

Abstract
Insulin-secreting pancreatic islet β-cells express a Group VIA Ca2+-independent phospholipase A2 (iPLA2β) that contains a calmodulin binding site and protein interaction domains. We identified Ca2+/calmodulin-dependent protein kinase IIβ (CaMKIIβ) as a potential iPLA2β-interacting protein by yeast two-hybrid screening of a cDNA library using iPLA2β cDNA as bait. Cloning CaMKIIβ cDNA from a rat islet library revealed that one dominant CaMKIIβ isoform mRNA is expressed by adult islets and is not observed in brain or neonatal islets and that there is high conservation of the isoform expressed by rat and human β-cells. Binary two-hybrid assays using DNA encoding this isoform as bait and iPLA2β DNA as prey confirmed interaction of the enzymes, as did assays with CaMKIIβ as prey and iPLA2β bait. His-tagged CaMKIIβ immobilized on metal affinity matrices bound iPLA2β, and this did not require exogenous calmodulin and was not prevented by a calmodulin antagonist or the Ca2+ chelator EGTA. Activities of both enzymes increased upon their association, and iPLA2β reaction products reduced CaMKIIβ activity. Both the iPLA2β inhibitor bromoenol lactone and the CaMKIIβ inhibitor KN93 reduced arachidonate release from INS-1 insulinoma cells, and both inhibit insulin secretion. CaMKIIβ and iPLA2β can be coimmunoprecipitated from INS-1 cells, and forskolin, which amplifies glucose-induced insulin secretion, increases the abundance of the immunoprecipitatable complex. These findings suggest that iPLA2β and CaMKIIβ form a signaling complex in β-cells, consistent with reports that both enzymes participate in insulin secretion and that their expression is coinduced upon differentiation of pancreatic progenitor to endocrine progenitor cells.
Phospholipases A2 (PLA2)1 are a diverse group of enzymes that catalyze hydrolysis of sn-2 fatty acid substituents from glycerophospholipid substrates to yield a free fatty acid and a 2-lysophospholipid (1). The Group VIA PLA2 designated iPLA2β has a molecular mass of 84–88 kDa and does not require Ca2+ for catalysis (2). Various splice variants of iPLA2β are expressed at high levels in testis (3), brain (4), and pancreatic islet β-cells (5), among other tissues.
Certain nutrients, hormones, neurotransmitters, and pharmacologic agents stimulate insulin secretion from β-cells, and the dominant physiologic insulin secretagogue is d-glucose. A series of signals that result from glucose-induced ATP production and alterations of intracellular redox potentials trigger insulin secretion via a rise in cytosolic [Ca2+], and Ca2+/calmodulin-dependent protein kinase II (CaMKII) is an important β-cell [Ca2+] sensor and mediator of Ca2+-dependent events in insulin secretion (6–11). Much evidence (12–22) suggests that iPLA2β also participates in insulin secretion, including the facts that the mechanism-based bromoenol lactone (BEL) inhibitor of iPLA2β suppresses glucose-induced hydrolysis of arachidonate from islet membrane phospholipids, the rise in β-cell cytosolic [Ca2+], and insulin secretion (19–22).
Depleting intracellular Ca2+ stores activates iPLA2β in β-cells and vascular smooth muscle cells (23, 24), and iPLA2β participates in store-operated entry of Ca2+ from the extracellular space (25), which is thought to be involved in glucose-induced insulin secretion (26–31). Regulating store-operated calcium (SOC) entry requires that intracellular Ca2+ stores communicate with plasma membrane ion channels, and calmodulin participates in cross-talk between Ca2+ stores and SOC channels (25, 32). Lipid signaling molecules (e.g. lysophospholipids) and Ca2+-sensitive kinases and phosphatases (e.g. CaMKIIβ and calcineurin) are also proposed to affect these interactions (9, 10, 25, 32). Mechanisms whereby iPLA2β participates in glucose-induced rises in β-cell cytosolic [Ca2+] and insulin secretion are likely to involve Ca2+-sensitive regulation of modulatory and effector proteins by phosphorylation-dephosphorylation events (9, 10), and iPLA2β activity is also affected by local [Ca2+] increments that relieve its tonic inhibition by Ca2+/calmodulin (2, 8, 25).
The amino acid sequence of iPLA2β contains an ankyrin repeat domain with eight strings of a repetitive motif of about 33 amino acid residues each (34). Ankyrin repeats link integral membrane proteins to the cytoskeleton and mediate protein-protein interactions in signaling (34–38). Ankyrin binds to inositol trisphosphate receptors (37), for example, which are located on Ca2+-containing vesicles that release intracellular Ca2+ when β-cells are stimulated with glucose (26–31). Ankyrin G also associates with skeletal muscle postsynaptic membranes and sarcoplasmic reticulum (38), and CaMKII participates in regulating local [Ca2+] gradients in subcellular zones involved in Ca2+ signaling. CaMKII is an important Ca2+ signaling effector and serves as a gauge that temporally integrates [Ca2+] signal intensities (39), and calmodulin participates in several Ca2+-dependent processes in insulin secretion by β-cells (40, 41). Calmodulin and iPLA2β interact functionally (2, 8, 23, 24, 33), and the iPLA2β domain from residues 650–722 contains a calmodulin binding site (2).
During cell signaling, iPLA2 translocates to membranes (22, 25, 32) where it interacts with regulatory proteins to effect cellular activation. To identify proteins that interact with iPLA2β to understand better its role in signaling, we performed yeast two-hybrid screening and have found that iPLA2β interacts with the specific CaMKIIβ isoform expressed in pancreatic islet β-cells. This interaction is demonstrated by multiple independent techniques, and the interaction affects both iPLA2β and CaMKIIβ activities, thereby defining a signaling complex.
EXPERIMENTAL PROCEDURES
Materials
The materials [γ-32P]ATP, 55 mCi/mmol (16:0/[14C]18:2)-glycerophosphocholine [1-palmitoyl-2-[14C]linoleoyl-sn-glycero-3-phosphocholine, rainbow molecular mass standards, enhanced chemiluminescence (ECL) reagent, and [3H]arachidonic acid were obtained from Amersham Biosciences. SDS-PAGE supplies were purchased from Bio-Rad. Coomassie reagent was obtained from Pierce. Alkaline phosphatase and peroxidase-conjugated goat anti-rabbit IgG antibodies were obtained from Roche Applied Science. Protease inhibitor mixture, kanamycin, ampicillin, ATP, calmodulin, autocamtide-3, arachidonic acid, lysophosphatidylcholine, calmodulin kinase II inhibitor KN93, common reagents, and salts were obtained from Sigma. Tetracycline was obtained from Invitrogen. Gentamicin and cell culture media were obtained from the Tissue Culture Support Center (Washington University, St. Louis, MO). TALON metal affinity resin, a rat brain cDNA library, AH109 yeast cells, and media for yeast two-hybrid screening were obtained from Clontech (Palo Alto, CA). Polyclonal antibodies to iPLA2β and CaMKII were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The calmodulin inhibitor W13 was obtained from Calbiochem. The iPLA2β suicide substrate BEL was obtained from Cayman Chemical Company (Ann Arbor, MI). Avian myeloblastosis virus reverse transcriptase was obtained from Roche Applied Science.
Screening of a Rat Brain cDNA Library in the Yeast Two-hybrid System
A rat brain cDNA library cloned into pACT2 vector (containing the LEU2 gene for selection) to produce fusion between protein-encoding DNA sequences and the DNA activation domain of GAL4 was used as prey. To produce the bait construct, full-length iPLA2β cDNA cloned from rat pancreatic islets was ligated into the SalI-EcoRI sites of the two-hybrid BD pAS2-1 vector, which contained a TRP1 gene for selection, resulting in in-frame fusion of iPLA2β with the DNA binding domain of the yeast GAL4 protein. The fidelity of constructs was confirmed by automated sequencing. The yeast strain AH109 was used for screening assays, and this strain contains HIS3 and lacZ reporter genes. Expression of each of these genes is regulated by a distinct GAL4-responsive promoter under control of a GAL4-responsive upstream activation site. Lack of autonomous activation by the iPLA2β/DNA binding domain fusion product was demonstrated by plating cells transformed with bait alone on media lacking histidine. In these assays, both bait and prey plasmids were transformed simultaneously into AH109 yeast cells, which were plated on medium lacking leucine, tryptophan, and histidine and allowed to grow at 30 °C for 4 days. Putative positive colonies were lifted onto filter paper and incubated with the chromogenic substrate X-gal. Interactions were confirmed when the blue β-galactosidase reaction product was evident after 4 h of incubation at room temperature. Plasmids were then recovered from yeast and transformed into DH5α bacterial cells using ampicillin for selection. Isolated plasmids were sequenced, and BLAST searches were performed against GenBank (NIH genetic sequence data base) to identify putative iPLA2β-interacting proteins.
Molecular Cloning of CaMKIIβ cDNA from a Rat Islet Library
Total RNA was isolated from adult rat islets as described previously (5). First strand cDNA was transcribed with avian myeloblastosis virus reverse transcriptase. PCR was performed using a pair of gene-specific primers designed from regions of cDNA sequence that are conserved in the mouse and rat brain CaMKIIβ cDNA sequences (sense, 5′-ATCGCCACCGCCATGGCCACC-3′; antisense, 5′-CAGGCGCAGCTCTCACTGCAG-3′). A PCR band of 1,650 bp was gel purified, ligated into pGEM-T vector, and transformed into DH5α cells for amplification. DNA was purified and sequenced using T3 and T7 primers and gene-specific primers.
Binary Yeast Two-hybrid Assays
The iPLA2β cDNA was cloned from an adult rat islet library (5). Full-length iPLA2β cDNA was ligated into BD vector pAS2-1 or AD vector pACT2 and used as bait or prey. Full-length CaMKIIβ cDNA was cloned into the AD vector pACT2 or BD vector pAS2-1 and used as prey or bait. Both bait and prey plasmids were transformed simultaneously into AH109 yeast cells, which were plated on restriction medium. After incubation (30 °C, 4 days), colonies were lifted onto filter paper and screened as described above. Colonies that produced the blue β-galactosidase reaction product were considered positive for the interaction between iPLA2β and CaMKIIβ.
Cloning and Expression of His-tagged CaMKIIβ, His-tagged iPLA2β, and FLAG-tagged Proteins in Sf9 Cells
Recombinant proteins were expressed in Spodoptera frugiperda (Sf9) cells using the Bac-to-Bac baculovirus expression system (Invitrogen) following the manufacturer's instructions, as described in detail elsewhere (2, 23, 33, 42). cDNA containing the entire coding sequence of His-tagged CaMKIIβ, His-tagged iPLA2β, or FLAG-tagged iPLA2β was cloned into the SalI-EcoRI site of the pFastBac-1 vector. The sequence of the insert was verified, and the plasmid was then transformed into DH10Bac cells. Recombinant bacmid DNA was isolated using an alkaline lysis protocol modified for high molecular weight plasmid purification. PCR analysis was performed with purified bacmid DNA and pUC/M13 forward and reverse primers to characterize the inserts in the recombinant bacmid DNA. The recombinant baculovirus was produced by transfecting the recombinant bacmid DNA into Sf9 cells. The baculovirus was amplified and used to infect Sf9 cell cultures to express the recombinant proteins (2, 23, 33, 42).
Immunoblotting Analyses
Proteins were analyzed by SDS-PAGE and transferred to a nylon membrane that was subsequently blocked with 5% nonfat dry milk for 1 h. The membrane was washed and incubated for 1 h with polyclonal antibody (1:200) to iPLA2β or CaMKII. The membrane was then incubated with secondary antibody (1:30,000) coupled to horseradish peroxidase, and the antibody complex was visualized by ECL.
Interaction of CaMKIIβ with iPLA2β and Protein Pull-down Assays
In some experiments, both iPLA2β and His-tagged CaMKIIβ proteins were coexpressed in Sf9 cells. The Sf9 cell cytosol containing iPLA2β and His-tagged CaMKIIβ proteins was mixed with TALON metal affinity resin in the presence or absence of added Ca2+/calmodulin, the calmodulin antagonist W13, or the Ca2+ chelator EGTA and incubated (room temperature, with shaking, for 1 h). The mixture was washed with 10 bed volumes of wash buffer (50 mm Na2HPO4, 500 mm NaCl, pH 7.8) twice and transferred onto a gravity-flow column. The His-tagged CaMKIIβ was eluted with elution buffer (50 mm Na2HPO4, 300 mm NaCl, and 200 mm imidazole, pH 7.8) and collected in 0.5-ml fractions. Desorbed proteins were visualized by immunoblotting analyses with antibodies to iPLA2β or CaMKIIβ.
In other experiments, iPLA2β protein was first expressed in Sf9 cells and purified as described previously (23, 33). Cytosol was prepared from Sf9 cells infected with baculovirus containing cDNA that encoded His-tagged CaMKIIβ and mixed with 1 ml of TALON metal affinity resin, as described above. The resin was washed and mixed with purified iPLA2β protein. The mixture was then incubated (30 min at room temperature with shaking), washed three times, and loaded onto a 5-ml gravity-flow column. Bound proteins were desorbed with elution buffer, collected in 0.5-ml fractions, and analyzed by immunoblotting with antibodies specific for iPLA2β or CaMKIIβ.
Immunoprecipitation of FLAG-tagged iPLA2β and CaMKIIβ Expressed in Sf9 Cells
Sf9 cells expressing FLAG-tagged iPLA2β, CaMKIIβ, or both were harvested by centrifugation, washed with phosphate-buffered saline, resuspended in cell lysis buffer supplemented with protease inhibitors, and homogenized by sonication. Cytosol was prepared by centrifugation (15,000 × g, 20 min) and incubated with 100 μl of anti-FLAG M2 affinity resin (2 h, 4 °C, gentle rotation) in the presence or absence of 10 mm Ca2+ chelator EGTA. Immunoprecipitated material was recovered by centrifugation and washed four times with wash buffer. Samples immunoprecipitated with anti-FLAG affinity resin were eluted with elution buffer (0.1 m glycine, pH 3.5). Aliquots (30 μl) were analyzed by 10% SDS-PAGE, transferred onto a nylon membrane, and blotted with iPLA2β or CaMKIIβ antibodies (1:200) followed by horseradish peroxidase-conjugated secondary antibodies (1:30,000).
Enzyme Activity Assays
For CaMKIIβ activity assays, sample buffer (50 mm Pipes, 10 mm MgCl2, 1 mm dithiothreitol, 0.1 mm ATP, 0.75 mm CaCl2, 20 μg/ml calmodulin, 20 μm autocamtide-3, 2 μCi of [γ-32P]ATP, pH 7.4) was mixed with CaMKIIβ (final volume 50 μl) and incubated (30 °C, 3 min). Assays were initiated by adding His-tagged CaMKIIβ and terminated by adding 100 mm EDTA. Aliquots (30 μl) of the mixture were placed on Whatman P-81 phosphocellulose paper, which was washed with 75 mm H3PO4 and air-dried. Phosphorylated autocamtide-3 (a CaMKIIβ model substrate) was quantified by liquid scintillation counting of 32P. Control assays were performed without added Ca2+/calmodulin and with 10 mm EGTA.
The iPLA2β activity assays were performed as described previously (5, 22). Briefly, 100 μl of sample was added to assay buffer containing 10 mm EGTA. Reactions were initiated by injecting 5 μl of 1-palmitoyl-2-[1-14C]palmitoyl-sn-glycerol-3-phosphorylcholine (specific activity 50 mCi/mmol, final concentration 5 μm) in ethanol. The assay mixture was incubated (37 °C, 5 min, with shaking), and the reaction was terminated by adding 100 μl of butanol. A 25-μl aliquot of the butanol layer was analyzed by Silica Gel G TLC as described previously (19). The amount of 14C-labeled free fatty acid was determined by liquid scintillation spectrometry.
[3H]Arachidonic Acid Release Measurements
INS-1 insulinoma cells (5 × 105 cells/well) were prelabeled for 20 h with 0.5 μCi/ml [3H]arachidonic acid and placed in serum-free medium for 1 h. The cells were washed three times with glucose-free RPMI 1640 medium to remove unincorporated radiolabel. Cells were treated with 20 μm BEL or 8 μm KN93 for 30 min before adding RPMI 1640 medium containing 0.5% bovine serum albumin and incubating for 1 h. The medium was then removed and replaced with fresh medium of identical composition, and the cells were incubated for 40 min. Supernatants and cells were separated by centrifugation (500 × g, 5 min) and assayed for 3H content by liquid scintillation spectrometry.
Coimmunoprecipitation of iPLA2β and CaMKIIβ from INS-1 Cells
Immunoprecipitation was performed with a protein A-agarose slurry that had been washed twice with phosphate-buffered saline, mixed with a 10-μl solution of antibody to CaMKIIβ or to iPLA2β, and incubated (room temperature, 40 min). The mixture was centrifuged, and the supernatant was discarded. The agarose-antibody complex in the precipitate was washed three times with phosphate-buffered saline, mixed with INS-1 cell cytosol, and incubated (overnight, 4 °C, with shaking). Immunoprecipitates were collected by centrifugation, washed, boiled for 5 min in SDS-PAGE sample loading buffer, and analyzed by SDS-PAGE. Proteins were transferred to nylon membranes, and immunoblotting was performed with primary antibody to iPLA2β or to CaMKIIβ and secondary antibody coupled to horseradish peroxidase, as described above.
RESULTS
Yeast Two-hybrid Screening Indicates That CaMKIIβ Is an iPLA2β-interacting Protein
To identify proteins that interact with iPLA2β, a yeast two-hybrid screen of a rat brain cDNA library was performed using iPLA2β cDNA cloned from a rat islet cDNA library as bait. The commercially available rat brain cDNA library was used for screening because there are many biochemical similarities between brain and islets, including high iPLA2β expression (19–22, 34). Colonies were identified that activated transcription of both the HIS3 gene (permitting autotrophic selection) and the lacZ reporter gene (permitting X-gal analysis) in the presence of bait. Such colonies were purified by culture after serial dilution, and the sequences of their cDNA inserts were determined. One colony contained cDNA that encoded 241 residues of rat brain CaMKIIβ N-terminal amino acid sequence (residues 34–274). Several other colonies also contained inserts with the CaMKIIβ sequence. This interaction was examined further because of its likely functional importance, which is suggested by the facts that calmodulin is an important β-cell Ca2+-binding protein (43) and that β-cells express high levels of CaMKIIβ (44, 48), which regulates voltage-operated Ca2+ channels involved in insulin secretion (7, 45–47). Insulinoma cell secretion is also potentiated by overexpressing CaMKIIβ (49) or iPLA2β (22), and iPLA2β binds calmodulin (2, 34).
Cloning CaMKIIβ from a Rat Islet cDNA Library Reveals Tissue Specificity and a Developmental Profile of CaMKIIβ Isoform Expression
Pancreatic islets express distinct CaMKII isoforms, and adult rat islets express predominantly the CaMKIIβ isoform (48, 49). To determine whether CaMKII isoform(s) expressed in rat islets, like those in rat brain, also interact with iPLA2β, we cloned CaMKIIβ cDNA from adult rat islets. Reverse transcription-PCR was performed using RNA isolated from rat islets as template and a pair of primers designed from regions of cDNA sequence which are conserved in rat and mouse CaMKIIβ. The PCR product was cloned. Sequencing the insert revealed a putative initiation codon (ATG) at the 5′-end, a stop codon (TGA) at the 3′-end, and the entire coding sequence in an intervening single open reading frame. Fig. 1 illustrates the nucleotide and deduced amino acid sequences of the CaMKIIβ isoform cloned from adult rat islets (ACaMKII). Fig. 2A illustrates sequence alignments for CaMKIIβ from rat brain (BCaMKII), adult rat islets (ACaMKII), human β-cells (HCaMKII), and neonatal rat islets (NCaMKII).
Fig. 1. Nucleotide and deduced amino acid sequences of the CaMKIIβ isoform cloned from an adult rat pancreatic islet cDNA library.

The autophosphorylation sites are displayed in bold type. The minimal CaM binding sequence is shaded.
Fig. 2. Alignment of deduced amino acid sequences of CaMKIIβ isoforms cloned from rat brain (B), adult rat islets (A), adult human β-cells (H), and neonatal rat islets (N).

A compares aligned sequences of CaMKIIβ isoforms from various sources. The variable region is shaded. B is a graphical alignment of the sequences. The region of difference in amino acid sequence between ACaMKIIβ and HCaMKIIβ is denoted by an asterisk (*).
The CaMKIIβ cDNA cloned from adult rat islet mRNA contained a complete coding sequence of 1,509 bp which encodes 503 amino acid residues (Fig. 1), and this CaMKIIβ isoform is distinct from previously (50, 51) described rat isoforms. Analysis of nucleotide sequences revealed that ACaMKIIβ differs from BCaMKIIβ (50) by the lack of sequence corresponding to the first (residues 316–339) and third (residues 379–393) variable domains (Fig. 2A). ACaMKIIβ differs from NCaMKIIβ (51) by the absence of the sequence from residues 370 to 456 in the association domain (Fig. 2B). Sequence alignments revealed 99.4% amino acid sequence identity between ACaMKIIβ and the HCaMKIIβ isoform cloned from human insulinoma cells (48). The ACaMKIIβ and HCaMKIIβ sequences differ only in 3 amino acid residues in variable domain 2 (Fig. 2B).
To search for other subtypes of CaMKIIβ in adult rat islets, we performed a series of reverse transcription-PCR experiments using RNA from adult rat islets as template and primers designed from various regions of the ACaMKIIβ sequence, but we observed no other CaMKIIβ subtype in adult rat islets (not shown). Adult rat pancreatic islets and adult human β-cells thus express mRNA that encodes a CaMKIIβ isoform that differs from those in adult brain or in neonatal islets, and the latter two isoforms also differ from each other. There is thus both tissue specificity and a developmental profile of CaMKIIβ isoform expression, but there is little rat-to-human species heterogeneity in the CaMKIIβ isoform expressed in adult pancreatic islet β-cells.
Binary Yeast Two-hybrid Assays Confirm the Interaction between iPLA2β and CaMKIIβ
To confirm the interaction between iPLA2β and ACaMKIIβ observed in yeast two-hybrid screening experiments, binary yeast two-hybrid assays were performed. We first used ACaMKIIβ as bait and iPLA2β as prey. When bait or prey alone was transformed into yeast cells, no colonies grew in medium lacking leucine, tryptophan, and histidine, but when both bait and prey were transformed simultaneously into yeast cells, colonies formed and produced blue reaction products when treated with the chromogenic substrate X-gal (Fig. 3B, left column) that reflect interaction between ACaMKIIβ and iPLA2β. When the bait and prey DNA were switched (so that iPLA2β was bait, and ACaMKIIβ was prey), similar results were obtained (Fig. 3B, right column). These results reflect a specific interaction between iPLA2β and CaMKIIβ.
Fig. 3. iPLA2β interacts with CaMKIIβ in yeast cells.

A illustrates binary yeast two-hybrid assays performed using full-length iPLA2β (or CaMKIIβ) as bait and full-length CaMKIIβ (or iPLA2β)as prey. B contains schematic structures of wild-type iPLA2β and CaMKIIβ and of constructs that correspond to N- or C-terminal fragments of each protein. An iPLA2β N-terminal fragment that contains the ankyrin repeat domain and a C-terminal fragment that contains the catalytic site are shown, as are a CaMKIIβ N-terminal fragment that contains the catalytic domain and a C-terminal fragment that contains the association domain. C illustrates binary yeast two-hybrid assays involving coexpression of N- or C-terminal fragments of iPLA2β and of CaMKIIβ as bait/prey pairs together (lanes 1–4) or, in control experiments, expression of an N- or C-terminal fragment of one of the proteins alone (lanes 5ȓ8). The blue colonies reflect specific interactions between two proteins that constitute bait-prey partners in the binary yeast two-hybrid assay. The arrow identifies such blue colonies formed by the β-galactosidase reaction product after incubation with the chromogenic substrate X-gal.
To identify domains of the proteins essential for their interaction, we performed binary yeast two-hybrid assays using N-or C-terminal fragments of iPLA2β as the bait or prey and N- or C-terminal fragments of CaMKIIβ as the prey or bait. Fig. 3B shows the schematic representation of wild-type iPLA2β and CaMKIIβ proteins and of their N- and C-terminal fragments. When the N-terminal fragment of iPLA2β (NiPLA2β) was used as the bait or prey and the N-terminal fragment of CaMKIIβ (NCaMKIIβ) was used as the prey or bait, large colonies formed after incubation at 30 °C for 4 days, and these colonies turned blue after incubation with the chromogenic substrate X-gal for 4 h at room temperature (Fig. 3C, lane 1). When an N-terminal fragment (NiPLA2β or NCaMKIIβ) was used as bait or prey and a C-terminal fragment (CiPLA2β or CCaMKIIβ)asthe prey or bait, only small colonies formed after incubation at 30 °C for 4 days (Fig. 3C, lanes 2 and 3). These colonies were lifted onto filter paper and incubated until they grew large enough to perform the X-gal assay. As illustrated in Fig. 3C (lanes 2 and 3), these colonies failed to turn blue after incubation with the chromogenic substrate X-gal, indicating that the interactions between the C-terminal domains of iPLA2β and CaMKIIβ are weak and nonspecific. No colonies formed when the C-terminal fragment CiPLA2β was used as bait or prey and CCaMKIIβ as prey or bait (Fig. 3C, lane 4). These results demonstrate that the N-terminal domains of iPLA2β and CaMKIIβ interact, but the C-terminal domains do not, in agreement with the initial library screening result that the N-terminal domain of CaMKIIβ (residues 34–271) participates in the interaction with iPLA2β. In control experiments, expression of N- or C-terminal fragments of either protein as bait or prey alone resulted in no colonies, as expected (Fig. 3C, lanes 5–8).
CaMKIIβ Can Be Expressed from Its DNA at High Levels in a Baculovirus-Sf9 Cell System and Retains Activity after Purification
Sf9 cells have been used to express iPLA2β (2, 23, 33), and we found that His-tagged ACaMKIIβ can also be expressed at high levels in Sf9 cells infected with baculovirus containing its cDNA. Cytosol from Sf9 cells infected with baculovirus containing DNA encoding His-tagged ACaMKIIβ was loaded onto TALON metal affinity columns, which were then washed to remove nonadsorbed proteins. Interaction of His-tagged ACaMKIIβ with metal ions on the column resin was then disrupted with imidazole-containing buffers, and this caused desorption of His-tagged ACaMKIIβ protein, which was collected in 0.5-ml fractions of column eluant. Proteins in eluant fractions were analyzed by SDS-PAGE and visualized by immunoblotting using a CaMKII antibody to demonstrate expression and purification of His-tagged ACaMKIIβ (Fig. 4A). Purified His-tagged ACaMKIIβ retained catalytic activity reflected by phosphorylation of the synthetic substrate autocamtide-3 in the presence of added Ca2+/CaM. In the absence of added Ca2+/CaM little activity was detected (Fig. 4B). The intensity of the immunochemical signal for CaMKIIβ in the eluant fractions (Fig. 4A) correlated well with CaMKIIβ activity in these fractions (Fig. 4B).
Fig. 4. Expression of His-tagged CaMKIIβ in Sf9 cells and its adsorption to and desorption from metal affinity columns.
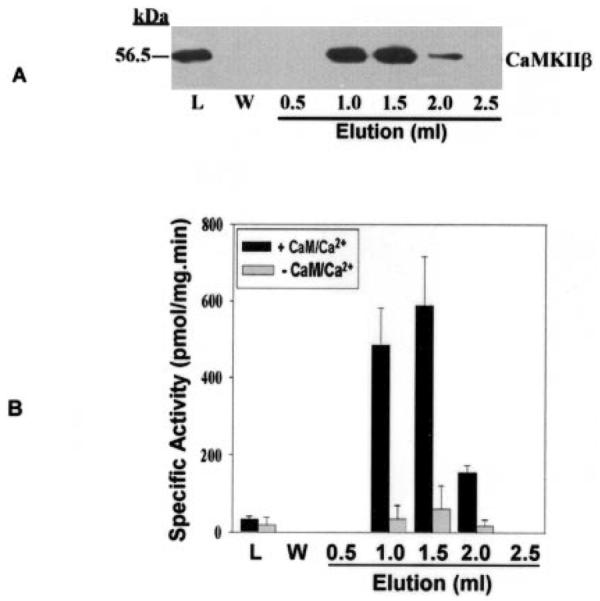
In A, cytosol from Sf9 cells that had been infected with baculovirus containing DNA that encodes His-tagged CaMKIIβ was incubated with TALON metal affinity resin, as described under “Experimental Procedures.” The resin was then loaded into a gravity-flow column and washed with buffer, and His-tagged CaMKIIβ was eluted with imidazole-containing buffer and collected in 0.5-ml fractions. Proteins in aliquots of the load (L), wash (W), and elution fractions were analyzed by SDS-PAGE, and immunoblotting was then performed with CaMKII antibody. In B, the protein content of each fraction was measured, and CaMKII activity was determined in the presence (+) or absence (−) of added Ca2+/CaM. When Ca2+/CaM was not added, 1 mm EGTA was added. For each assay, an aliquot of each eluant fraction was mixed with assay buffer, peptide substrate (autocamtide-3), and [γ-32P]ATP, as described under “Experimental Procedures.” Displayed values represent the means, and error bars denote S.E. (n = 6).
ACaMKIIβ and iPLA2β Interact with Each Other When Co-expressed in Sf9 Cells
To characterize further the interaction between the two proteins, His-tagged ACaMKIIβ and full-length, untagged iPLA2β (hereafter designated “native” iPLA2β) were coexpressed in Sf9 cells to determine whether His-tagged ACaMKIIβ could pull down native iPLA2β from cell cytosol. Sf9 cells were coinfected with baculovirus that contained DNA encoding His-tagged ACaMKIIβ and with baculovirus that contained DNA encoding native iPLA2β. Cytosol was loaded onto TALON metal affinity columns, which were then washed as described above. Imidazole-containing buffer was used to desorb His-tagged CaMKIIβ and any proteins associated with it. Aliquots of eluant fractions were analyzed by SDS-PAGE and immunoblotting with antibodies specific for CaMKIIβ or iPLA2β. His-tagged ACaMKIIβ (Fig. 5A, lower panel) and native iPLA2β (Fig. 5A, upper panel) proteins eluted in the same fractions, as detected by immunoblotting. Activity assays for iPLA2β (Fig. 5B) and ACaMKIIβ (Fig. 5C) indicate that both proteins retain activity after elution. The intensity of the immunochemical signals (Fig. 5A) correlated well with the activities of iPLA2β (Fig. 5B) and CaMKIIβ (Fig. 5C) in the eluant fractions. Similar results were obtained using purified proteins from Sf9 cells (Fig. 6A). These findings support the conclusions from yeast two-hybrid assays that these two proteins interact with each other.
Fig. 5. iPLA2β interacts with CaMKIIβ when the two proteins are coexpressed in Sf9 insect cells.
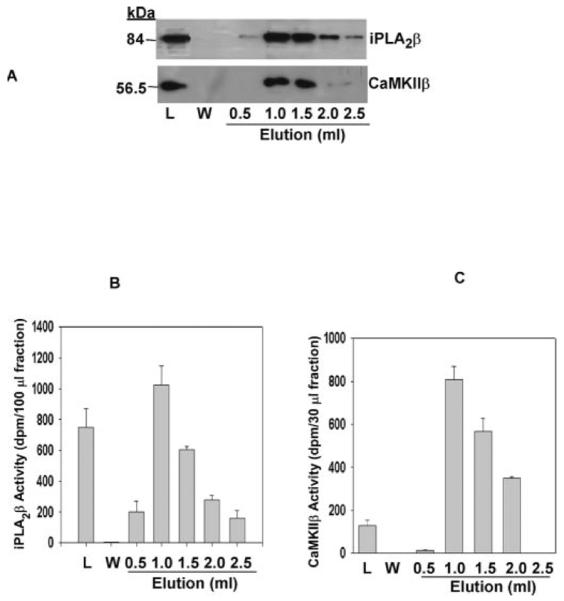
In A, Sf9 cells were infected simultaneously with baculovirus containing full-length HisCaMKIIβ and iPLA2β DNAs, cultured, and then homogenized, as described under “Experimental Procedures.” Cytosol prepared from homogenates was loaded onto a TALON metal affinity column and washed with buffer. HisCaMKIIβ was eluted with imidazole-containing buffer and collected in 0.5-ml fractions. The proteins in aliquots of load (L), wash (W), and elution fractions were analyzed by SDS-PAGE, and immunoblotting was performed with antibodies to iPLA2β (upper panel) or HisCaMKIIβ (lower panel). In B, an aliquot of load, wash, or elution fractions was added to assay buffer containing 10 mm EGTA, 1 mm ATP, and 1-palmitoyl-2-[14C]linoleoyl-sn-glycero-3-phosphocholine substrate. Reactions to measure iPLA2β activity were performed and terminated as described under “Experimental Procedures,” and released [14C]linoleic acid was isolated by TLC and measured by liquid scintillation spectrometry. Displayed values represent the means, and error bars denote S.E. (n = 6). In C, an aliquot of load, wash, or elution fractions was mixed with assay buffer containing 0.1 mm ATP, 0.75 mm CaCl2, 20 μg/ml calmodulin, 20 μm autocamtide-3, and 2 μCi of [γ-32P]ATP and incubated at 30 °C for 3 min to determine CaMKII activity. An aliquot of the reaction mixture was applied to phosphocellulose paper, which was then washed. CaMKII activity was calculated from the amount of phosphorylated autocamtide-3, as determined by liquid scintillation spectrometric measurement of 32P content. Displayed values represent the means, and error bars denote S.E. (n = 6).
Fig. 6. The stoichiometry of the interaction between iPLA2β and CaMKIIβ.
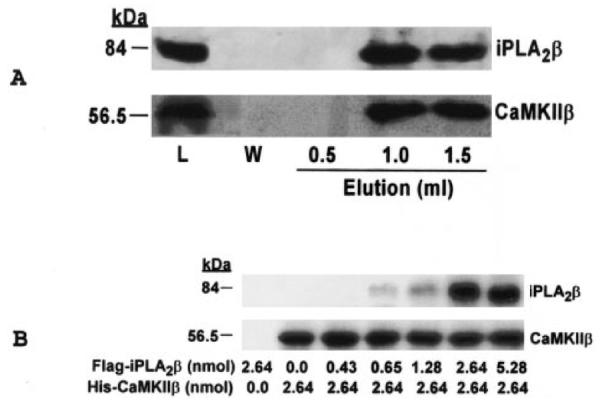
In A, purified, recombinant, His-tagged CaMKIIβ from Sf9 cells was mixed with TALON metal affinity resin, and the resin was then washed. Bound CaMKIIβ was measured with a Coomassie protein assay kit. iPLA2β protein expressed in Sf9 cells was purified as described previously (33), and 850 μg (10 nmol) of the protein was mixed with metal affinity resin to which 570 μg (10 nmol) of His-tagged CaMKIIβ had been adsorbed. The mixture was incubated at room temperature for 30 min with shaking, and the resin was washed and loaded onto a gravity-flow column. Bound proteins were eluted with imidazole-containing buffer and collected in 0.5-ml fractions. Proteins in aliquots of the load (L), wash (W), and elution fractions were analyzed by 10% SDS-PAGE, and immunoblotting was then performed with antibodies specific for iPLA2β (upper panel) or CaMKIIβ (lower panel). In B, 200 μl of metal affinity resin slurry to which 150 μg (2.64 nmol) of His-tagged CaMKIIβ had been adsorbed was mixed with FLAG-tagged iPLA2β in amounts that varied from 0 to 5.28 nmol. The mixture was incubated at 4 °C overnight with shaking, and the resin was then washed to remove noncomplexed proteins. Proteins were eluted from the metal affinity resin and analyzed by 10% SDS-PAGE. Immunoblotting was then performed with primary antibodies specific for iPLA2β (upper panel) or CaMKIIβ (lower panel).
The Stoichiometry of the Interaction between iPLA2β and CaMKIIβ
To characterize further the interaction of iPLA2β with ACaMKIIβ, His-tagged ACaMKIIβ was adsorbed onto TALON metal affinity resin, and purified iPLA2β was incubated with the resin. The resin was then washed and loaded into a gravity-flow column, and the interaction between the His tag and the immobilized metal ions was disrupted by elution with imidazole-containing buffer. Proteins in eluant fractions were analyzed by SDS-PAGE and immunoblotting. Fig. 6A illustrates that His-tagged ACaMKIIβ (lower panel) and iPLA2β (upper panel) eluted from the column in the same fractions, which provides additional evidence that these two proteins interact with each other. To determine the molar ratio of the two enzymes in the complex, the dose-response studies illustrated in Fig. 6B were performed. The amount of iPLA2β enzyme pulled down by His-tagged ACaMKIIβ increases as the molar ratio increases up to 1:1 but does not increase further at a ratio of 2:1. This suggests that the two enzymes form a complex with 1:1 stoichiometry.
The Calmodulin Antagonist W13 Does Not Prevent the Interaction of CaMKIIβ with iPLA2β
Because both iPLA2β and CaMKIIβ have calmodulin binding domains, calmodulin might mediate the interaction between these two proteins by forming a ternary complex. To evaluate this possibility, the interaction between iPLA2β and CaMKIIβ was examined in the presence and absence of added calmodulin. FLAG-tagged iPLA2β was expressed in Sf9 cells and purified with a FLAG M kit (Sigma). FLAG-tagged iPLA2β was then mixed with TALON metal affinity resin that had previously been loaded with His-tagged ACaMKIIβ in the presence or absence of calmodulin and then washed. When calmodulin was not added, the calmodulin antagonist W13 was added to block binding of any contaminating calmodulin to the target proteins. Adsorbed proteins were eluted from the metal affinity resin with imidazole-containing buffer, and proteins in eluant fractions were analyzed by SDS-PAGE and immunoblotting with iPLA2β-specific antibody. Fig. 7A illustrates that added calmodulin is not required for the interaction between iPLA2β and CaMKIIβ and that this interaction is not prevented by the calmodulin antagonist W13. These results are consistent with the findings that the CaM binding site(s) of iPLA2β reside in its C-terminal domain (2) and that the interaction of iPLA2β and CaMKIIβ occurs between their N-terminal domains (Fig. 3C).
Fig. 7. The interaction between iPLA2β and CaMKIIβ does not require added calmodulin and is not prevented by a calmodulin antagonist or the Ca2+ chelator EGTA.
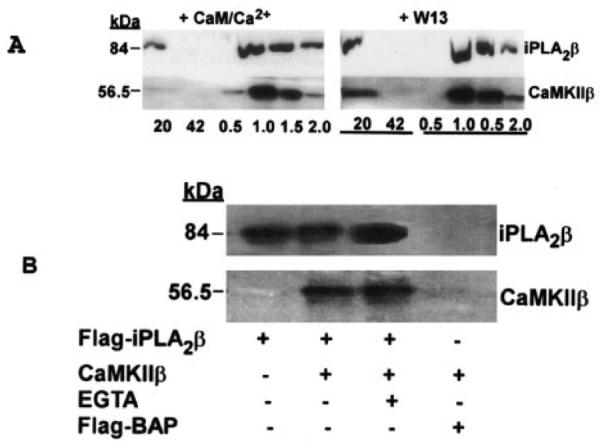
In A, FLAG-tagged iPLA2β expressed in Sf9 cells was purified with a FLAG M kit. Recombinant, purified, His-tagged CaMKIIβ was adsorbed to TALON metal affinity resin. The FLAG-tagged iPLA2β was incubated with the resin to which His-tagged CaMKIIβ had been adsorbed at room temperature for 30 min with shaking in the presence of 0.25 mm Ca2+ and 1 mm CaM (upper left panel) or 0.4 mm calmodulin antagonist W13 (upper right panel). The resin was then washed, and adsorbed proteins were eluted with imidazole-containing buffer. Aliquots of wash and elution fractions were analyzed by SDS-PAGE and immunoblotting with antibody specific for iPLA2β or CaMKII. In B, cytosol was prepared from baculovirus-infected Sf9 cells that expressed FLAG-iPLA2β, CaMKIIβ without a FLAG tag, or the control fusion protein N-terminal FLAG-tagged alkaline phosphatase (Flag-BAP). Binary mixtures of cytosols were prepared and incubated with anti-FLAG M2 affinity resin for 2 h at 4 °C in the presence or absence of 10 mm EGTA. Immunoprecipitated material was recovered by centrifugation and washed four times with wash buffer. Samples immunoprecipitated with anti-FLAG affinity resin were eluted with buffer containing FLAG peptide. Proteins in the eluant were analyzed by 10% SDS-PAGE and transferred onto a nylon membrane, and immunoblotting was performed with iPLA2β or CaMKII antibodies.
The Ca2+ Chelator EGTA Does Not Prevent the Interaction between iPLA2β and CaMKIIβ
The ability of iPLA2β to bind calmodulin causes iPLA2β preparations purified from cytosol to contain calmodulin, as detected by immunoblotting with calmodulin antibody (data not shown). Previous studies demonstrate that iPLA2β dissociates from calmodulin-agarose in the presence of EGTA (23, 39). To determine the role of calmodulin in the interaction between iPLA2β and CaMKIIβ, we performed an immunoprecipitation study of the interaction of FLAG-tagged iPLA2β with CaMKIIβ in the presence and absence of EGTA. Fig. 7B illustrates that in the presence of 10 mm EGTA, FLAG-tagged iPLA2β can still pull down CaMKIIβ from cytosol. The immunoblotting results in Fig. 7B illustrate that the amount of CaMKIIβ pulled down by FLAG-tagged iPLA2β is unaffected by EGTA and suggest that calmodulin is not directly involved in the interaction between iPLA2β and CaMKIIβ. In control experiments, the N-terminal FLAG-tagged alkaline phosphatase fusion protein was found not to pull down CaMKIIβ from cytosol, as expected.
The Activities of Both iPLA2β and CaMKIIβ Increase When the Proteins Associate with Each Other
Because results from yeast two-hybrid assays and protein pull-down experiments indicate that the ACaMKIIβ and iPLA2β proteins interact with each other, we next determined whether this interaction affects the catalytic activity of either enzyme. PLA2 activity assays involved measuring radiolabeled free fatty acid release from phospholipid substrates and were performed in buffer supplemented with 10 mm EGTA and 10 mm ATP with no added Ca2+. Under these conditions, adding purified, recombinant, His-tagged ACaMKIIβ to purified, recombinant, His-tagged iPLA2β resulted in a statistically significant increase in PLA2 activity (Fig. 8A). Results from dose-response studies under conditions where [iPLA2β] was constant and [CaMKIIβ] was varied indicate that the maximal iPLA2β activity is achieved at a 1:1 molar ratio of the two enzymes (Fig. 8B), which is consistent with the finding in Fig. 6B that iPLA2β and CaMKIIβ form a complex with 1:1 stoichiometry.
Fig. 8. Influence of CaMKIIβ on iPLA2β activity.
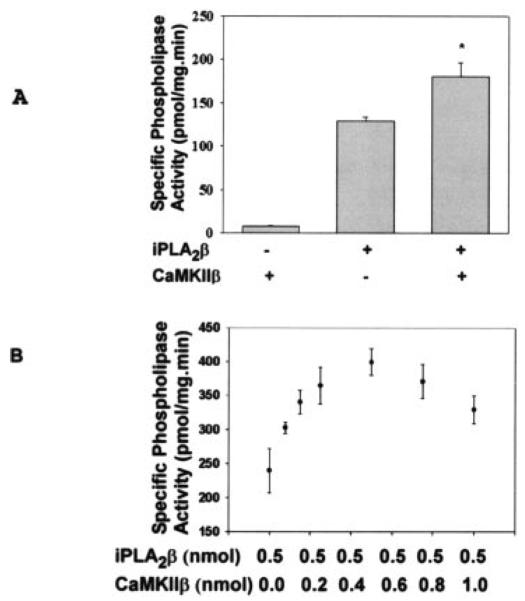
In A, His-tagged CaMKIIβ and His-tagged iPLA2β were purified with TALON metal affinity columns. Purified, His-tagged CaMKIIβ, His-tagged iPLA2β, or both were then added to buffer containing 10 mm ATP, 10 mm EGTA, and the radiolabeled substrate 1-palmitoyl-2-[14C]linoleoyl-sn-glycero-3-phosphocholine. The iPLA2 activity was then calculated from released [14C]linoleate as in Fig. 5. Values are represented as the mean ± S.E. (n = 4). Statistical significance is denoted by an asterisk (*), which indicates a p value < 0.05. In B, iPLA2β activity assays were performed in the presence of 0.5 nmol of iPLA2β and the indicated amounts of CaMKIIβ. Displayed values represent the means ± S.E. (n = 3).
CaMKII activity assays involved measurement of [32PO4] incorporation from [γ-32P]ATP into a model peptide substrate. Fig. 9 illustrates that adding purified, recombinant, His-tagged iPLA2β to purified, recombinant, His-tagged CaMKIIβ resulted in a statistically significant increase in CaMKII activity in the presence of added Ca2+/CaM. Without added Ca2+ or CaM, CaMKII activity was low, and it was little affected by adding iPLA2β.
Fig. 9. Influence of iPLA2β on CaMKIIβ activity.
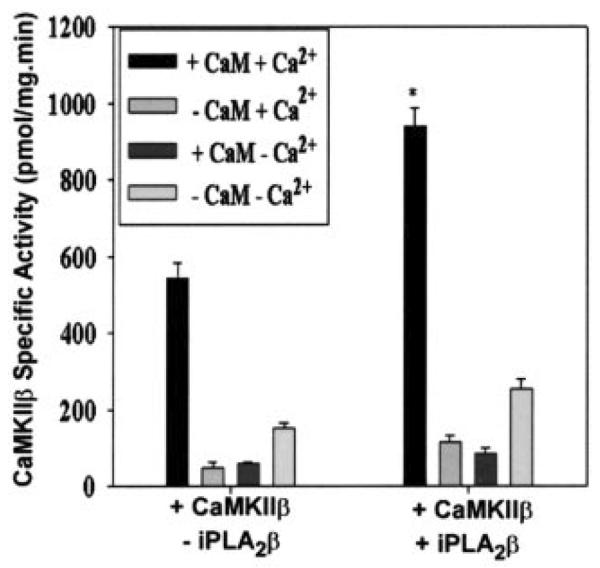
CaMKIIβ activity was measured in the presence (+) or absence (−) of iPLA2β, Ca2+, and CaM. For each assay, His-tagged CaMKIIβ was mixed with assay buffer containing ATP, autocamtide-3 substrate, and [γ-32P]ATP. CaMKII activity was calculated from the amount of phosphorylated autocamtide-2 as in Fig. 4. Values are represented as the mean ± S.E. (n = 5). Statistical significance is denoted by an asterisk, which indicates a p value < 0.01 compared with the group to which no iPLA2β was added (−iPLA2β).
Arachidonic Acid and 2-Lysophosphatidylcholine Inhibit CaMKIIβ Activity
The above results suggest that iPLA2β and CaMKIIβ form a complex and that this affects activities of both enzymes. To examine further the functional relationship between the two enzymes, we measured effects of the iPLA2β reaction products arachidonic acid and 2-lysophosphatidylcholine on CaMKIIβ activity. Fig. 10 illustrates that both arachidonic acid and 2-lysophosphatidylcholine inhibit CaMKIIβ activity in a concentration-dependent manner.
Fig. 10. Inhibition of CaMKIIβ activity by arachidonic acid (AA) and lysophosphatidylcholine (LPC).
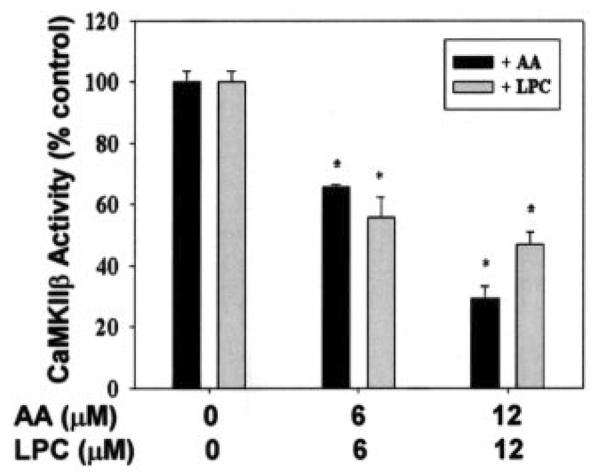
CaMKIIβ activity was measured in the presence or absence of arachidonic acid or lysophosphatidylcholine as in Fig. 4. For each assay, two separate measurements were performed simultaneously, one in the presence and the other in the absence of added Ca2+/CaM. Activity values were calculated from the difference between these two measurements and are represented as the mean ± S.E. (n = 3). Statistical significance is denoted by an asterisk (*), which indicates a p value < 0.05 compared with control.
Arachidonic Acid Release from INS-1 Insulinoma Cells Is Suppressed by Inhibitors of CaMKIIβ and iPLA2β
To determine whether evidence for a signaling complex between iPLA2β and CaMKIIβ could be observed in intact β-cells, we examined the effects of the CaMKII inhibitor KN93 and the iPLA2β inhibitor BEL on [3H]arachidonic acid release from prelabeled INS-1 insulinoma cells. Both KN93 and BEL are known to suppress insulin secretion from β-cells (9, 10, 19–22). Fig. 11 illustrates that both the CaMKII inhibitor and the iPLA2β inhibitor suppress [3H]arachidonic acid release from INS-1 cells, which is consistent with an interaction of CaMKIIβ and iPLA2β in β-cells to form a signaling complex.
Fig. 11. Arachidonic acid (AA) release from INS-1 cells is suppressed by inhibitors of iPLA2β and CaMKIIβ.
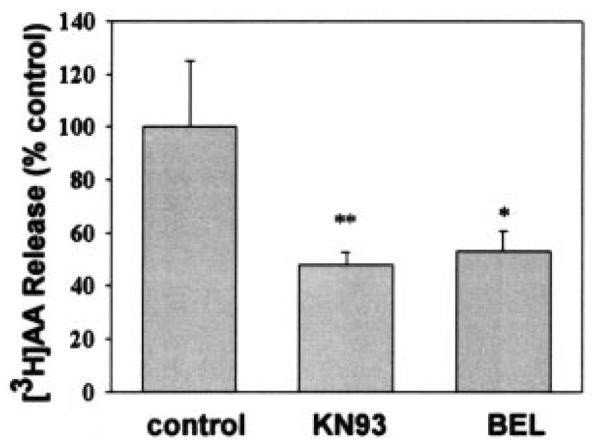
INS-1 cells were prelabeled with [3H]arachidonic acid and then washed free of unincorporated radiolabel. The labeled cells were then treated without or with 20 μm BEL or 8 μm KN93. [3H]Arachidonic acid release was then measured as described under “Experimental Procedures.” Release values are represented as the mean ± S.E. (n = 3). Statistical significance is denoted by an asterisk (*) or a double asterisk (**), which indicates a p value < 0.05 or 0.01, respectively, compared with control.
CaMKIIβ and iPLA2β Form a Complex in Insulin-secreting β Cells
To confirm the formation of an iPLA2β·CaMKIIβ complex in β-cells, we determined whether the two enzymes can be coimmunoprecipitated from INS-1 insulinoma cells. Fig. 12A illustrates that both enzymes can be coimmunoprecipitated from parental INS-1 cells and from a stably transfected INS-1 cell line that overexpresses iPLA2β (22) using antibodies against CaMKII (left panel). Similar results were obtained in coimmunoprecipitation experiments using antibodies against iPLA2β (right panel). This demonstrates the existence of an iPLA2β·CaMKIIβ complex in intact β-cells. Fig. 12B illustrates that forskolin, which is an adenylyl cyclase activator that amplifies insulin secretion (22), increases the intensity of the immunochemical signal for iPLA2β that coimmunoprecipitates with CaMKIIβ in INS-1 cells. This suggests that forskolin promotes formation of the iPLA2β·CAMKIIβ complex, and forskolin is also known to induce subcellular redistribution of iPLA2β in INS-1 cells (22).
Fig. 12. Forskolin stimulates complex formation between iPLA2β and CaMKIIβ in INS-1 insulinoma cells.
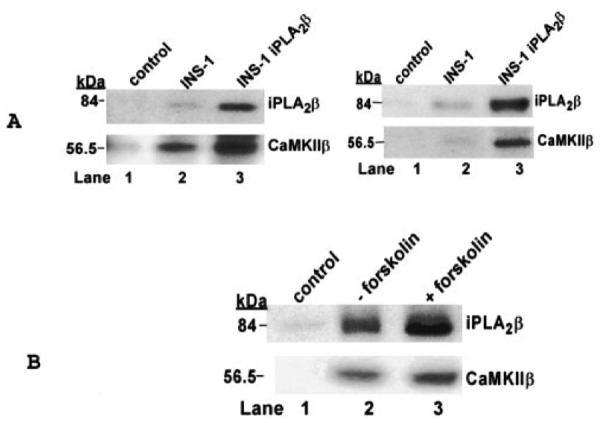
A illustrates coimmunoprecipitation of iPLA2β and CaMKIIβ. In lane 1 of the left panel, control preimmune serum was used for sham immunoprecipitation of INS-1 cell cytosol as a negative control. In lanes 2 and 3 of the left panel, cytosol from INS-1 cells (lane 2) or from INS-1 cells that overexpress iPLA2β (lane 3) were incubated with anti-CaMKIIβ antibody attached to protein A-agarose. The immunoprecipitate was collected by centrifugation, washed, boiled in SDS-PAGE sample loading buffer, and analyzed by SDS-PAGE. After transfer of proteins to nylon membranes, immunoblotting was performed with antibodies against iPLA2β (upper blot) or CaMKIIβ (lower blot). Similar results were obtained from the reverse immunoprecipitation experiment (right panel of A), in which cytosol from INS-1 cells (lane 2) or INS-1 cells that overexpress iPLA2β (lane 3) was immunoprecipitated with iPLA2β antibody-protein A-agarose. In lane 1 of B, control preimmune serum was used in sham immunoprecipitation of INS-1 cell cytosol as a negative control. In lanes 2 and 3 of B, INS-1 cells that overexpress iPLA2β were incubated without (lane 2) or with (lane 3) 4 μm forskolin. The cytosol was then immunoprecipitated with CaMKIIβ antibody-protein A-agarose. After SDS-PAGE analyses of the immunoprecipitates, immunoblotting was performed with iPLA2β antibody (upper blot) or with CaMKIIβ antibody (lower blot).
DISCUSSION
Major PLA2 activities in pancreatic islet β-cells and insulinoma cells are Ca2+-independent, and much evidence indicates that iPLA2β participates in signaling events involved in glucose-induced insulin secretion (19–22, 34, 52). The iPLA2β enzyme is also the predominant PLA2 activity in hippocampus, where it catalyzes arachidonic acid release that is required for long term potentiation (4), which is an electrophysiologic analog of learning. CaMKII is also involved in both insulin secretion (6, 7, 9–11, 45–49, 53) and long term potentiation (54–56). The physiological functions of iPLA2β and CaMKII thus appear to be linked in some cells, such as β-cells and neurons. Another isoform of CaMKII (CaMKIIα) interacts with Group IVA PLA2 (cPLA2) in vascular smooth muscle cells (57), and our findings indicate that CaMKIIβ interacts similarly with iPLA2β to form a complex. Because β-cells express both CaMKIIβ and iPLA2β, a complex of these enzymes could affect β-cell function.
We first observed the complex between iPLA2β and CaMKIIβ by using iPLA2β as bait in yeast two-hybrid screening of a rat brain cDNA library. Formation of a complex between the two enzymes was confirmed in binary yeast two-hybrid assays in which iPLA2β was bait and CaMKIIβ was prey and in the converse assay configuration in which CaMKIIβ was bait and iPLA2β was prey. Pull-down assays with recombinant, His-tagged proteins adsorbed to metal affinity matrices also provided direct evidence for the physical association of CaMKIIβ and iPLA2β. These findings clearly demonstrate that iPLA2β and CaMKIIβ interact with each other. We have demonstrated here that an immunoprecipitatable complex of these two enzymes exists in insulinoma cells and that the amount of the complex increases upon stimulation of intact β-cells with forskolin, which is an adenylyl cyclase activator that amplifies insulin secretion and induces subcellular redistribution of iPLA2β in β-cells (22).
We have demonstrated previously that depletion of internal Ca2+ stores causes activation of iPLA2β in β-cells (23) and in vascular smooth muscle cells (24). It has been demonstrated recently that iPLA2β participates in SOC entry from the extra-cellular space (25, 32), and this process is required for insulin secretion (26–31). Lysophospholipid products of iPLA2β activate SOC channels that mediate capacitative Ca2+ influx (25, 32), and CaMKII also affects Ca2+ fluxes by potentiating SOC channel activity (58) and regulating T-type voltage-operated calcium channels (59). Our findings indicate that iPLA2β interacts with the specific isoform of CaMKIIβ that is expressed in β-cells and that this interaction affects activities of both iPLA2β and CaMKIIβ. This suggests that CaMKIIβ and iPLA2β form a signaling complex, and this complex represents a potential means to regulate SOC entry.
Such a complex could orchestrate bidirectional signals that result in Ca2+ influx into β-cells and insulin secretion. Upon complexation with iPLA2β, CaMKIIβ could displace CaM from iPLA2β (2, 23) and increase iPLA2β activity by relieving tonic inhibition of the enzyme by CaM (2, 8, 23, 24). Lysophospholipids activate SOC channels (32) and are produced by iPLA2 action. Both the CaMKII inhibitor KN93 and the iPLA2β inhibitor BEL inhibit insulin secretion (9, 10, 19–22), and both compounds are also demonstrated here to inhibit arachidonate release from INS-1 insulinoma cells, which supports the possibility that iPLA2β and CaMKIIβ form a signaling complex in β-cells. CaMKIIβ is capable of decoding the frequency of oscillations in intracellular [Ca2+] by its autophosphorylation (54, 61). Autophosphorylated CaMKIIβ has 1,000-fold greater affinity for Ca2+/CaM than does nonphosphorylated CaMKIIβ (62). CaMKIIβ activity is affected by association with iPLA2β (Fig. 9) and by products of iPLA2β action (Fig. 10), including lysophospholipids that also modulate Ca2+ channel activities (63, 64). The interaction between CaMKIIβ and iPLA2β at the β-cell plasma membrane could thus affect Ca2+ influx and cytosolic [Ca2+], which is a key determinant of insulin secretion (26–31).
Alignment of the deduced amino acid sequences of HCaMKIIβ (48) and ACaMKIIβ, which have been cloned from adult human β-cells and adult rat islets, respectively, reveals more than 99% sequence conservation, and this indicates that there is little species-to-species variation in pancreatic islet β-cell expression of CaMKIIβ isoforms. The expression pattern of CaMKII isoforms does change with development in islets, as reflected by the difference in isoforms expressed in neonatal and adult islets, and there is also tissue-to-tissue heterogeneity in CaMKII isoform expression, as reflected by the different isoforms expressed by islets and brain. The high degree of CaMKIIβ sequence conservation between rat and human islets and the fact that islets express only a single, predominant CaMKIIβ isoform is consistent with the possibility that the islet isoform has a special function in β-cells and that iPLA2β and other proteins that interact with this enzyme modulate that function. It is thus of interest that expression of both iPLA2β and of CaMKIIβ has recently been found to occur at the same stage of differentiation of pancreatic progenitor cells to endocrine progenitor cells during development (60).
Acknowledgments
We thank Dr. Mary Wohltmann, Sheng Zhang, Wu Jin, and Alan Bohrer for excellent technical assistance, and we are grateful to Denise Kampwerth for secretarial assistance.
Footnotes
This work was supported by United States Public Health Service Grants R37-DK34388 (to J. T.), P01-HL57278 (to R. W. G.), P41-RR00954, P60-DK20579, and P30-DK56341, by an award (to S. R.) from the American Diabetes Association, and by an award (to Z. A. M.) from the Juvenile Diabetes Research Foundation (No. 1-2002-646).
The abbreviations used are: PLA2, phospholipase A2; BEL, (E)-6-(bromomethylene)tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one; CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase II; iPLA2, calcium-independent phospholipase A2; Pipes, 1,4-piperazinediethanesulfonic acid; SOC, store-operated calcium channel; X-gal: 5-bromo-4-chloro-3-indolyl-β-d-galactoside.
REFERENCES
- 1.Dennis EA. Trends Biochem. Sci. 1997;22:1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. J. Biol. Chem. 2001;276:7129–7135. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- 3.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J. Biol. Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 4.Wolf MJ, Izumi Y, Zorumski CF, Gross RW. FEBS Lett. 1995;377:358–362. doi: 10.1016/0014-5793(95)01371-7. [DOI] [PubMed] [Google Scholar]
- 5.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson JL, Turk J. J. Biol. Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 6.Wenham RM, Easom RA. J. Biol. Chem. 1994;269:4947–4952. [PubMed] [Google Scholar]
- 7.Wenham RM, Landt M, Walters SM, Hidaka H, Easom RA. Biochem. Biophys. Res. Commun. 1992;189:128–133. doi: 10.1016/0006-291x(92)91534-w. [DOI] [PubMed] [Google Scholar]
- 8.Wolf MJ, Gross RW. J. Biol. Chem. 1996;271:20989–20992. doi: 10.1074/jbc.271.35.20989. [DOI] [PubMed] [Google Scholar]
- 9.Bhatt HS, Conner BP, Prasanna G, Yorio T, Easom RA. Biochem. Pharmacol. 2000;60:1655–1663. doi: 10.1016/s0006-2952(00)00483-4. [DOI] [PubMed] [Google Scholar]
- 10.Easom RA. Diabetes. 1999;48:675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 11.Krueger KA, Ings EI, Brun AM, Landt M, Easom RA. Diabetes. 1999;48:499–506. doi: 10.2337/diabetes.48.3.499. [DOI] [PubMed] [Google Scholar]
- 12.Laychock SG. Cell Calcium. 1982;3:43–54. doi: 10.1016/0143-4160(82)90036-7. [DOI] [PubMed] [Google Scholar]
- 13.Fujimoto WY, Metz SA. Endocrinology. 1987;120:1750–1757. doi: 10.1210/endo-120-5-1750. [DOI] [PubMed] [Google Scholar]
- 14.Jolly YC, Major C, Wolf BA. Biochemistry. 1993;32:12209–12217. doi: 10.1021/bi00096a034. [DOI] [PubMed] [Google Scholar]
- 15.Konrad RJ, Jolly C, Wolf BA. Biochem. J. 1992;297:283–290. doi: 10.1042/bj2870283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konrad RJ, Jolly C, Wolf BA. Biochim. Biophys. Acta. 1992;1135:215–220. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- 17.Loweth AC, Scarpello JH, Morgan NG. Biochim. Biophys. Res. Commun. 1996;218:423–427. doi: 10.1006/bbrc.1996.0075. [DOI] [PubMed] [Google Scholar]
- 18.Simonsson E, Karlsson S, Ahren B. Diabetes. 1998;47:1436–1443. doi: 10.2337/diabetes.47.9.1436. [DOI] [PubMed] [Google Scholar]
- 19.Ramanadham S, Gross RW, Han X, Turk J. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 20.Ramanadham S, Bohrer A, Gross RW, Turk J. Biochemistry. 1993;32:13499–13509. doi: 10.1021/bi00212a015. [DOI] [PubMed] [Google Scholar]
- 21.Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Biochemistry. 1993;32:5339–5351. doi: 10.1021/bi00071a009. [DOI] [PubMed] [Google Scholar]
- 22.Ma Z, Ramanadham S, Wohltmann M, Zhang S, Turk J. J. Biol. Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 23.Nowatzke W, Ramanadham S, Ma Z, Hsu FF, Bohrer A, Turk J. Endocrinology. 1998;139:4073–4085. doi: 10.1210/endo.139.10.6225. [DOI] [PubMed] [Google Scholar]
- 24.Wolf MJ, Wang J, Turk J, Gross RW. J. Biol. Chem. 1997;272:1522–1526. doi: 10.1074/jbc.272.3.1522. [DOI] [PubMed] [Google Scholar]
- 25.Smani T, Zakharov SI, Leno N, Csutora P, Trepakova ES, Bolotina VM. J. Biol. Chem. 2003;278:11909–11915. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]
- 26.Woly JF, McIntyre MS, Spencer B, Mertz RJ, Roe MW, Dukes ID. J. Biol. Chem. 1994;269:14359–14362. [PubMed] [Google Scholar]
- 27.Dukes ID, Roe MW, Woly JF, Philipson LH. Curr. Opin. Endocrinol. Diabetes. 1997;4:262–271. [Google Scholar]
- 28.Roe MW, Worley JF, Qian F, Tamarina N, Mittal AA, Dralyuk F, Blair NT, Mertz RJ, Philipson LH, Dukes ID. J. Biol. Chem. 1998;273:10402–10410. doi: 10.1074/jbc.273.17.10402. [DOI] [PubMed] [Google Scholar]
- 29.Herson PS, Lee K, Pinnock RD, Hughes J, Ashford ML. J. Biol. Chem. 1999;274:833–841. doi: 10.1074/jbc.274.2.833. [DOI] [PubMed] [Google Scholar]
- 30.Gilon P, Arredouani A, Gailly P, Gromada J, Henquin JC. J. Biol. Chem. 1999;274:20197–20205. doi: 10.1074/jbc.274.29.20197. [DOI] [PubMed] [Google Scholar]
- 31.Straub SG, Kornreich B, Oswald RE, Nemeth EF, Sharp GWG. J. Biol. Chem. 2000;275:18777–18784. doi: 10.1074/jbc.M000090200. [DOI] [PubMed] [Google Scholar]
- 32.Smani T, Zakharov SI, Csutora P, Leon E, Trepakova ES, Bolotina VM. Nat. Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 33.Wolf MJ, Gross RW. J. Biol. Chem. 1996;271:30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]
- 34.Ma Z, Turk J. Prog. Nucleic Acids Res. Mol. Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 35.Lux ES, John KM, Bennett V. Nature. 1990;344:36–42. doi: 10.1038/344036a0. [DOI] [PubMed] [Google Scholar]
- 36.Peters LL, Lux SE. Semin. Hematol. 1993;30:85–118. [PubMed] [Google Scholar]
- 37.Bourguignon LYW, Jin H. J. Biol. Chem. 1995;270:7257–7260. doi: 10.1074/jbc.270.13.7257. [DOI] [PubMed] [Google Scholar]
- 38.Kordeli E, Ludosky M-A, Deprette V, Frappier T, Cartaud J. J. Cell Sci. 1998;111:2197–2207. doi: 10.1242/jcs.111.15.2197. [DOI] [PubMed] [Google Scholar]
- 39.Yang HC, Mosior M, Ni B, Dennis EA. J. Neurochem. 1999;73:1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- 40.Schöfl C, Mader T, Kraämer C, Waring M, Krippeit-Drews P, Prank K, Mühlen AVZ, Draws G, Brabant G. Endocrinology. 1999;140:5516–5523. doi: 10.1210/endo.140.12.7180. [DOI] [PubMed] [Google Scholar]
- 41.Jan C-R, Ribarm TJ, Means AR, Augustine GJ. J. Biol. Chem. 1996;271:15478–15485. doi: 10.1074/jbc.271.26.15478. [DOI] [PubMed] [Google Scholar]
- 42.O'Reilly DR, Miller LK, Luckow VA. Baculovirus Expression Vector: A Laboratory Manual. W. H. Freeman and Co.; New York: 1992. [Google Scholar]
- 43.Sugden MC, Christie MR, Ashcroft SJ. FEBS Lett. 1979;105:95–100. doi: 10.1016/0014-5793(79)80894-7. [DOI] [PubMed] [Google Scholar]
- 44.Landt M, McDaniel ML, Bry CG, Kotagal N, Colca JR, Lacy PE, McDonald JM. Arch. Biochem. Biophys. 1982;213:148–154. doi: 10.1016/0003-9861(82)90449-0. [DOI] [PubMed] [Google Scholar]
- 45.Niki I, Okazaki K, Saitoh M, Niki A, Niki H, Tamagawa T, Iguchi A, Hidaka H. Biochem. Biophys. Res. Commun. 1993;191:255–261. doi: 10.1006/bbrc.1993.1210. [DOI] [PubMed] [Google Scholar]
- 46.Smith MK, Colbran RJ, Brickey DA, Soderling TR. J. Biol. Chem. 1992;267:1761–1768. [PubMed] [Google Scholar]
- 47.Ammala C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. J. Physiol. (Lond.) 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rochlitz H, Voigt A, Lankat-Buttgereit B, Göke B, Heimberg H, Nauck MA, Schiemann U, Schatz H, Pfeiffer AFH. Diabetologia. 2000;43:465–473. doi: 10.1007/s001250051330. [DOI] [PubMed] [Google Scholar]
- 49.Tabuchi H, Yamamoto H, Matsumoto K, Ebihara K, Takeuchi Y, Fukunaga K, Hiraoka H, Sasaki Y, Shichiri M, Miyamoto E. Endocrinology. 2000;141:2350–2360. doi: 10.1210/endo.141.7.7553. [DOI] [PubMed] [Google Scholar]
- 50.Bulleit RF, Bennett MK, Molloy SS, Hurley JB, Kennedy MB. Neuron. 1988;1:63–72. doi: 10.1016/0896-6273(88)90210-3. [DOI] [PubMed] [Google Scholar]
- 51.Urquidi V, Ashcroft SJH. FEBS Lett. 1995;358:23–26. doi: 10.1016/0014-5793(94)01381-a. [DOI] [PubMed] [Google Scholar]
- 52.Ramanadham S, Hsu FF, Zhang S, Bohrer A, Turk J. J. Biol. Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 53.Easom RA, Filler NR, Ings EM, Tarpley J, Landt M. Endocrinology. 1997;138:2359–2364. doi: 10.1210/endo.138.6.5179. [DOI] [PubMed] [Google Scholar]
- 54.Koninck PD, Schulman H. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 55.Lisman J, Malenka RC, Nicoll RA, Malinow R. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- 56.Lisman J, Schulman H, Cline H. Nat. Rev. Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 57.Muthalif MM, Hefner Y, Canaan S, Harper J, Zhou H, Parmentier JH, Aebersold R, Gelb MH, Malik KU. J. Biol. Chem. 2001;276:39653–39660. doi: 10.1074/jbc.M103136200. [DOI] [PubMed] [Google Scholar]
- 58.Machaca K. J. Biol. Chem. 2003;278:33730–33737. doi: 10.1074/jbc.M305023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQJ. Neuroscience. 2003;23:10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, Melton DA. Development. 2004;131:165–179. doi: 10.1242/dev.00921. [DOI] [PubMed] [Google Scholar]
- 61.Hudmon A, Schulman H. Annu. Bev. Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 62.Myer T, Hanson PI, Stryer L, Schulman H. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 63.Shen K, Meyer T. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 64.Wang J, Best PM. Nature. 1992;359:739–741. doi: 10.1038/359739a0. [DOI] [PubMed] [Google Scholar]