of a TAA-induced IH-CCA nodule. Tumour shows an intense desmoplastic reaction, stained in red, similar to human disease (original magnification 20x, right). (From: Marzioni and Nilsson, unpublished observations, 2011). (C) Mz-ChA-1 cells (a CCA cell line) implanted subcutaneously in the flank of a nude mouse give rise to a clearly visible tumour. Tumour changes in size can be easily measured over time (top). Enlarged view of the same nodule (bottom). (From: Francis and Alpini, unpublished observations (2009)).
There have been attempts to modify the above-mentioned protocol by increasing the daily dose of TAA. Al-Bader et al., in a dose-response study, observed the anticipation of the development of CCA to weeks 11–13 if TAA was increased by 0.05–0.1%, whereas high mortality, before CCA development, was seen in animals receiving 0.15% TAA [82]. More recently, these data were confirmed by Mansuroglu et al., who showed the consistent development of CCA nodules in 100% of the animals in 18 weeks in 0.05% TAA treated rats [88].
The TAA model reproduces several features of human CCA, such as the association with chronic liver injury and fibrosis, the intense tumoural desmoplastic reaction and, most importantly, the persistent inflammation of liver parenchyma and bile ducts (Fig. 2B) [5,7]. The molecular phenotype of malignant cells in this model is similar to that of human disease, being positive for COX-2, EGFR, MUC1, MMP-2, MMP-9, c-Met, c-erb-B2, c-Kit and oestrogen receptors [83,84,87,88].
From an experimental point of view, the model has the advantage of requiring any abdominal manipulation or surgery; in addition, the simple TAA-enriched water induces a consistent development of CCA nodules. As a confirmation of its reproducibility and feasibility, this model has been employed in several pre-clinical studies in order to test novel diagnostic or therapeutic approaches for CCA. [18F]fluoro-2-deoxyglucose, a positron emission tomography tracer, accumulates in TAA-induced CCA and it is able to distinguish tumoural nodules from liver cirrhosis [89,90]. Administration of an oestrogen receptor-β selective agonists inhibits TAA-induced CCA development and reduces its progression after tumour full establishment [86].
The major limit of this model is that it is currently standardized only in rats. Besides the handling and care issues, the animals' marked increase in size and weight after 16–24 weeks of treatment implies the employment of greater amounts of compounds to be tested as novel therapeutic tools, especially when compared to mice. The limited availability of rats with genetic knock down of specific genes hampers the chances of studying the specific role of molecules involved in CCA pathophysiology.
3.2. Mouse models of CCA
3.2.1. The “Smad4-Pten knock out” model
The “Smad4-Pten knock out” mouse model of CCA was proposed by Xu et al. [91]. The authors used an elegant approach, the conditional disruption of both Smad4 and Pten, using the Cre-loxP. They crossbred mice carrying the Smad4 conditional allele (Smad4Co) and/or the Pten conditional allele (PtenCo), which were then crossed with albumin-Cre mice (Alb-Cre). Hyperplastic foci of the biliary epithelium were observed at 2–3 months of age in the so generated Smad4Co/CoPtenCo/CoAlb-Cre mice. Full and consistent development of CCA was observed in all the animals at 4–7 months of age, followed by a progressive increase of tumoural intra-hepatic nodules (Table 3).
This model is of major relevance for the understanding of the genetic and molecular mechanisms underlying disease development. SMAD4 is a tumour suppressor gene frequently altered in CCA [92]. PTEN (phosphatase and tensin homolog deleted chromosome 10) has been involved in the pathogenesis of several cancers [93]. PTEN loss induces a constitutive activation of the pro-proliferative and anti-apoptotic PI3K pathway, known to play a major role in human CCA development [77,94,95]. As a confirmation, tumoural cells of the Smad4Co/CoPtenCo/CoAlb-Cre mice were found to have ERK1/2 hyperphosphorylation, nuclear overexpression of cyclin D1, AKT hyperphosphorylation and nuclear translocation. This led the authors to investigate human CCA samples, finding PTEN inactivation by epigenetic modification and loss of expression of SMAD in 71% and 48% (respectively) of the phosphorylated-AKT positive tumours. Another point in favour of this model is that it allows the consistent development of tumours already at 4–5 months of age, without any further manipulation.
The limitations of this model reside in the absence of chronic liver injury and inflammation, the absence of metastases (even in older animals) and the concomitant development of tumours of the salivary glands, although in a limited number of mice. Another aspect of the study, is the utilization of the Alb-Cre mice, as a mean for delivering conditional gene knock out. By crossing these mice with Rosa-26, the authors observed that the Cre-mediated recombination was detected not only, as expected, in hepatocytes but also in cholangiocytes, thus justifying the knock down of Smad4 and Pten in cholangiocytes as well. However, recent studies showed that in conditional knock-out in Alb-Cre mice is highly specific for hepatocytes, being minimal in other liver cells [96,97]. How hepatocyte specific mutations may contribute to CCA development remains thus to be understood.
3.2.2. The “p53 knock out-carbon tetrachloride” model
This model was proposed by Farazi et al. [98], and consisted of CCl4 administration three times per week for 4 months to p53 knockout mice. Mutations of the p53 gene are frequent genomic alterations observed in human intra-hepatic CCA (IH-CCA) [5,99–101]. As expected, mice developed progressive liver injury and fibrosis, with associated bile duct proliferation. At early time points cholangiocyte death by apoptosis was observed only in p53+/+ and +/− mice, but not in −/− mice. Cytological abnormalities and, shortly after the end of CCl4 administration, foci of early carcinoma were detected only in p53−/− animals.
A cohort of mice was followed up for a longer term after the end of CCl4 administration. In time, fully developed IH-CCA nodules became detectable. Tumours were formed by deranged, infiltrating CK-19-positive ducts and tubules with a dense collagenous stroma. The p53 genotype had a major impact on tumour development: IH-CCA was detected only in p53−/− and +/− mice (54% and 18%, respectively), with a consistent reduction of tumour latency (29 weeks for −/− mice and 52 weeks for +/− mice) (Table 3).
From a pathophysiological point of view, the positive aspect of this model is that of combining a genetic susceptibility with a toxic chronic liver injury, a condition postulated to be similar to that leading to CCA development in humans [5]. As confirmation, tumoural nodules showed iNOS, COX-2, c-Met and cErbB2 positive malignant cholangiocytes [7,102–104]. From an experimental point of view, the model is limited by the length of time needed for tumours establishment (29–52 weeks) and by the lack of consistency in IH-CCA development.
3.2.3. The “xenograft” model
The first application of this model in the study of CCA was in 1985, when a cell line derived from a human CCA metastasis was injected subcutaneously into the flank of nude mice [105]. Detectable changes in tumour size in different experimental sets begin after 2 weeks from cell implantation (Fig. 2C), with studies following up to 11 weeks (Table 3). Besides patho-physiological studies [106–113], this model is suitable for testing the efficacy of novel therapeutic approaches for CCA. Molecules like tannic acid, resveratrol, caffeic acid, anandamide, tamoxifen, felodipine, melatonin, and clobenpropit were shown to inhibit CCA xenograft tumour growth as did hematoporphyrin derivative-mediated photodynamic therapy, and oncolytic gene therapy,[114–126]. Similarly, targeting CCA cells with Slug si-RNA increased tumour sensitivity to cisplatin [127].
Besides the species-specific differences, the micro-environment and pharmacodynamics of this model are critically different from the tumour developing within the liver [49]. One solution, proposed by Yokomuro et al., is to inject CCA cells directly into the livers of nude mice, although it carries the drawback of an abdominal incision [128].
3.2.4. The “DEN-left median bile duct ligation” model
This is the newest rodent model of CCA, being proposed by Yang et al. [129]. To achieve tumour development, the authors subjected young adult Balb/c mice to two separate weekly IP injections of DEN (diethylnitrosamine). Two weeks later, animals were subjected to left median bile duct ligation (LMBDL) and then, 1 week later DEN feeding by oral gavage, the total duration of the experiment being 28 weeks (Table 3). The overall survival of the animals was around 70% at the end of the 28 weeks. At week 8, livers showed multifocal cystic hyperplasia of the intra-hepatic bile ducts and multifocal cyst formation. At week 12, the biliary epithelium of the hyperplastic foci, and the epithelium lining the cysts showed elongated nuclei. Cholangiomas and biliary adenomas developed at week 16, with full development of CCA in these areas at week 28. CCA did not develop in control animals, i.e. those subjected to either DEN injection or feeding or to LMBDL, although the biliary epithelium was found to be abnormal. The number of liver c-Myc positive cells increased and remained persistently high in animals that developed CCA, whereas it increased and then tended to decrease in control animals.
The advantage of this model is that it allows the development of CCA in wild type mice, thus being the only one standardized for tumoural development in non-engineered mice. Other advantages are the induction of oncogenes such as c-Myc and the association with biliary obstruction, features thought to be important for the development of human primary liver cancers [130]. From a pathophysiological point of view, c-Myc overexpression was observed not only in cholangiocytes, but also in hepatocytes and inflammatory cells, which does not clarify the actual role of the molecule in the malignant transformation of cholangiocytes.
From the experimental point of view, the merit of this model is the short time required for tumour development (i.e. 28 weeks). On the other hand, the model is quite complex, needing subtle abdominal manipulation and long-term weekly gavage of the mice.
4. Conclusions
Animal models represent essential tools in cancer research, since they allow scientists to reproduce genetic, pathophysiological or environmental abnormalities thought to be important for cancer development. Novel therapeutic approaches can also be assayed in pre-clinical sets by employing oncologic models of diseases. It is common to use rodents for such studies, given their light weight, easy breeding and limited expense, as compared to other animals. Mice are widely used in these studies due to the availability of genetically altered mice [131].
Over the last few years, a number of HCC and CCA rodent models have been developed. With their heterogeneity, they all represent valuable tools to study and understand several pathophysiological aspects of these two malignancies.
In many cases it is difficult to determine to what extent mouse models reproduce features observed in corresponding human conditions. This issue has been elegantly evaluated by Prof. Thorgeirsson's group, who compared the global gene expression patterns of 68 HCCs from seven different mouse models and 91 human HCCs from predefined subclasses: the gene expression patterns in HCCs from Myc, E2f1 and Myc E2f1 transgenic mice were most similar to those of the human HCCs better survival group, whereas the expression patterns in HCCs from Myc Tgfα transgenic mice and in DEN-induced mouse HCCs were most similar to those of the human HCCs poorer survival group. Gene expression patterns in HCCs from Acox1−/− mice and in ciprofibrate-induced HCCs were least similar to those observed in human HCCs [132]. A similar study of the differences in gene expression between human disease and experimental models of CCA is still lacking. However, a synoptic view shows us that key features of human disease (such as genetic background, chronic liver injury and cholestasis) are inconsistently represented in the different models (Table 4). In addition, no models of extra-hepatic CCA (EH-CCA) are as yet available.
Table 4.
Synopsis of the main experimental features of rodent models of CCA.
| Model | Species | Orthotopic | Genetic | Toxic | Abdominal surgery | Inflammation | Intra/Extra-hepatic | References |
|---|---|---|---|---|---|---|---|---|
| Syngenic | Rat | √ | √ | Intra | [76,78,79] | |||
| TAA | Rat | √ | √ | √ | Intra | [80–88] | ||
| Smad4-Pten knock out | Mouse | √ | √ | Intra | [91] | |||
| p53 knock out-CCl4 | Mouse | √ | √ | √ | √ | Intra | [98] | |
| Xenograft | Mouse | Intra | [105–113] | |||||
| DEN-LMBDL | Mouse | √ | √ | √ | √ | Intra | [129] |
TAA = thioacetamide; DEN = N-nitrosodiethylamine; LMBDL = left median bile duct ligation.
To adequately interpret the significance of rodent models and to employ them properly for future studies, it is thus important to have a proper perception of their experimental features and similarities/differences with the corresponding human disease. Differences, in particular, stand as an “imperative” for researchers and the scientific community to pursue and develop the “ideal” model for studying primary liver cancers.
Acknowledgments
Funding This work was supported by a MIUR Grant PRIN 2009 – prot 2009X84L84 003 to Dr. Marzioni; by MIUR Grant PRIN 2009 – prot 2009YNERCE 002, FIRB 2010 – prot RBAP10MY35 001 to Dr. Svegliati Baroni.
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. HEALTH-F2-2009-241762 for the project FLIP.
Portions of the work presented here were supported by the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White Hospital, a VA Research Career Scientist Award, a VA Merit award and the NIH Grants DK58411 and DK76898 to Dr. Alpini.
Portions of the work presented here were supported by the NIH Grants GM41804 and AA15055.
List of abbreviations
- CCA
cholangiocarcinoma
- CCl4
carbon tetrachloride
- CDD
choline deficient diet
- DEN
N-nitrosodiethylamine
- HCC
hepatocellular carcinoma
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PB
phenobarbital
- PH
partial hepatectomy
- PPAR α
peroxisome proliferator activated receptor α
- PP
speroxisome proliferators
- ROS
reactive oxygen species
- TA
Athioacetamide
Footnotes
Conflict of interest Authors state that they have no conflict of interest.
References
- [1].El-Serag HB. Hepatocellular carcinoma. New England Journal of Medicine. 2011;365:1118–27. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- [2].Papatheodoridis GV, Lampertico P, Manolakopoulos S, et al. Incidence of hepatocellular carcinoma in chronic hepatitis B patients receiving nucleos(t)ide therapy: a systematic review. Journal of Hepatology. 2010;53:348–56. doi: 10.1016/j.jhep.2010.02.035. [DOI] [PubMed] [Google Scholar]
- [3].Roncalli M, Terracciano L, Di Tommaso L, et al. Liver precancerous lesions and hepatocellular carcinoma: the histology report. Digestive and Liver Disease. 2011;43(Suppl 4):S361–72. doi: 10.1016/S1590-8658(11)60592-6. [DOI] [PubMed] [Google Scholar]
- [4].Alves RC, Alves D, Guz B, et al. Advanced hepatocellular carcinoma. Review of targeted molecular drugs. Annal of Hepatology. 2011;10:21–7. [PubMed] [Google Scholar]
- [5].Khan SA, Thomas HC, Davidson BR, et al. Cholangiocarcinoma. Lancet. 2005;366:1303–14. doi: 10.1016/S0140-6736(05)67530-7. [DOI] [PubMed] [Google Scholar]
- [6].Wolpin BM, Mayer RJ. A step forward in the treatment of advanced biliary tract cancer. New England Journal of Medicine. 2010;362:1335–7. doi: 10.1056/NEJMe1001183. [DOI] [PubMed] [Google Scholar]
- [7].Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–21. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pitot HC, Dragan YP. Facts and theories concerning the mechanisms of carcinogenesis. FASEB Journal. 1991;5:2280–6. [PubMed] [Google Scholar]
- [9].Binato M, Kruel Schmidt M, Silveira Volkweis B, et al. Mouse model of diethylnitrosamine-induced gastric cancer. Journal of Surgical Research. 2008;148:152–7. doi: 10.1016/j.jss.2007.12.748. [DOI] [PubMed] [Google Scholar]
- [10].Gray R, Peto R, Brantom P, et al. Chronic nitrosamine ingestion in 1040 rodents: the effect of the choice of nitrosamine, the species studied, and the age of starting exposure. Cancer Research. 1991;51:6470–91. [PubMed] [Google Scholar]
- [11].Kawanishi S, Hiraku Y, Murata M, et al. The role of metals in site-specific DNA damage with reference to carcinogenesis. Free Radical Biology and Medicine. 2002;32:822–32. doi: 10.1016/s0891-5849(02)00779-7. [DOI] [PubMed] [Google Scholar]
- [12].Valko M, Rhodes CJ, Moncol J, et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- [13].Frey S, Buchmann A, Bursch W, et al. Suppression of apoptosis in C3H mouse liver tumors by activated Ha-ras oncogene. Carcinogenesis. 2000;21:161–6. doi: 10.1093/carcin/21.2.161. [DOI] [PubMed] [Google Scholar]
- [14].Park TJ, Kim JY, Oh SP, et al. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology. 2008;47:1533–43. doi: 10.1002/hep.22212. [DOI] [PubMed] [Google Scholar]
- [15].Teoh NC, Dan YY, Swisshelm K, et al. Defective DNA. strand break repair causes chromosomal instability and accelerates liver carcinogenesis in mice. Hepatology. 2008;47:2078–88. doi: 10.1002/hep.22194. [DOI] [PubMed] [Google Scholar]
- [16].Williams GM, Iatropoulos MJ, Jeffrey AM. Mechanistic basis for nonlinearities and thresholds in rat liver carcinogenesis by the DNA-reactive carcinogens 2-acetylaminofluorene and diethylnitrosamine. Toxicologic Pathology. 2000;28:388–95. doi: 10.1177/019262330002800306. [DOI] [PubMed] [Google Scholar]
- [17].Zimmers TA, Jin X, Gutierrez JC, et al. Effect of in vivo loss of GDF-15 on hepatocellular carcinogenesis. Journal of Cancer Research and Clinical Oncology. 2008;134:753–9. doi: 10.1007/s00432-007-0336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Finnberg N, Stenius U, Hogberg J. Heterozygous p53-deficient (+/−) mice develop fewer p53-negative preneoplastic focal liver lesions in response to treatment with diethylnitrosamine than do wild-type (+/+) mice. Cancer Letters. 2004;207:149–55. doi: 10.1016/j.canlet.2003.11.013. [DOI] [PubMed] [Google Scholar]
- [19].Shiota G, Harada K, Ishida M, et al. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis. 1999;20:59–63. doi: 10.1093/carcin/20.1.59. [DOI] [PubMed] [Google Scholar]
- [20].Hsu HC, Jeng YM, Mao TL, et al. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. American Journal of Pathology. 2000;157:763–70. doi: 10.1016/s0002-9440(10)64590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mao TL, Chu JS, Jeng YM, et al. Expression of mutant nuclear beta-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. Journal of Pathology. 2001;193:95–101. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH720>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- [22].Farber E, Solt D, Cameron R, et al. Newer insights into the pathogenesis of liver cancer. American Journal of Pathology. 1977;89:477–82. [PMC free article] [PubMed] [Google Scholar]
- [23].Klinman NR, Erslev AJ. Cellular response to partial hepatectomy. Proceedings of the Society for Experimental Biology and Medicine. 1963;112:338–40. doi: 10.3181/00379727-112-28037. [DOI] [PubMed] [Google Scholar]
- [24].Rignall B, Braeuning A, Buchmann A, et al. Tumor formation in liver of conditional beta-catenin-deficient mice exposed to a diethylnitrosamine/phenobarbital tumor promotion regimen. Carcinogenesis. 2011;32:52–7. doi: 10.1093/carcin/bgq226. [DOI] [PubMed] [Google Scholar]
- [25].Puatanachokchai R, Kakuni M, Wanibuchi H, et al. Lack of promoting effects of phenobarbital at low dose on diethylnitrosamine-induced hepatocarcinogenesis in TGF-alpha transgenic mice. Asian Pacific Journal of Cancer Prevention. 2006;7:274–8. [PubMed] [Google Scholar]
- [26].Hasmall SC, James NH, Macdonald N, et al. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: role of PPARalpha and TNFalpha. Mutation Research. 2000;448:193–200. doi: 10.1016/s0027-5107(99)00236-5. [DOI] [PubMed] [Google Scholar]
- [27].Reddy JK, Rao S, Moody DE. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Research. 1976;36:1211–7. [PubMed] [Google Scholar]
- [28].Hays T, Rusyn I, Burns AM, et al. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26:219–27. doi: 10.1093/carcin/bgh285. [DOI] [PubMed] [Google Scholar]
- [29].Zhao W, Iskandar S, Kooshki M, et al. Knocking out peroxisome proliferator-activated receptor (PPAR) alpha inhibits radiation-induced apoptosis in the mouse kidney through activation of NF-kappaB and increased expression of IAPs. Radiation Research. 2007;167:581–91. doi: 10.1667/RR0814.1. [DOI] [PubMed] [Google Scholar]
- [30].Yin PH, Lee HC, Chau GY, et al. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. British Journal of Cancer. 2004;90:2390–6. doi: 10.1038/sj.bjc.6601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu SP, Li YS, Chen YJ, et al. Glycine N-methyltransferase −/− mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology. 2007;46:1413–25. doi: 10.1002/hep.21863. [DOI] [PubMed] [Google Scholar]
- [32].Woo LL, Egner PA, Belanger CL, et al. Aflatoxin B1-DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxicological Sciences. 2011;122:38–44. doi: 10.1093/toxsci/kfr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hulla JE, Chen ZY, Eaton DL. Aflatoxin B1-induced rat hepatic hyperplastic nodules do not exhibit a site-specific mutation within the p53 gene. Cancer Research. 1993;53:9–11. [PubMed] [Google Scholar]
- [34].McGlynn KA, Hunter K, LeVoyer T, et al. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Research. 2003;63:4594–601. [PubMed] [Google Scholar]
- [35].Weisburger EK. Carcinogenicity studies on halogenated hydrocarbons. Environmental Health Perspectives. 1977;21:7–16. doi: 10.1289/ehp.77217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Campo GM, Avenoso A, Campo S, et al. The antioxidant activity of chondroitin-4-sulphate, in carbon tetrachloride-induced acute hepatitis in mice, involves NF-kappaB and caspase activation. British Journal of Pharmacology. 2008;155:945–56. doi: 10.1038/bjp.2008.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Domenicali M, Caraceni P, Principe A, et al. A novel sodium overload test predicting ascites decompensation in rats with CCl4-induced cirrhosis. Journal of Hepatology. 2005;43:92–7. doi: 10.1016/j.jhep.2005.01.034. [DOI] [PubMed] [Google Scholar]
- [38].Sheweita SA, Abd El-Gabar M, Bastawy M. Carbon tetrachloride-induced changes in the activity of phase II drug-metabolizing enzyme in the liver of male rats: role of antioxidants. Toxicology. 2001;165:217–24. doi: 10.1016/s0300-483x(01)00429-2. [DOI] [PubMed] [Google Scholar]
- [39].Farazi PA, Glickman J, Horner J, et al. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. Cancer Research. 2006;66:4766–73. doi: 10.1158/0008-5472.CAN-05-4608. [DOI] [PubMed] [Google Scholar]
- [40].Frezza EE, Gerunda GE, Farinati F, et al. CCL4-induced liver cirrhosis and hepatocellular carcinoma in rats: relationship to plasma zinc, copper and estradiol levels. Hepato-Gastroenterology. 1994;41:367–9. [PubMed] [Google Scholar]
- [41].Knight B, Yeoh GC, Husk KL, et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. Journal of Experimental Medicine. 2000;192:1809–18. doi: 10.1084/jem.192.12.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nakano T, Cheng YF, Lai CY, et al. Impact of artificial sunlight therapy on the progress of non-alcoholic fatty liver disease in rats. Journal of Hepatology. 2011;55:415–25. doi: 10.1016/j.jhep.2010.11.028. [DOI] [PubMed] [Google Scholar]
- [43].de Lima VM, Oliveira CP, Alves VA, et al. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. Journal of Hepatology. 2008;49:1055–61. doi: 10.1016/j.jhep.2008.07.024. [DOI] [PubMed] [Google Scholar]
- [44].Zhong B, Zhou Q, Toivola DM, et al. Organ-specific stress induces mouse pancreatic keratin overexpression in association with NF-kappaB activation. Journal of Cell Science. 2004;117:1709–19. doi: 10.1242/jcs.01016. [DOI] [PubMed] [Google Scholar]
- [45].Guest I, Ilic Z, Sell S. Age dependence of oval cell responses and bile duct carcinomas in male fischer 344 rats fed a cyclic choline-deficient, ethionine-supplemented diet. Hepatology. 2010;52:1750–7. doi: 10.1002/hep.23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kodama Y, Kisseleva T, Iwaisako K, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–77. e1465. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang MC, Chang CP, Lei HY. Induction of liver fibrosis in a murine hepatoma model by thioacetamide is associated with enhanced tumor growth and suppressed antitumor immunity. Laboratory Investigation. 2010;90:1782–93. doi: 10.1038/labinvest.2010.139. [DOI] [PubMed] [Google Scholar]
- [48].Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta Pathologica et Microbiologica Scandinavica. 1969;77:758–60. doi: 10.1111/j.1699-0463.1969.tb04520.x. [DOI] [PubMed] [Google Scholar]
- [49].Newell P, Villanueva A, Friedman SL, et al. Experimental models of hepatocellular carcinoma. Journal of Hepatology. 2008;48:858–79. doi: 10.1016/j.jhep.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sun FX, Tang ZY, Lui KD, et al. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. International Journal of Cancer. 1996;66:239–43. doi: 10.1002/(SICI)1097-0215(19960410)66:2<239::AID-IJC17>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [51].Huynh H, Soo KC, Chow PK, et al. Xenografts of human hepatocellular carcinoma: a useful model for testing drugs. Clinical Cancer Research. 2006;12:4306–14. doi: 10.1158/1078-0432.CCR-05-2568. [DOI] [PubMed] [Google Scholar]
- [52].Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Molecular Cancer Therapeutics. 2003;2:557–61. [PubMed] [Google Scholar]
- [53].Kornek M, Raskopf E, Tolba R, et al. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver International. 2008;28:509–18. doi: 10.1111/j.1478-3231.2008.01670.x. [DOI] [PubMed] [Google Scholar]
- [54].Tang TC, Man S, Xu P, et al. Development of a resistance-like phenotype to sorafenib by human hepatocellular carcinoma cells is reversible and can be delayed by metronomic UFT chemotherapy. Neoplasia. 2010;12:928–40. doi: 10.1593/neo.10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shao DM, Wang QH, Chen C, et al. N-acetylglucosaminyltransferase V activity in metastatic models of human hepatocellular carcinoma in nude mice. Journal of Experimental and Clinical Cancer Research. 1999;18:331–5. [PubMed] [Google Scholar]
- [56].Hollingshead MG, Alley MC, Camalier RF, et al. In vivo cultivation of tumor cells in hollow fibers. Life Sciences. 1995;57:131–41. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- [57].Suggitt M, Bibby MC. 50 years of preclinical anticancer drug screening: empirical to target-driven approaches. Clinical Cancer Research. 2005;11:971–81. [PubMed] [Google Scholar]
- [58].Shnyder SD, Cooper PA, Scally AJ, et al. Reducing the cost of screening novel agents using the hollow fibre assay. Anticancer Research. 2006;26:2049–52. [PubMed] [Google Scholar]
- [59].Frese KK, Tuveson DA. Maximizing mouse cancer models. Nature Reviews Cancer. 2007;7:645–58. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- [60].Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Current Opinion in Genetics and Development. 2002;12:105–10. doi: 10.1016/s0959-437x(01)00272-6. [DOI] [PubMed] [Google Scholar]
- [61].Koo JS, Seong JK, Park C, et al. Large liver cell dysplasia in hepatitis B virus × transgenic mouse liver and human chronic hepatitis B virus-infected liver. Intervirology. 2005;48:16–22. doi: 10.1159/000082090. [DOI] [PubMed] [Google Scholar]
- [62].Lakhtakia R, Kumar V, Reddi H, et al. Hepatocellular carcinoma in a hepatitis B `x' transgenic mouse model: a sequential pathological evaluation. Journal of Gastroenterology and Hepatology. 2003;18:80–91. doi: 10.1046/j.1440-1746.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- [63].Xiong J, Yao YC, Zi XY, et al. Expression of hepatitis B virus X protein in transgenic mice. World Journal of Gastroenterology. 2003;9:112–6. doi: 10.3748/wjg.v9.i1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Naas T, Ghorbani M, Alvarez-Maya I, et al. Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. Journal of General Virology. 2005;86:2185–96. doi: 10.1099/vir.0.80969-0. [DOI] [PubMed] [Google Scholar]
- [65].Kamegaya Y, Hiasa Y, Zukerberg L, et al. Hepatitis C. virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology. 2005;41:660–7. doi: 10.1002/hep.20621. [DOI] [PubMed] [Google Scholar]
- [66].Baek HJ, Lim SC, Kitisin K, et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128–37. doi: 10.1002/hep.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Beraza N, Malato Y, Sander LE, et al. Hepatocyte-specific NEMO. deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. Journal of Experimental Medicine. 2009;206:1727–37. doi: 10.1084/jem.20082152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. Journal of Clinical Investigation. 2004;113:1774–83. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Inokuchi S, Aoyama T, Miura K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:844–9. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- [71].Seki E, Brenner DA. The role of NF-kappaB in hepatocarcinogenesis: promoter or suppressor? Journal of Hepatology. 2007;47:307–9. doi: 10.1016/j.jhep.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Harada N, Oshima H, Katoh M, et al. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Research. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- [73].Merle P, Kim M, Herrmann M, et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. Journal of Hepatology. 2005;43:854–62. doi: 10.1016/j.jhep.2005.05.018. [DOI] [PubMed] [Google Scholar]
- [74].Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. American Journal of Pathology. 2002;160:2295–307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Watanabe S, Horie Y, Kataoka E, et al. Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. Journal of Gastroenterology and Hepatology. 2007;22(Suppl 1):S96–100. doi: 10.1111/j.1440-1746.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- [76].Sirica AE, Zhang Z, Lai GH, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–90. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- [77].Fava G, Marzioni M, Benedetti A, et al. Molecular pathology of biliary tract cancers. Cancer Letters. 2007;250:155–67. doi: 10.1016/j.canlet.2006.09.011. [DOI] [PubMed] [Google Scholar]
- [78].Fingas CD, Blechacz BR, Smoot RL, et al. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology. 2010;52:550–61. doi: 10.1002/hep.23729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Blechacz BR, Smoot RL, Bronk SF, et al. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861–70. doi: 10.1002/hep.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Praet MM, Roels HJ. Histogenesis of cholangiomas and cholangiocarcinomas in thioacetamide fed rats. Experimental Pathology. 1984;26:3–14. doi: 10.1016/s0232-1513(84)80063-8. [DOI] [PubMed] [Google Scholar]
- [81].Dashti H, Jeppsson B, Hagerstrand I, et al. Thioacetamide- and carbon tetrachloride-induced liver cirrhosis. European Surgical Research. 1989;21:83–91. doi: 10.1159/000129007. [DOI] [PubMed] [Google Scholar]
- [82].Al-Bader A, Mathew TC, Abul H, et al. Cholangiocarcinoma and liver cirrhosis in relation to changes due to thioacetamide. Molecular and Cellular Biochemistry. 2000;208:1–10. doi: 10.1023/a:1007082515548. [DOI] [PubMed] [Google Scholar]
- [83].Jan YY, Yeh TS, Yeh JN, et al. Expression of epidermal growth factor receptor, apomucins, matrix metalloproteinases, and p53 in rat and human cholangiocarcinoma: appraisal of an animal model of cholangiocarcinoma. Annals of Surgery. 2004;240:89–94. doi: 10.1097/01.sla.0000129492.95311.f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yeh CN, Maitra A, Lee KF, et al. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis. 2004;25:631–6. doi: 10.1093/carcin/bgh037. [DOI] [PubMed] [Google Scholar]
- [85].Fava G, Alpini G, Rychlicki C, et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Research. 2008;68:6752–61. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Marzioni M, Torrice A, Saccomanno S, et al. An oestrogen receptor beta-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Digestive and Liver Disease. 2012;44:132–42. doi: 10.1016/j.dld.2011.06.014. [DOI] [PubMed] [Google Scholar]
- [87].Liu KH, Liao LM, Ro LS, et al. Thalidomide attenuates tumor growth and preserves fast-twitch skeletal muscle fibers in cholangiocarcinoma rats. Surgery. 2008;143:375–83. doi: 10.1016/j.surg.2007.09.035. [DOI] [PubMed] [Google Scholar]
- [88].Mansuroglu T, Ramadori P, Dudas J, et al. Expression of stem cell factor and its receptor c-Kit during the development of intrahepatic cholangiocarcinoma. Laboratory Investigation. 2009;89:562–74. doi: 10.1038/labinvest.2009.15. [DOI] [PubMed] [Google Scholar]
- [89].Laverman P, Blokx WA, Te Morsche RH, et al. [(18)F]FDG accumulation in an experimental model of multistage progression of cholangiocarcinoma. Hepatology Research. 2007;37:127–32. doi: 10.1111/j.1872-034X.2007.00016.x. [DOI] [PubMed] [Google Scholar]
- [90].Yeh CN, Lin KJ, Hsiao IT, et al. Animal PET for thioacetamide-induced rat cholangiocarcinoma: a novel and reliable platform. Molecular Imaging and Biology. 2008;10:209–16. doi: 10.1007/s11307-008-0141-8. [DOI] [PubMed] [Google Scholar]
- [91].Xu X, Kobayashi S, Qiao W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. Journal of Clinical Investigation. 2006;116:1843–52. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kang YK, Kim WH, Jang JJ. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Human Pathology. 2002;33:877–83. doi: 10.1053/hupa.2002.127444. [DOI] [PubMed] [Google Scholar]
- [93].Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. Journal of Clinical Oncology. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- [94].Kobayashi S, Werneburg NW, Bronk SF, et al. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054–65. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- [95].Tanno S, Yanagawa N, Habiro A, et al. Serine/threonine kinase AKT is frequently activated in human bile duct cancer and is associated with increased radioresistance. Cancer Research. 2004;64:3486–90. doi: 10.1158/0008-5472.CAN-03-1788. [DOI] [PubMed] [Google Scholar]
- [96].Taura K, Miura K, Iwaisako K, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–36. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. Journal of Biological Chemistry. 2007;282:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- [98].Farazi PA, Zeisberg M, Glickman J, et al. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Research. 2006;66:6622–7. doi: 10.1158/0008-5472.CAN-05-4609. [DOI] [PubMed] [Google Scholar]
- [99].Momoi H, Itoh T, Nozaki Y, et al. Microsatellite instability and alternative genetic pathway in intrahepatic cholangiocarcinoma. Journal of Hepatology. 2001;35:235–44. doi: 10.1016/s0168-8278(01)00106-4. [DOI] [PubMed] [Google Scholar]
- [100].Furubo S, Harada K, Shimonishi T, et al. Protein expression and genetic alterations of p53 and ras in intrahepatic cholangiocarcinoma. Histopathology. 1999;35:230–40. doi: 10.1046/j.1365-2559.1999.00705.x. [DOI] [PubMed] [Google Scholar]
- [101].Tullo A, D'Erchia AM, Honda K, et al. New p53 mutations in hilar cholangiocarcinoma. European Journal of Clinical Investigation. 2000;30:798–803. doi: 10.1046/j.1365-2362.2000.00717.x. [DOI] [PubMed] [Google Scholar]
- [102].Okuda K, Nakanuma Y, Miyazaki M. Cholangiocarcinoma: recent progress. Part 2: molecular pathology and treatment. Journal of Gastroenterology & Hepatology. 2002;17:1056–63. doi: 10.1046/j.1440-1746.2002.02780.x. [DOI] [PubMed] [Google Scholar]
- [103].Okuda K, Nakanuma Y, Miyazaki M. Cholangiocarcinoma: recent progress. Part 1: epidemiology and etiology. Journal of Gastroenterology and Hepatology. 2002;17:1049–55. doi: 10.1046/j.1440-1746.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- [104].Endo K, Yoon BI, Pairojkul C, et al. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36:439–50. doi: 10.1053/jhep.2002.34435. [DOI] [PubMed] [Google Scholar]
- [105].Hudd C, Euhus DM, LaRegina MC, et al. Effect of cholecystokinin on human cholangiocarcinoma xenografted into nude mice. Cancer Research. 1985;45:1372–7. [PubMed] [Google Scholar]
- [106].Wang M, Xiao J, Shen M, et al. Isolation and characterization of tumorigenic extrahepatic cholangiocarcinoma cells with stem cell-like properties. International Journal of Cancer. 2011;128:72–81. doi: 10.1002/ijc.25317. [DOI] [PubMed] [Google Scholar]
- [107].Lu D, Han C, Wu T. Microsomal prostaglandin E synthase-1 inhibits PTEN and promotes experimental cholangiocarcinogenesis and tumor progression. Gastroenterology. 2011;140(7):2084–94. doi: 10.1053/j.gastro.2011.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rozich RA, Mills DR, Brilliant KE, et al. Accumulation of neoplastic traits prior to spontaneous in vitro transformation of rat cholangiocytes determines susceptibility to activated ErbB-2/Neu. Experimental and Molecular Pathology. 2010;89:248–59. doi: 10.1016/j.yexmp.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Shiraso S, Katayose Y, Yamamoto K, et al. Overexpression of adenovirus-mediated p27kip1 lacking the Jab1-binding region enhances cytotoxicity and inhibits xenografted human cholangiocarcinoma growth. Anticancer Research. 2009;29:2015–24. [PubMed] [Google Scholar]
- [110].Fava G, Demorrow S, Gaudio E, et al. Endothelin inhibits cholangiocarcinoma growth by a decrease in the vascular endothelial growth factor expression. Liver International. 2009;29:1031–42. doi: 10.1111/j.1478-3231.2009.01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Fava G, Marucci L, Glaser S, et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Research. 2005;65:11437–46. doi: 10.1158/0008-5472.CAN-05-1470. [DOI] [PubMed] [Google Scholar]
- [112].Francis H, Onori P, Gaudio E, et al. H3 histamine receptor-mediated activation of protein kinase Calpha inhibits the growth of cholangiocarcinoma in vitro and in vivo. Molecular Cancer Research. 2009;7:1704–13. doi: 10.1158/1541-7786.MCR-09-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology. 2010;51:881–90. doi: 10.1002/hep.23381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jing G, Yuan K, Turk AN, et al. Tamoxifen enhances therapeutic effects of gemcitabine on cholangiocarcinoma tumorigenesis. Laboratory Investigation. 2011;91:896–904. doi: 10.1038/labinvest.2011.60. [DOI] [PubMed] [Google Scholar]
- [115].Huang L, Ramirez JC, Frampton GA, et al. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Laboratory Investigation. 2011;91(7):1007–117. doi: 10.1038/labinvest.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Demorrow S, Onori P, Venter J, et al. Neuropeptide Y inhibits cholangiocarcinoma cell growth invasion. American Journal of Physiology Cell Physiology. 2011;300:C1078–89. doi: 10.1152/ajpcell.00358.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].DeMorrow S, Francis H, Gaudio E, et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. American Journal of Physiology Cell Physiology. 2008;295:G1150–8. doi: 10.1152/ajpgi.90455.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lang M, Henson R, Braconi C, et al. Epigallocatechin-gallate modulates chemotherapy-induced apoptosis in human cholangiocarcinoma cells. Liver International. 2009;29:670–7. doi: 10.1111/j.1478-3231.2009.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Onori P, DeMorrow S, Gaudio E, et al. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF-kappaB and induction of apoptosis. International Journal of Cancer. 2009;125:565–76. doi: 10.1002/ijc.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kojima Y, Honda K, Hamada H, et al. Oncolytic gene therapy combined with double suicide genes for human bile duct cancer in nude mouse models. Journal of Surgical Research. 2009;157:e63–70. doi: 10.1016/j.jss.2008.12.016. [DOI] [PubMed] [Google Scholar]
- [121].Cao LQ, Xue P, Lu HW, et al. Hematoporphyrin derivative-mediated photodynamic therapy inhibits tumor growth in human cholangiocarcinoma in vitro and in vivo. Hepatology Research. 2009;39:1190–7. doi: 10.1111/j.1872-034X.2009.00569.x. [DOI] [PubMed] [Google Scholar]
- [122].Frampton GA, Lazcano EA, Li H, et al. Resveratrol enhances the sensitivity of cholangiocarcinoma to chemotherapeutic agents. Laboratory Investigation. 2010;90:1325–38. doi: 10.1038/labinvest.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Marienfeld C, Tadlock L, Yamagiwa Y, et al. Inhibition of cholangiocarcinoma growth by tannic acid. Hepatology. 2003;37:1097–104. doi: 10.1053/jhep.2003.50192. [DOI] [PubMed] [Google Scholar]
- [124].Braconi C, Swenson E, Kogure T, et al. Targeting the IL-6 dependent phenotype can identify novel therapies for cholangiocarcinoma. PLoS One. 2010;5:e15195. doi: 10.1371/journal.pone.0015195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Han Y, Demorrow S, Invernizzi P, et al. Melatonin exerts by an autocrine loop antiproliferative effects in cholangiocarcinoma; its synthesis is reduced favoring cholangiocarcinoma growth. American Journal of physiology Gastrointestinal and Liver Physiology. 2011;301(4):G623–33. doi: 10.1152/ajpgi.00118.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Meng F, Han Y, Staloch D, et al. The H4HR agonist, clobenpropit, suppresses human cholangiocarcinoma progression by disruption of EMT and tumor metastasis. Hepatology. 2011;54(5):1718–28. doi: 10.1002/hep.24573. [DOI] [PubMed] [Google Scholar]
- [127].Zhang K, Chen D, Wang X, et al. RNA interference targeting slug increases cholangiocarcinoma cell sensitivity to cisplatin via upregulating PUMA. International Journal of Molecular Sciences. 2011;12:385–400. doi: 10.3390/ijms12010385. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [128].Yokomuro S, Tsuji H, Lunz JG, 3rd, et al. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor beta1, and activin A: comparison of a cholangiocarcinoma cell line with primary cultures of non-neoplastic biliary epithelial cells. Hepatology. 2000;32:26–35. doi: 10.1053/jhep.2000.8535. [DOI] [PubMed] [Google Scholar]
- [129].Yang H, Li TW, Peng J, et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology. 2011;141(1):378–88. doi: 10.1053/j.gastro.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Marquardt JU, Raggi C, Andersen JB, et al. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology. 2011;54(3):1031–42. doi: 10.1002/hep.24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].de Jong M, Maina T. Of mice and humans: are they the same? – Implications in cancer translational research. Journal of Nuclear Medicine. 2010;51:501–4. doi: 10.2967/jnumed.109.065706. [DOI] [PubMed] [Google Scholar]
- [132].Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nature Genetics. 2004;36:1306–11. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]