

Abstract
Background
The sequences of the 16S rRNA genes extracted from fecal samples provide insights into the dynamics of fecal microflora. This potentially gives valuable etiological information for patients whose conditions have been ascribed to unknown pathogens, which cannot be accomplished using routine culture methods. We studied 33 children with diarrhea who were admitted to the Children’s Hospital in Shanxi Province during 2006.
Results
Nineteen of 33 children with diarrhea could not be etiologically diagnosed by routine culture and polymerase chain reaction methods. Eleven of 19 children with diarrhea of unknown etiology had Streptococcus as the most dominant fecal bacterial genus at admission. Eight of nine children whom three consecutive fecal samples were collected had Streptococcus as the dominant fecal bacterial genus, including three in the Streptococcus bovis group and three Streptococcus sp., which was reduced during and after recovery. We isolated strains that were possibly from the S. bovis group from feces sampled at admission, which were then identified as Streptococcus lutetiensis from one child and Streptococcus gallolyticus subsp. pasteurianus from two children. We sequenced the genome of S. lutetiensis and identified five antibiotic islands, two pathogenicity islands, and five unique genomic islands. The identified virulence genes included hemolytic toxin cylZ of Streptococcus agalactiae and sortase associated with colonization of pathogenic streptococci.
Conclusions
We identified S. lutetiensis and S. gallolyticus subsp. pasteurianus from children with diarrhea of unknown etiology, and found pathogenic islands and virulence genes in the genome of S. lutetiensis.
Keywords: Microbial communities, 16S rRNA gene analysis, Streptococcus lutetiensis, Genome analysis, Pathogenic island
Background
In the developing world, every child under 5 years of age experiences approximately three episodes per year of diarrhea [1]. Although more than 200 viral, bacterial, and parasitic causes of diarrhea have been identified to date, only a few etiological agents cause the vast majority of diarrheal diseases in children in the developing world. These include rotavirus, diarrheagenic Escherichia coli, Campylobacter jejuni, Shigella spp., non-typhoidal Salmonella, Giardia lamblia, Cryptosporidium spp. and Entamoeba histolytica[2]. Unfortunately, a large proportion of cases of diarrheal disease are of unknown etiology. There are many reasons for this problem, including fragility of causative agents, exacting growth requirements, and lack of recognition of some organisms as enteric pathogens.
Here, we used the previously described strategy of 16S rRNA gene polymerase chain reaction (PCR) and sequencing technology [3] to analyze quantitatively the densities of different bacterial species in fecal samples of patients with diarrhea of unknown etiology at different times relative to hospital admission, and analyzed the features of the dominant species.
Methods
Study design
Children with diarrhea without antibiotic treatment who were admitted to the Children’s Hospital, Shanxi Province, China from August 17 to 30, 2006 were screened for enteric pathogens, including Shigella, Salmonella, enterotoxigenic E. coli, enteroinvasive E. coli (EIEC), enteropathogenic E. coli (EPEC), Shiga-toxin-producing E. coli, enteroaggregative adherence E. coli (EAEC), and common diarrhea viruses, including group A rotavirus, human calicivirus (HuCV), enteric adenovirus (Adv) and human astrovirus (HAstV). The targeted virulence genes of enteric bacterial pathogens included heat-labile (LT), heat-stable (ST) enterotoxins, Shiga-like toxin (SLT), bundle forming pili (bfpA), enteric attaching and effacing locus (eaeA), EAEC specific probe, and the genes encoding invasive plasmid antigens (ipaBCD) [4-7]. The group A rotaviruses were detected by commercially available enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (Oxoid, Basingstoke, UK). Total viral DNA and RNA were extracted from fecal specimens prepared in phosphate-buffered saline at 10%(wt/vol) using the QIAamp MinElute Virus Spin Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. HuCV, enteric Adv and HAstV were detected by PCR as described previously [8-10]. G. lamblia and Ent. histolytica were detected using direct microscopy with a saline preparation of the specimen. The clinical history and physiological findings of each patient were documented on standardized case report forms. Fecal samples from five healthy and five hospitalized children at the same location but with no apparent diarrhea were analyzed as controls. Libraries of the 16S rRNA gene were constructed for each fecal sample, with a minimum size of 100 analyzable sequences [11].
Analyzing dominant fecal bacterial species by 16S rRNA gene sequence technology
All fecal samples were collected in triplicate; one for timely isolation and detection of the enteric pathogens; one stored at −20°C for 16S rRNA sequence analysis; and one stored in 20% glycerol at −80°C for isolation of the putative pathogens suggested by the 16S rRNA gene analysis.
The DNA was extracted from a 200-mg fecal sample, which was measured and adjusted to 100 ng/μl of each sample for PCR. The universal eubacterial primers 27 F-519R (5’-agagtttgatcmtggctcag-3’ and 5’-gwattaccgcggckgctg-3’) were used to amplify a 500-bp region of the 16S rRNA gene. LaTaq polymerase (TaKaRa, Dalian, China) was used for PCR under the following conditions: 95°C for 5 min, followed by 20 cycles of: 95°C for 30 s, 52°C for 30 s, and 72°C for 1 min; and a final elongation step at 72°C for 10 min.
The PCR products were extracted from sliced gels and cloned into the pGEMR-T Easy Vector System (Promega, Madison, WI, USA). They were then transformed into competent E. coli JM109. A total of 130 white clones for each fecal sample were randomly selected for enrichment. The purified plasmid DNA was used for sequence analysis. To verify the repeatability, we repeated the 16S rRNA gene analysis of feces at admission for nine children with diarrhea of unknown etiology. The 16S rRNA gene sequences were analyzed for chimeric constructs using the Chimera Check program within the Ribosomal Database Project.
Species-level identification was performed using a 16S rRNA gene sequence similarity of ≥99% compared with the prototype strain sequence in the GenBank. Identification at the genus level was defined as a 16S rRNA gene sequence similarity of ≥97% with that of the prototype strain sequence in the GenBank, and the sequences were listed by genus. The sequences matched attributable to either E. coli or Shigella sp. were listed as E. coli/Shigella sp.
Isolation of suggested fecal-dominant Streptococcus
Strains of Streptococcus were isolated from fecal samples using KF Streptococcus Agar(Oxiod, Hampshire, United Kingdom), and identified using the MicroScan WalkAway SI 40 system(Dade Behring,West Sacramento, CA, USA). The full length of the 16S rRNA gene sequence was obtained for confirmation of identification. Pulsed-field gel electrophoresis was performed according to the protocol for Streptococcus suis[12]. The DNA was digested with 40 U SmaI (TaKaRa, Dalian, China). A dendrogram of isolates was drawn using BioNumerics software (version 4.0, Applied Maths BVBA, Belgium). Clustering of patterns was performed using the unweighted pair group with arithmetic averaging (UPGMA).
Genome sequencing and analysis of Streptococcus lutetiensis
The genome of S. lutetiensis 033 isolated from Patient 033 was sequenced using a combination of 454 sequencings with a Roche 454 FLX and paired end sequencing derived from the pUC18 library using an ABI 3730 Automated DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The genome was predicted using Glimmer software [13]. All putative open reading frames (ORFs) were annotated using non-redundant nucleotides and proteins in the NCBI, Swissport and KEGG databases. BLASTN and Artemis Comparison Tool (ACT) were used for the pair alignment. Orthologous gene clusters were searched for using the orthoMCL pipeline. We clustered these orthologous genes according to their presence or absence in different genome sequences among Streptococcus spp., and then a phylogenic tree was constructed using the neighbor-joining method. Genome islands were defined as having abnormal GC content with at least five continuous genes. The homologous genes within each island were compared with the references using BLASTN with an e-value cutoff at 1×10–5.
Nucleotide sequence accession numbers
The GenBank accession numbers reported in this study are CP003025 for the genome sequence of S. lutetiensis strain 033; and JN581988 and JN581989 for the 16S rRNA gene sequences of S. gallolyticus subsp. pasteurianus strains 017 and 035, respectively.
Ethics statement
Feces samples were acquired with the written informed consent from the parents of the children with diarrhea and normal children. This study was reviewed and approved by the ethics committee of the National Institute for Communicable Disease Control and Prevention, China CDC, according to the medical research regulations of the Ministry of Health, China (permit number 2006-16-3).
Results
Detection of enteric pathogens in feces of children with diarrhea
From August 17 to 30, 2006, fecal samples were obtained from 33 children with diarrhea admitted to the Children’s Hospital, Shanxi Province, China (Additional file 1: Table S1). Thirty-two of 33 children with diarrhea yielded negative culture for common enteric bacterial pathogens, such as Salmonella, Vibrio or diarrheagenic E. coli. Shigella sonnei was isolated from one patient (Figure 1). The 16S rRNA gene sequences of fecal samples were also negative for Salmonella, Vibrio or Yersinia spp. Eleven children with diarrhea were diagnosed with Shigella or diarrheagenic E. coli, including two with EAEC, one with EPEC, and eight with EIEC/Shigella, according to virulence gene detection results (Figure 1). These 11 children belonged to a group of 26 who had the 16S rRNA gene sequence of E. coli/Shigella sp.
Figure 1.
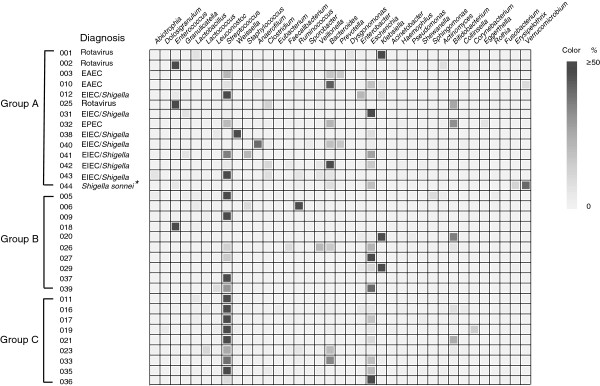
Microbiota in the feces of children with diarrhea at admission. Each block represents a bacterial genus. The color value changes from red to yellow and displays the percentage value (50% to 0%) of a given bacterial genus. The bacterial genera with fewer than five determined sequences, or <1% in a given sample, or unrecognized bacteria are not shown. The patients were divided into three groups. Group A were patients with diarrheagenic E. coli and Shigella. Group B were patients with diarrhea of unknown etiology and fecal samples collected only at admission. Group C were patients with diarrhea of unknown etiology and fecal samples collected at admission, during recovery, and after recovery. *S. sonnei was isolated from patient 044.
The 16S rRNA gene sequence of Bacteroides fragilis was detected in five children with diarrhea, but its virulence gene heat-labile protein toxin was not detected. Twelve of 33 children with diarrhea were positive for the Clostridium 16S rRNA gene sequence, but the virulence gene toxin A or B of Clostridium difficile was not detected. Three samples were positive for group A rotavirus by ELISA and none tested positive for HuCV, Adv and HastV (Figure 1).
Dominant fecal bacteria in children with diarrhea of unknown etiology
We divided the 33 children with diarrhea into three groups based on the etiological diagnosis. Group A included 14 children who were infected with diarrheagenic E. coli or Shigella species and rotaviruses. Group B included 10 children with diarrhea of unknown etiology with only one fecal sample collected at admission. Group C included nine children with diarrhea of unknown etiology from whom three fecal samples were collected, including one at admission, one during recovery, and one after recovery (Figure 1). The 16S rRNA gene sequencing data revealed that 11 of 19 children with diarrhea of unknown etiology had Streptococcus as the dominant fecal bacterial genus at admission. Among the remaining eight children, Escherichia (n = 4), Klebsiella (n = 2), Enterococcus (n = 1) or Ruminococcus (n = 1) was the most dominant bacterial genus (Figure 1).
We analyzed fecal samples from five healthy and five hospitalized children at the same location but with no apparent diarrhea as controls. None of the genera Escherichia, Enterococcus, Klebsiella, Ruminococcus and Streptococcus was dominant within the control fecal samples taken from five healthy children. None of five hospitalized children at the same location but with no apparent diarrhea had Streptococcus as the dominant genus, although one of them had the percent of Streptococcus to 34.96% in fecal microbiota. There were no species in the Streptococcus bovis group in the 10 children in the control group (Additional file 2: Figure S1).
In nine children with diarrhea of unknown etiology in Group C, eight had Streptococcus as the most dominant fecal bacterial genus at admission, one with S. lutetiensis, two with S. gallolyticus subsp. pasteurianus, two with Streptococcus salivarius, and three with Streptococcus sp. (Figures 1 and 2, Table 1). We divided these nine children in Group C into two, according to the most dominant fecal bacterial species at admission. Group C1 included one child whose most dominant species was E. coli. The percentage of E. coli in the fecal microflora of Patient 036 (age 7 months) was increased from 87.10% at admission to 90.91% during treatment, and then dropped to 28.90% after recovery (Figure 2B), based on 445 analyzed 16 s rRNA gene sequences.
Figure 2.
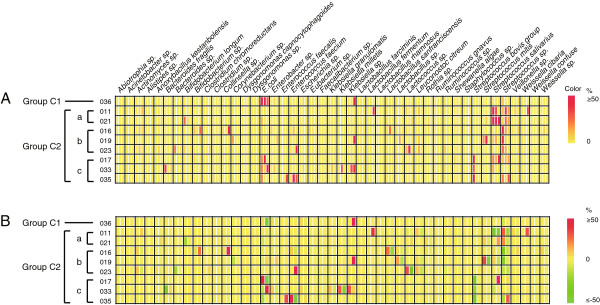
Percentage changes in fecal bacteria in children with diarrhea at admission and during and after recovery. Only patients who had unknown etiology and provided three fecal samples were included. The bacterial species with fewer than five determined sequences, or <1% in a given sample, or unrecognized species are not shown. (A) The true percentage value of individual bacterial species in fecal samples of patients with diarrhea sampled at admission and during and after recovery. Each block was divided into three columns by white vertical lines, representing the fecal samples at admission and during and after recovery, respectively. The color value from red to yellow displayed the percentage (50% to 0%) of a given bacterial species in each sample. (B) The percentage changes in individual bacterial species in fecal samples from patients with diarrhea during and after recovery compared with that at admission. Each block was divided into two columns by white vertical lines, representing the relative percentage changes of given bacterial species during and after recovery, compared with that in feces at admission. The color value from red to yellow to green displayed the percentage (50% to 0% to −50%) of a given bacterial species in each sample. The negative percentage shows that the percentage of a given bacterial species was reduced compared with that detected at admission.
Table 1.
Features of study samples from children with diarrhea of unknown etiology
|
Patient information |
Clinical presentation |
Stool routine analysis |
|||||
|---|---|---|---|---|---|---|---|
| Patient and feces number | Sampling date (after onset) | Times of stool/day | Characteristics of stool | Temperature (°C) | WBC* | RBC* | Occult blood |
| 011-1 |
1 |
5 |
Watery |
Normal |
+ |
++ |
+ |
| 011-3 |
3 |
5 |
Loose |
|
|
|
|
| 011-4 |
5 |
2 |
Formed |
|
|
|
|
| 016-1 |
1 |
3-4 |
Bloody and mucoid |
39.0°C |
++ |
+ |
+/− |
| 016-3 |
3 |
3 |
Watery |
|
|
|
|
| 016-6 ** |
12 |
2 |
Formed |
|
|
|
|
| 017-1 |
16 |
10 |
Watery |
Normal |
+ |
++ |
+/− |
| 017-3 |
18 |
6 |
Watery |
|
|
|
|
| 017-5 |
20 |
6 |
Watery |
|
|
|
|
| 019-1 |
133 |
8-9 |
Bloody and mucoid |
Normal |
++ |
++ |
+ |
| 019-6 |
138 |
3 |
Loose |
|
|
|
|
| 019-7** |
143 |
3 |
Loose |
|
|
|
|
| 021-1 |
33 |
6 |
Watery |
Normal |
+ |
+ |
- |
| 021-4 |
35 |
5 |
Watery |
|
|
|
|
| 021-7 |
38 |
4-5 |
Loose |
|
|
|
|
| 023-1 |
20 |
6 |
Loose |
38.7°C |
++ |
- |
- |
| 023-5 |
24 |
3 |
Formed |
|
|
|
|
| 023-6** |
28 |
2-3 |
Formed |
|
|
|
|
| 033-1 |
5 |
5 |
Watery |
Normal |
- |
- |
- |
| 033-3 |
7 |
2 |
Formed |
|
|
|
|
| 033-5 |
9 |
2 |
Formed |
|
|
|
|
| 035-1 |
1 |
5 |
Bloody and mucoid |
38.3°C |
+ |
++ |
+ |
| 035-4 |
4 |
2-3 |
Formed |
|
|
|
|
| 035-6 ** |
9 |
2-3 |
Formed |
|
|
|
|
| 036-1 |
7 |
6 |
Loose |
Normal |
++ |
+ |
+ |
| 036-2 |
8 |
3 |
Loose |
|
|
|
|
| 036-3 | 9 | 2 | Loose | ||||
* +: 6–10/ high power field (HPF) ++: >10/HPF.
**: Fecal samples collected at patient discharge from hospital.
Group C2 included eight children with diarrhea, who were further divided into three subgroups, based on the most dominant fecal bacterial species at admission. Group C2a included two children who had S. salivarius as the most dominant fecal bacterial species. Group C2b included three children who had Streptococcus sp. as the most dominant species. Group C2c included three children who had S. bovis group as the most dominant species (Figure 2A and B).
For Patient 011 (age 2.5 years) in Group C2a, the percentage of S. salivarius in the fecal microflora was reduced from 78.95% at admission to 31.43% during recovery (Figure 2B), based on 442 sequences analyzed. Patient 021 (age 8 months) had the percentage of S. salivarius in the fecal microflora of 58.56% at admission, which increased to 60.0% during recovery and then to 76.67% after recovery (Figure 2B).
Group C2b had Streptococcus sp. as the dominant fecal species at admission. For Patient 016 (age 9 months), the percentage of Streptococcus sp. in fecal microflora was reduced from 51.28% to 15.65% during recovery (3 days of treatment), and then to 4.67% after recovery (12 days of treatment) (Figure 2B), based on 456 16S rRNA gene sequences analyzed. For Patient 019 (age 4 months), the percentage of Streptococcus sp. in fecal microflora was reduced from 40.54% at admission to 7.08% during recovery (6 days of treatment) and then to 1.77% after recovery (11 days of treatment) (Figure 2A and B), based on 448 16S rRNA gene sequences analyzed. For Patient 023 (age 5 months), the percentage of Streptococcus sp. in fecal microflora was reduced from 26.05% at admission to 13.56% during recovery (5 days of treatment) and then to zero after recovery (9 days of treatment) (Figure 2B), based on 440 16S rRNA gene sequences analyzed.
All three patients in Group C2c had S. bovis group as their most dominant fecal bacterial species at admission. For Patient 033 (age 2 months), the percentage of S. bovis group in fecal microflora was reduced from 26.84% at admission to zero during recovery (3 days of treatment) (Figure 2B). It was not detected in feces sampled at discharge from the hospital, after 5 days of treatment. For Patient 017 (age 1.5 years), the percentage of S. bovis group in fecal microflora was reduced from 39.82% at admission to zero during recovery (3 days of treatment) (Figure 2B). It was not detected in feces sampled at discharge from hospital, after 5 days of treatment. For Patient 035 (age 8 months), the percentage of S. bovis group in fecal microflora was reduced from 42.73% at admission to zero after 4 days of treatment (Figure 2B). It was not detected in the feces sampled at discharge from hospital, after 9 days of treatment.
Isolation and identification of the S. bovis group from feces
We attempted to culture the dominant bacterial species as identified by the 16S rRNA gene analysis from the feces of all nine patients in Group C (Figures 1 and 2). Four patients (016, 019, 021 and 023) had negative cultures even on non-selective blood agar; possibly because antibiotics had been given before the hospital consultation.
Patient 017 had seven isolates belonging to the S. bovis group in the feces samples collected at admission, Patient 033 had 19, and Patient 035 had 10. According to the results of the MicroScan WalkAway SI 40 system, all isolates of the S. bovis group were identified as biotype II (mannitol fermentation negative). We then amplified, cloned, and sequenced the major portion of the 16S rRNA gene from each isolate. The strains isolated from Patient 033 were identified as S. lutetiensis and those from Patients 017 and 035 were S. gallolyticus subsp. pasteurianus. A dendrogram comparing representative 16S rRNA gene sequences of the isolated S. bovis group strains with other Streptococcus species mapped our isolates within the S. bovis group (Figure 3).
Figure 3.

Phylogenetic analysis of isolated strains of the S. bovis group and other major streptococcal species based on complete 16S rRNA gene sequences. The multiple sequence alignment of 16S rRNA genes was performed using ClustalW. The conserved tree was constructed using the neighbor-joining method. Bootstrap values are shown above each branch. All 16S rRNA gene sequences were derived from the NCBI and validated using genome sequences. The strains with complete genomes are marked with a star to the right of the species name. Staphylococcus aureus subsp. aureus MRSA252 was included as an out-group. The strains in red were isolated in this study.
Chromosomal DNA from the 36 strains of the S. bovis group from the three patients were digested with restriction enzyme SmaI and analyzed using pulsed-field gel electrophoresis (PFGE). Strains from each patient (seven from Patient 017, 19 from Patient 033 and 10 from Patient 035) were found to have unique restriction patterns.
Genome sequence and comparison of the S. bovis group with S. lutetiensisstrain 033
We sequenced the entire genome of the S. lutetiensis strain 033 and compared it withits close relatives, S. gallolyticus subsp. pasteurianus and S. gallolyticus subsp. gallolyticus [14]. To the best of our knowledge, this is the first time the genome of S. lutetiensis has been completely sequenced. The genome of strain 033 contained 1,975,547 bp with a GC content of 37.7%. It had 60 tRNAs and 18 rRNAs (six operons). Fifty-five tandem repeated regions were identified in the genome with the highest number of tandem repeats duplicated 104 times (at 3,744 bp, genome position from 844,798 to 848,542). A total of 2,015 ORFs >300 bp (100 aa) were identified. Of these, 86.2% matched clusters of orthologous groups (COGs) in the database with e-values <1×10–5 (Figure 4).
Figure 4.
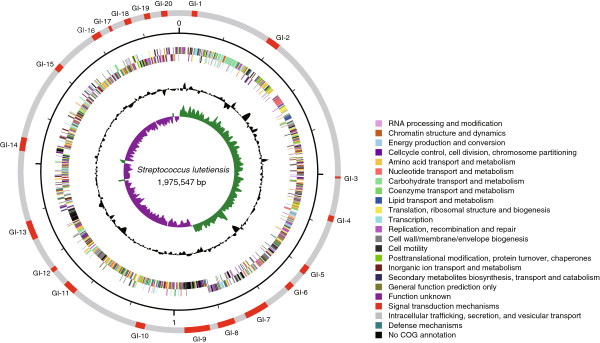
Genome sequence of S. lutetiensis strain 033. Key to the circular diagram (outer to inner): (1) GI found in the chromosome. (2) S. lutetiensis strain 033 COG categories on the forward strand (+) and the reverse strand (−). (3) G + C content and GC skew (G-C/G + C) of 033, respectively, with a window size of 10 kb.
Twenty genomic islands (GIs) in the genome of S. lutetiensis 033 were identified. Of these, five were antibiotic-resistance islands and two were putative pathogenicity islands (Figure 4). Notably, GI-7 was found to contain four glycosyl transferase genes, four pilin-related genes, and >10 transposase genes or putative transposase genes that have been reported to be associated with virulence in Streptococcus pneumoniae,Neisseriaceae, and others [15-17]. GI-18 encodes a colonization-associated adhesion factor previously described in S. suis[18]. GI-6 encodes the capsule polysaccharide (CPS) genes that are associated with the virulence of pathogenic streptococci; for example, S. pneumoniae and S. suis (Figure 5C) [19-21]. Five GIs were unique to S. lutetiensis and have not been identified in other species of this genus. Two were phage related, one encoded a cellobiose phosphorylase-like protein, one encoded an ATPase, and one had an unknown function. We found the hemolytic toxin cylZ in S. lutetiensis that activates the neutrophil signaling pathways in the brain endothelium and contributes to the development of meningitis identified in S. agalactiae[22]. The gene for sortase (SrtA), also identified in the genome of S. lutetiensis, was found to be associated with adhesion to epithelial cells and with colonization of pathogenic streptococci [23-25] (Table 2).
Figure 5.

Genome analysis of S. lutetiensis strain 033. Comparative analysis of all completed genomes of the S. bovis group (S. gallolyticus subsp. gallolyticus BAA-2069, S. gallolyticus subsp. gallolyticus ATCC43143, and S. gallolyticus subsp. pasterurianus ATCC43144). (A) Venn diagram of homologous genes in four complete genomes. The number of homologous genes is noted in each circle: red for BAA-2069, blue for 033, green for ATCC43143, and purple for ATCC43144. (B) Local collinear block of the chromosome sequences of four genomes. The red blocks represent similar regions within nucleotide sequences, and the blue block represents a region similar to the complementary strands. GIs in our 033 genome are shown in the green block near the genome. (C) Organization of GI-6 encoding CPS. GC contents calculated using each 1 kb with a 500-bp step. The direction of the arrows represents the coding strand of the ORFs. The genes in the GIs are marked with blue (unknown functions) and yellow (known functions).
Table 2.
Putative virulence genes detected in the genome of S. lutetiensis strain 033
| Virulence factors | Genes related | Putative function | References |
|---|---|---|---|
| Pneumcoccal cell surface adherence protein A |
pavA |
Fibronectin binding |
[26] |
| Laminin-binding protein |
lmb |
Colonization |
[22] |
| Pilus-associated adhesin |
rrgA |
Colonization |
[18] |
| Sortase A |
srtA |
Adhesion to epithelial cells |
[27] |
| Streptococcal lipoprotein rotamase A |
slrA |
Colonization |
[28] |
| Streptococcal enolase |
eno |
Plasminogen binding |
[29] |
| Pneumococcal surface antigen |
psaA |
Adhesin |
[30] |
| C3-degrading protease |
cppA |
Evasion of innate immunity |
[31] |
| Serine protease |
htrA/degP |
Biogenesis of Streptolysin S |
[14] |
| Trigger factor | tig/ropA | Stress tolerance and biofilm formation | [32] |
Together with the biochemical tests, the results of 16S rRNA gene sequencing with 48 complete genomes for streptococci, two draft genomes (Streptococcus infantarius subsp. infantarius BAA-102 and S. gallolyticus UCN34), and four segment 16S rRNA genes (EU163500, EU163502, EU163503, and EU163504) in the S. bovis group were selected for an evolutionary study. The reference strain of S. lutetiensis (accession number: EU163503) was found to be the nearest strain to the S. lutetiensis genome sequence in our study, showing the same 16S rRNA gene sequences. Compared with the nearest species S. infantarius subsp. infantarius BAA-102 and EU163504, strain 033 had two and three nucleotide differences in the 16S rRNA genes, respectively.
An entire genome comparative analysis was performed on the four completed genomes of S. gallolyticus subsp. gallolyticus BAA-2069, S. gallolyticus subsp. gallolyticus ATCC43143, and S. gallolyticus subsp. pasterurianus ATCC43144 in the S. bovis group. The S. lutetiensis sequenced genome in our study was found to be phylogenetically related to the genome of S. gallolyticus subsp. pasterurianus ATCC43144; and 94.1% of the genes were found in the homologous genes in ATCC43144 (Figure 5A) [14]. Although large-scale genome rearrangements, inversions and deletions were observed, the four genomes displayed the same collinear structure (Figure 5B). We found 15.2% of the genes of S. gallolyticus subsp. pasterurianus and 34.9% of the genes of S. gallolyticus subsp. gallolyticus were not present in S. lutetiensis, suggesting that the genome of S. lutetiensis strain 033 was similar to that of S. gallolyticus subsp. pasterurianus (Figure 5A).
Discussion
Selective media are routinely used to isolate particular pathogens from mixtures of bacterial species from the feces of patients with diarrhea. However, they cannot be used to isolate putative bacterial agents of diarrhea of unknown etiology. The important feature of the direct sequencing of the 16S rRNA gene in the fecal samples is the ability to identify most of the existing bacterial species [33]. Using this technique, we analyzed the dynamics of the fecal bacteria flora in nine patients with diarrhea of unknown etiology. We examined three fecal samples per patient, at admission, during recovery, and after recovery. We speculated that the putative causative enteric pathogens were dominant in the intestine when infection was established. The number of causative pathogens in the intestine may decrease during treatment and after recovery.
Eight of nine patients (Group C2) who provided all three specimens with unknown etiology at admission had as the dominant Streptococcus genus in their fecal samples. There is a report of a child who developed hemolytic uremic syndrome with group A beta hemolytic streptococcus-positive diarrhea [34]. Streptococci are also numerous in the fecal microflora of patients with irritable bowel syndrome patients [35]. So, the role of streptococci in the fecal microflora of children with diarrhea deserved further research.
Three patients from Group C2 had Streptococcus as the dominant genus, and all showed a reduced the percentage of Streptococcus sp. in fecal microflora of during and after recovery. Two patients had S. salivarius as the dominant species with one showing a reduced the percentage of Streptococcus sp. in fecal microflora during and after recovery. The other patient showed an increase. Three patients had the S. bovis group as the dominant species, and all showed a reduced the percentage of S. bovis group in fecal microflora during and after recovery. This observation suggests that the association of the S. bovis group with diarrhea is worthy of further investigation.
S. bovis is divided into three biotypes, I (S. gallolyticus subsp. gallolyticus), II/1 (S. lutetiensis and S. infantarius), and II/2 (S. gallolyticus subsp. pasteurianus), based upon mannitol fermentation and β-glucuronidase activities. S. gallolyticus subsp. gallolyticus is known to be associated with endocarditis and colon carcinoma. S. infantarius, S. lutetiensis and S. gallolyticus subsp. pasteurianus are associated with non-colonic cancer and meningitis. Children with signs of gastrointestinal disturbance at presentation associated with S. bovis were also reported [36].
The dominant species from the nine patients of group C were cultured and four showed that they were negative. Thirty-six strains of the S. bovis group were isolated from three patients, and PFGE analysis showed that they had their own unique restriction pattern, indicating that the strains within individual patients were identical. The isolates were identified as S. lutetiensis and S. gallolyticus subsp. pasteurianus.
We determined and analyzed the full genome sequence of the S. lutetiensis strain isolated from a child with diarrhea. Two previously recognized pathogenicity islands were identified in the genome. GI-6 was found to encode a CPS gene cluster involved in the pathogenicity of S. suis[21]. GI-7 was found to encode glycosyl transferase, the virulence factor in S. pneumoniae[17]. Eight additional virulence factors were identified in the S. bovis group. These included the putative hemolytic toxin cylZ and the sortase gene associated with adhesion and colonization [22,24,25].
Conclusions
We studied the dynamics of the fecal microbial community in children with diarrhea of unknown etiology and found for the first time that strains of the S. bovis group were among the predominant bacteria in some of the patients at admission, and showed a reduction in numbers during treatment and recovery. In addition, we report the first genome sequence of a S. lutetiensis isolate, identifying putative pathogenic islands and virulence genes. However, it was hard to detect all the infectious agents and there were many non-infectious factors that may cause diarrhea; therefore, additional studies are needed to clarify the potential contribution of these bacteria to diarrhea in children.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
DJ, LL, ZL, HJ, CY and JX conceived and designed the experiments.DJ, SL,YXu and XB performed the experiments.CC, ZZ, PD, HW, YXiong, HZ and LW carried out the molecular genetic studies and participated in the sequence alignment.HS contributed reagents and materials. DJ, MG and JX wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by grants (2011CB504901, 2008ZX10004-001, 2008ZX10004-009, 2009ZX10004-101, 2011SKLID209) from the Ministry of Science and Technology, the National Key Programs for Infectious Diseases of China; and by grants from the State Key Laboratory for Infectious Disease Prevention and Control, People’s Republic of China.
Supplementary Material
Characteristics of patients and clinical presentation of diarrhea among children included in this study.
Dominant bacterial species in the feces of the control group.
Contributor Information
Dong Jin, Email: jindong@icdc.cn.
Chen Chen, Email: chenchen@icdc.cn.
Lianqing Li, Email: sxllq@tom.com.
Shan Lu, Email: lushan@icdc.cn.
Zhenjun Li, Email: Lizhenjun@icdc.cn.
Zhemin Zhou, Email: jemmings@gmail.com.
Huaiqi Jing, Email: jinghuaiqi@icdc.cn.
Yanmei Xu, Email: xyanmei2006@126.com.
Pengcheng Du, Email: dupengcheng@icdc.cn.
Haiyin Wang, Email: wanghaiyin@icdc.cn.
Yanwen Xiong, Email: xiongyanwen@icdc.cn.
Han Zheng, Email: zhenghan@icdc.cn.
Xuemei Bai, Email: BXMTS@yahoo.com.cn.
Hui Sun, Email: sunhui@icdc.cn.
Lei Wang, Email: wanglei@nankai.edu.cn.
Changyun Ye, Email: yechangyun@icdc.cn.
Marcelo Gottschalk, Email: marcelo.gottschalk@umontreal.ca.
Jianguo Xu, Email: xujianguo@icdc.cn.
References
- Kosek M, Bern C, Guerrant RL. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ. 2003;81(3):197–204. [PMC free article] [PubMed] [Google Scholar]
- O’Ryan M, Prado V, Pickering LK. A millennium update on pediatric diarrheal illness in the developing world. Semin Pediatr Infect Dis. 2005;16(2):125–136. doi: 10.1053/j.spid.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Vidal M, Lagos R, Levine M, Prado V. Multiplex PCR for diagnosis of enteric infections associated with diarrheagenic Escherichia coli. J Clin Microbiol. 2004;42(4):1787–1789. doi: 10.1128/JCM.42.4.1787-1789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2(2):123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11(1):142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruque SM, Khan R, Kamruzzaman M, Yamasaki S, Ahmad QS, Azim T, Nair GB, Takeda Y, Sack DA. Isolation of Shigella dysenteriae type 1 and S. flexneri strains from surface waters in Bangladesh: comparative molecular analysis of environmental Shigella isolates versus clinical strains. Appl Environ Microbiol. 2002;68(8):3908–3913. doi: 10.1128/AEM.68.8.3908-3913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Kageyama T, Fukushi S, Hoshino FB, Shinohara M, Uchida K, Natori K, Takeda N, Katayama K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J Virol Methods. 2002;100(1–2):107–114. doi: 10.1016/s0166-0934(01)00404-9. [DOI] [PubMed] [Google Scholar]
- Xu W, McDonough MC, Erdman DD. Species-specific identification of human adenoviruses by a multiplex PCR assay. J Clin Microbiol. 2000;38(11):4114–4120. doi: 10.1128/jcm.38.11.4114-4120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JS, Lee TW, Kurtz JB, Glass RI, Monroe SS. Typing of human astroviruses from clinical isolates by enzyme immunoassay and nucleotide sequencing. J Clin Microbiol. 1995;33(4):797–801. doi: 10.1128/jcm.33.4.797-801.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R. et al. Evolution of mammals and their gut microbes. Science. 2008;320(5883):1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Zhu X, Jing H, Du H, Segura M, Zheng H, Kan B, Wang L, Bai X, Zhou Y. et al. Streptococcus suis sequence type 7 outbreak, Sichuan. China. Emerg Infect Dis. 2006;12(8):1203–1208. doi: 10.3201/eid1708.060232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27(23):4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin IH, Liu TT, Teng YT, Wu HL, Liu YM, Wu KM, Chang CH, Hsu MT. Sequencing and comparative genome analysis of two pathogenic Streptococcus gallolyticus subspecies: genome plasticity, adaptation and virulence. PLoS One. 2011;6(5):e20519. doi: 10.1371/journal.pone.0020519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein DC, Miller CJ, Bhoopalan SV, Sommer DD. Sequence-based predictions of lipooligosaccharide diversity in the Neisseriaceae and their implication in pathogenicity. PLoS One. 2011;6(4):e18923. doi: 10.1371/journal.pone.0018923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole PW, Snelling WJ, Canchaya C, Forde BM, Hardie KR, Josenhans C, Graham R, McMullan G, Parkhill J, Belda E. et al. Comparative genomics and proteomics of Helicobacter mustelae, an ulcerogenic and carcinogenic gastric pathogen. BMC Genomics. 2010;11:164. doi: 10.1186/1471-2164-11-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkman MA, Morrison DA, Van Der Zeijst BA, Nuijten PJ. The capsule polysaccharide synthesis locus of streptococcus pneumoniae serotype 14: Identification of the glycosyl transferase gene cps14E. J Bacteriol. 1996;178(13):3736–3741. doi: 10.1128/jb.178.13.3736-3741.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Nishino H, Ishiji T, Ishii J, Osaki M, Fittipaldi N, Gottschalk M, Tharavichitkul P, Takai S, Sekizaki T. Genetic organization and preferential distribution of putative pilus gene clusters in Streptococcus suis. Vet Microbiol. 2009;138(1–2):132–139. doi: 10.1016/j.vetmic.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Wang Q, Xu Y, Perepelov AV, Xiong W, Wei D, Shashkov AS, Knirel YA, Feng L, Wang L. Characterization of the CDP-2-glycerol biosynthetic pathway in Streptococcus pneumoniae. J Bacteriol. 2010;192(20):5506–5514. doi: 10.1128/JB.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llull D, Lopez R, Garcia E. Genetic bases and medical relevance of capsular polysaccharide biosynthesis in pathogenic streptococci. Curr Mol Med. 2001;1(4):475–491. doi: 10.2174/1566524013363618. [DOI] [PubMed] [Google Scholar]
- Smith HE, Damman M, van der Velde J, Wagenaar F, Wisselink HJ, Stockhofe-Zurwieden N, Smits MA. Identification and characterization of the cps locus of Streptococcus suis serotype 2: the capsule protects against phagocytosis and is an important virulence factor. Infect Immun. 1999;67(4):1750–1756. doi: 10.1128/iai.67.4.1750-1756.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Schnitzler N, Lutticken R, Podbielski A. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect Immun. 1999;67(2):871–878. doi: 10.1128/iai.67.2.871-878.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan SA, Kurian D, Ward PN, Hunt L, Leigh JA. Identification of sortase A (SrtA) substrates in Streptococcus uberis: evidence for an additional hexapeptide (LPXXXD) sorting motif. J Proteome Res. 2010;9(2):1088–1095. doi: 10.1021/pr901025w. [DOI] [PubMed] [Google Scholar]
- Nobbs AH, Rosini R, Rinaudo CD, Maione D, Grandi G, Telford JL. Sortase A utilizes an ancillary protein anchor for efficient cell wall anchoring of pili in Streptococcus agalactiae. Infect Immun. 2008;76(8):3550–3560. doi: 10.1128/IAI.01613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Terao Y, Ogawa T, Takahashi T, Hamada S, Kawabata S. Role of Streptococcus sanguinis sortase A in bacterial colonization. Microbes Infect. 2006;8(12–13):2791–2796. doi: 10.1016/j.micinf.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Holmes AR, McNab R, Millsap KW, Rohde M, Hammerschmidt S, Mawdsley JL, Jenkinson HF. The pavA gene of Streptococcus pneumoniae encodes a fibronectin-binding protein that is essential for virulence. Mol Microbiol. 2001;41(6):1395–1408. doi: 10.1046/j.1365-2958.2001.02610.x. [DOI] [PubMed] [Google Scholar]
- Rusniok C, Couve E, Da Cunha V, El Gana R, Zidane N, Bouchier C, Poyart C, Leclercq R, Trieu-Cuot P, Glaser P. Genome sequence of Streptococcus gallolyticus: insights into its adaptation to the bovine rumen and its ability to cause endocarditis. J Bacteriol. 2010;192(8):2266–2276. doi: 10.1128/JB.01659-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD. et al. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;99(15):10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. 2007;189(1):38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajam G, Anderton JM, Carlone GM, Sampson JS, Ades EW. Pneumococcal surface adhesin A (PsaA): a review. Crit Rev Microbiol. 2008;34(3–4):163–173. doi: 10.1080/10408410802383610. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, Paulsen IT, Nelson KE, Margarit I, Read TD. et al. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci USA. 2002;99(19):12391–12396. doi: 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, Richter EW, Nagiec MJ, Sumby P, Porcella SF, DeLeo FR, Musser JM. Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A Streptococcus. Proc Natl Acad Sci USA. 2006;103(18):7059–7064. doi: 10.1073/pnas.0510279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte T, De Preter V, De Brandt E, Verbeke K, Swings J, Huys G. Molecular monitoring of the fecal microbiota of healthy human subjects during administration of lactulose and Saccharomyces boulardii. Appl Environ Microbiol. 2006;72(9):5990–5997. doi: 10.1128/AEM.00233-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan-Sanchez F, Guerrero-Lozano I, Rubio-Quinones F, Rodriguez-Iglesias M. Haemolytic uraemic syndrome associated with bloody diarrhoea caused by Streptococcus dysgalactiae. Enferm Infecc Microbiol Clin. 2013;31(1):60–61. doi: 10.1016/j.eimc.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Kassinen A, Krogius-Kurikka L, Makivuokko H, Rinttila T, Paulin L, Corander J, Malinen E, Apajalahti J, Palva A. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology. 2007;133(1):24–33. doi: 10.1053/j.gastro.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Gerber JS, Glas M, Frank G, Shah SS. Streptococcus bovis infection in young Infants. Pediatr Infect Dis J. 2006;25(11):1069–1073. doi: 10.1097/01.inf.0000240334.91713.48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of patients and clinical presentation of diarrhea among children included in this study.
Dominant bacterial species in the feces of the control group.