

Abstract
Introduction
Structural plasticity with synapse formation and elimination is a key component of memory capacity and may be critical for functional recovery after brain injury. Here we describe in detail two surgical techniques to create a cranial window in mice and show crucial points in the procedure for long-term repeated in vivo imaging of synaptic structural plasticity in the mouse neocortex.
Methods
Transgenic Thy1-YFP(H) mice expressing yellow-fluorescent protein (YFP) in layer-5 pyramidal neurons were prepared under anesthesia for in vivo imaging of dendritic spines in the parietal cortex either with an open-skull glass or thinned skull window. After a recovery period of 14 days, imaging sessions of 45–60 min in duration were started under fluothane anesthesia. To reduce respiration-induced movement artifacts, the skull was glued to a stainless steel plate fixed to metal base. The animals were set under a two-photon microscope with multifocal scanhead splitter (TriMScope, LaVision BioTec) and the Ti-sapphire laser was tuned to the optimal excitation wavelength for YFP (890 nm). Images were acquired by using a 20×, 0.95 NA, water-immersion objective (Olympus) in imaging depth of 100–200 μm from the pial surface. Two-dimensional projections of three-dimensional image stacks containing dendritic segments of interest were saved for further analysis. At the end of the last imaging session, the mice were decapitated and the brains removed for histological analysis.
Results
Repeated in vivo imaging of dendritic spines of the layer-5 pyramidal neurons was successful using both open-skull glass and thinned skull windows. Both window techniques were associated with low phototoxicity after repeated sessions of imaging.
Conclusions
Repeated imaging of dendritic spines in vivo allows monitoring of long-term structural dynamics of synapses. When carefully controlled for influence of repeated anesthesia and phototoxicity, the method will be suitable to study changes in synaptic structural plasticity after brain injury.
Keywords: 2-photon microscopy, Fluorescence, In vivo imaging, Neurons, Cranial window, Mouse model
Introduction
Since its introduction in the 1990’s [1], 2-photon microscopy (2-PM) soon proved its enormous benefit for intravital imaging, especially in the field of neuroscience [2-8]. The possibility of penetrating tissue in depths up to 1 mm [5,7,9] and, therefore visualization of neural structures such as neurons, glial cells, and blood vessels led to new insights in developmental and degenerative neurobiology as well as neuronal plasticity after trauma, ischemia and inflammation [3,10] To obtain high-resolution in vivo images even in deeper areas of the brain (> 500 μm), highly ambitious surgical techniques and even use of fluorescence microendoscopy were developed [11-13]. In addition, combination of high speed, low power 2-PM calcium imaging with patch recodings allow monitoring of spine function [14] and long term neuronal network activity [15]. The availability of various transgenic mice expressing fluorescent proteins in particular cell types [2] enables selective observation of neurons, their axons, and dendrites in different layers [16], while simultaneously monitoring glial cells and blood vessels [4,10,17-20]. By creating a permanent entrance to the brain via a cranial window [21,22] in transgenic mice, even repeated and long-term 2-PM imaging became feasible [7,8,23]. The microstructure of neuronal tissue, e.g. dendritic spines and synapses, was shown to be a dynamic, highly delicate process of formation and elimination [16]. These new imaging methods will help us to better understand the role of synaptic plasticity after traumatic head injuries or degenerative disease.
Materials and methods
For all experiments, we used male C57/Bl6 transgenic Thy1-YFP (H) mice expressing yellow-fluorescent protein (YFP) in layer 5 pyramidal neurons. All experiments required an appropriate animal experimentation facility and needed to be conducted in accordance with the laws and regulations of the regulatory authorities for animal care. The animal experiments presented here were approved by and conducted in accordance with the laws and regulations of the regulatory authorities for animal care and use in Lower Franconia (Regierung von Unterfranken, Würzburg, Germany; file number: 54–2531.01-20/07).
Experimental Setup
1. Operating microscope (Carl Zeiss AG, Jena, Germany).
2. Stereotactic frame (TSE, Bad Homburg, Germany, Figure 1).
Figure 1.

Stereotactic frame.
3. Heating device.
4. 2-Photon microscope (Figure 2) with multifocal scanhead splitter (TriMScope, LaVision Biotec, Bielefeld, Germany).
Figure 2.

2-photon microscope.
5. Anesthesia unit.
6. Custom-made head holding device (Figure 3).
Figure 3.

Custom-made head-holding device.
Surgical instruments and materials
1. Scalpel No. 15 (Aesculap, Tuttlingen, Germany).
2. Microsurgical blade (Surgistar #38-6961; Surgistar, Vista CA, USA).
3. Scissors (delicate curved sharp scissors; Aesculap, Tuttlingen, Germany).
4. Microdrill with diamond tip (diameter 1.5 - 3 mm; Figure 4).
Figure 4.

Diamond micro-drill tip.
5. Cyanoacrylate (Sigma-Aldrich Chemie GmbH, Steinheim, Germany).
6. Dental acrylic (Dentsply, York, PA, USA).
7. Custom-made cover slips (diameter 5 mm, thickness 1 mm).
8. Low-melting point agarose (1%, Sigma Type III; Sigma-Aldrich Chemie GmbH, Steinheim, Germany).
9. Sterile irrigation (e.g. sodium-chloride 0.9%, B.Braun, Melsungen, Germany).
10. Forceps (anatomical tips, straight or curved; Aesculap, Tuttlingen, Germany).
11. Needle holder (Aesculap, Tuttlingen, Germany).
12. Sterile suture material (Prolene 4.0, Vicryl 4.0; Ethicon, Norderstedt, Germany).
13. Anesthetics (xylazine/ketamine, isoflurane).
14. Cottonoids, swabs, gloves, and eye ointment (e.g. dexpanthenol).
Methods and results
In the following section, we describe two different techniques to create a cranial window and illustrate the imaging setup. For surgery, all mice were anaesthetized with intra-peritoneal injection of 0.1 mg/g ketamine (Ketanest-S 25 mg/ml; Pfizer, New York, NY, USA) and 0.005 mg/g xylazine (Rompun 2%; Bayer Health Care, Leverkusen, Germany). The depth of surgical anesthesia was verified before starting surgery and the mouse head was fixed in a stereotactic frame (Figure 1). For in vivo imaging, mice were anaesthetized with isoflurane (Isofluran, Baxter, Deerfild, IL, USA) via a facial mask and the head was restrained in a custom-made head-holding device (Figure 3).
Open-skull window
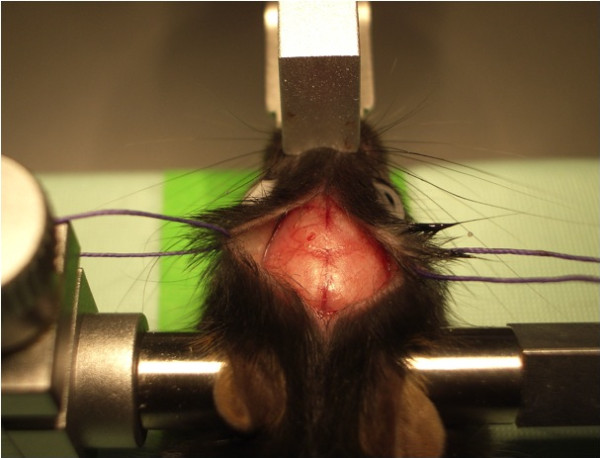
After fixation of the anaesthetized mouse in a stereotactic frame and application of eye ointment, a midline incision of the scalp was performed. Scalp and underlying periosteum were gently removed from skull bone with cotton swabs and the scalp was fixed laterally with two tack-up sutures (Figure 5). After localization of the region of interest (−1.5 mm bregma, 1.5 mm lateral), a craniectomy with the microdrill was carried out under the microscope and intermittent irrigation with sterile saline. Special care had to be taken before drilling away the last bone layer to avoid inadvertent injuries of the dura mater (Figure 6). Then, the exposed dura was covered then with fresh and sterile low-melting point agarose and a custom-made glass cover slip (diameter 5 mm, thickness 1 mm) was gently placed over the craniectomy and fixed with dental acrylic and cyanoacrylate (Figure 7). The crucial point here was to create a smooth agarose surface to prevent air bubbles between the agarose and the cover slip as well as averting fluid and sticky cyanoacrylate getting on the cover slip. Dental acrylic should be applied also on the exposed skull surface and the wound margins (the skin was not closed after the surgery). A strong micro magnet could be fixed in the dental acrylic for an alternative way to fix the head at the custom-made head holder instead of gluing it with cyanoacrylate repeatedly for long-term imaging (this could prevent cracking of glass cover while disconnecting the head from the head holder). After a recovery period of 14 days, imaging session could be started.
Figure 5.
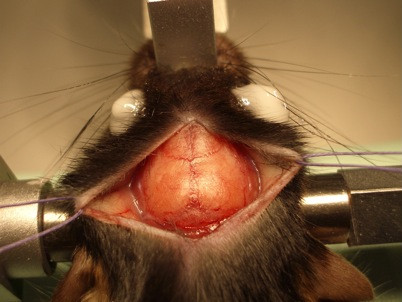
Exposed mouse skull.
Figure 6.
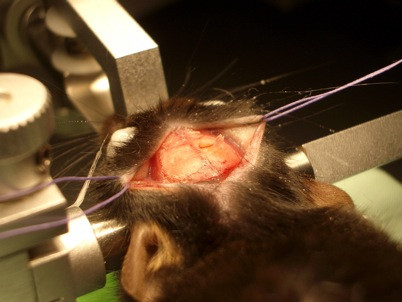
Craniotomy over right parietal bone, bone flap still in situ.
Figure 7.
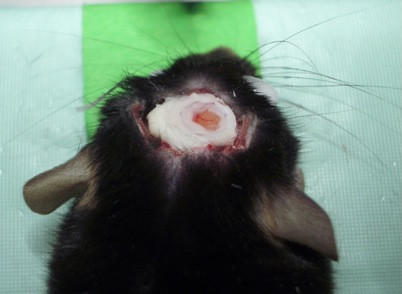
Glass cover slip fixed with cyanoacrylate and dental acrylic.
Thinned-skull window
After restraining the mouse head in a stereotactic frame, the scalp was incised in the midline. Periosteum was softly separated from underlying bone with cotton swabs.
The selected skull area (center of window 1.5 mm dorsolaterally of the bregma and the midline) was now carefully thinned in a circular area with the microdrill under the microscope until internal compact bone layer was reached (Figure 8). Generous irrigation is recommended for a clear view and to minimize the risk of heat-induced tissue injury. In the following step, the bone was continuously thinned in a cautious way with a microsurgical blade until the bone get so far thinned that cortex and vessels became visible. This procedure requires patience and dexterity because pushing and scraping to hard could damage brain and vessels, which leads to bleeding and inflammation or could even break the bone. Afterwards, one can start in vivo imaging immediately. Otherwise, the skin was sutured and the mouse was allowed to recover. For imaging sessions it is necessary to re-thin the skull with the microsurgical blades or to remove scar tissue.
Figure 8.
Thinned-skull cranial window.
In-vivo imaging
For the imaging sessions, the mice were anaesthetized and the head was fixed in the custom-made head holder by gluing the skull to the triple razor blades with cyanoacrylate to reduce respiration-induced movement artifacts. The animal was placed on a heating plate under the two-photon microscope with multifocal scan head splitter (Figure 9). To facilitate relocation of the imaged area, a high-quality picture of the cortex surface with meningeal blood vessels was obtained with a CCD camera (Figure 10). The Ti-sapphire laser was then tuned to optimal excitation wavelength for yellow fluorescence protein (890 nm). Images were acquired by using a 20x, 0.95 NA, water-immersion objective (Olympus, Tokyo, Japan) in an imaging depth of 100–200 μm from the pial surface (Figure 11 and Figure 12). Two-dimensional projections of three-dimensional image stacks containing dendritic segments of interest were saved for further analysis. One of the difficulties in repeated imaging lies in preserving the cranial window in appropriate condition and to ensure exactly the same region of interest.
Figure 9.

Mouse in head-holder under 2-photon microscope.
Figure 10.
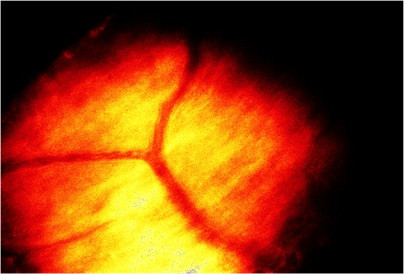
Meningeal blood vessels (video camera image).
Figure 11.
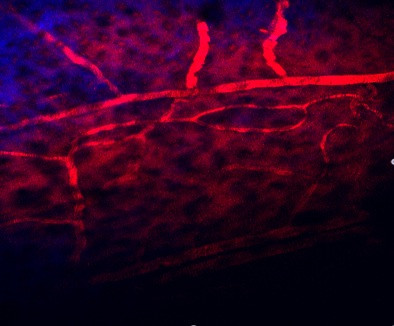
Cortical vessels under the 2-photon microscope.
Figure 12.
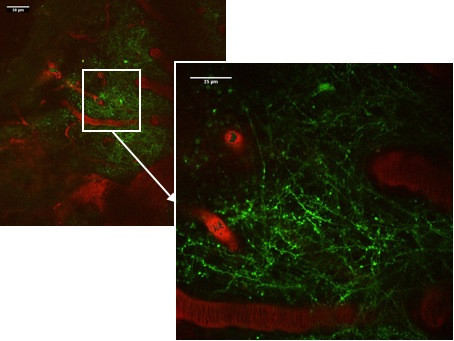
Rhodamine-dextran (red) filled cortical microvessels and Thy1-YFP labeled dendritic processes (green) in parietal cortex as viewed through a thinned skull cranial window.
Outlook
In this article, we provide a thorough methodological description of in vivo imaging of neuronal and vascular structures via two types of cranial windows. In experienced hands and with an established setup of two-photon microscopy, this method is a suitable tool for highly ambitious in vivo research, especially in the field of neurotrauma, neurodegenerative disorders, and neurovascular disease. The LaVision system was optimized for our application but the method is applicable for all two photon microscope systems. One of the advantages of the open skull method is that there is only one single surgery compared to multiple re-thinning procedures of the skull (and therefore multiple re-openings of the skin), an easier re-location of the same region of interest, and a higher penetration depth. However, the preparation of the open-skull window is demanding and bears a higher risk of dural tears and cortical injuries due to pressure or direct penetration. In addition, a damaged cover slip or opaque agarose layer could impair imaging results. Xu et al. reported a higher inflammation rate in neuronal tissue in the open-skull window with an increased turnover rate of dendritic spines [22]. Both models allow a “live” view on intracranial structures, not only on the surface of the brain, but even in deeper regions of neuronal tissue, and the possibility of long-term imaging.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CS carried out the surgical and imaging experiments, performed data analysis and drafted the manuscript. MHi designed and made custom-made devices and supported CS in performing imaging experiments. BN participated in the design and coordination of the study and supervised the experiments. RIE participated in the design of the study and edited the manuscript. MHe participated in the design and coordination of the study and supervised the experiments. ALS initiated, designed, supervised and coordinated the study and finalized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Christian Stetter, Email: Stetter_C@nch.uni-wuerzburg.de.
Markus Hirschberg, Email: markus.hirschberg@zeiss.com.
Bernhard Nieswandt, Email: nieswandt@uni-wuerzburg.de.
Ralf-Ingo Ernestus, Email: Ernestus.R-I@nch.uni-wuerzburg.de.
Manfred Heckmann, Email: heckmann@uni-wuerzburg.de.
Anna-Leena Sirén, Email: Siren.A@nch.uni-wuerzburg.de.
Acknowledgements
The authors thank Prof. Jens Eilers, Department of Physiology, University of Leipzig, for his expert advice. This work was supported by the German Research Council (DFG) SFB581/TP B27, the Interdisciplinary Center for Clinical Research (IZKF), University of Würzburg (TP N-229) and the University of Würzburg in the funding programme Open Access Publishing.
References
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Fraser SE. The neuronal naturalist: watching neurons in their native habitat. Nat Neurosci. 2001;4(Suppl):1215–1220. doi: 10.1038/nn754. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nat Rev Neurosci. 2006;7:449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Sigler A, Murphy TH. In vivo 2-photon imaging of fine structure in the rodent brain: before, during, and after stroke. Stroke. 2010;41:S117–S123. doi: 10.1161/STROKEAHA.110.594648. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Tian GF, Takano T, Lin JH, Wang X, Bekar L, Nedergaard M. Imaging of cortical astrocytes using 2-photon laser scanning microscopy in the intact mouse brain. Adv Drug Deliv Rev. 2006;58:773–787. doi: 10.1016/j.addr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Scheibe S, Dorostkar MM, Seebacher C, Uhl R, Lison F, Herms J. 4D in in vivo 2-photon laser scanning fluorescence microscopy with sample motion in 6 degrees of freedom. J Neurosci Methods. 2011;200:47–53. doi: 10.1016/j.jneumeth.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Jung CK, Herms J. Structural Dynamics of Dendritic Spines are Influenced by an Environmental Enrichment: An In Vivo Imaging Study. Cereb Cortex. 2012. first published online: October 18, 2012. [DOI] [PubMed]
- Niesner R, Andresen V, Neumann J, Spiecker H, Gunzer M. The power of single and multibeam two-photon microscopy for high-resolution and high-speed deep tissue and intravital imaging. Biophys J. 2007;93:2519–2529. doi: 10.1529/biophysj.106.102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzmaier SM, Zimmermann R, McGarry NB, Trabold R, Kim SW, Plesnila N. In vivo temporal and spatial profile of leukocyte adhesion and migration after experimental traumatic brain injury in mice. J Neuroinflammation. 2013;10:32. doi: 10.1186/1742-2094-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JC, Mehta AD, Aksay E, Stepnoski R, Schnitzer MJ. In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol. 2004;92:3121–3133. doi: 10.1152/jn.00234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW. In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol. 2004;91:1908–1912. doi: 10.1152/jn.01007.2003. [DOI] [PubMed] [Google Scholar]
- Mizrahi A, Crowley JC, Shtoyerman E, Katz LC. High-resolution in vivo imaging of hippocampal dendrites and spines. J Neurosci. 2004;24:3147–3151. doi: 10.1523/JNEUROSCI.5218-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Varga Z, Jia H, Deca D, Rochefort NL, Konnerth A. LOTOS-based two-photon calcium imaging of dendritic spines in vivo. Nat Protoc. 2012;7:1818–1829. doi: 10.1038/nprot.2012.106. [DOI] [PubMed] [Google Scholar]
- Margolis DJ, Lutcke H, Schulz K, Haiss F, Weber B, Kugler S, Hasan MT, Helmchen F. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci. 2012;15:1539–1546. doi: 10.1038/nn.3240. [DOI] [PubMed] [Google Scholar]
- De Paola V, Holtmaat A, Knott G, Song S, Wilbrecht L, Caroni P, Svoboda K. Cell type-specific structural plasticity of axonal branches and boutons in the adult neocortex. Neuron. 2006;49:861–875. doi: 10.1016/j.neuron.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Hechler D, Nitsch R, Hendrix S. Green-fluorescent-protein-expressing mice as models for the study of axonal growth and regeneration in vitro. Brain Res Rev. 2006;52:160–169. doi: 10.1016/j.brainresrev.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465:478–482. doi: 10.1038/nature09001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Lubischer JL, Kang H, Tian L, Mikesh M, Marks A, Scofield VL, Maika S, Newman C, Krieg P, Thompson WJ. Fluorescent proteins expressed in mouse transgenic lines mark subsets of glia, neurons, macrophages, and dendritic cells for vital examination. J Neurosci. 2004;24:10999–11009. doi: 10.1523/JNEUROSCI.3934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Piston DW. Imaging living cells and tissues by two-photon excitation microscopy. Trends Cell Biol. 1999;9:66–69. doi: 10.1016/S0962-8924(98)01432-9. [DOI] [PubMed] [Google Scholar]