

Abstract
Diabetes mellitus is a metabolic disease with multiple complications that causes serious diseases over the years. The condition leads to severe economic consequences and is reaching pandemic level globally. Much research is being carried out to address this disease and its underlying molecular mechanism. This review focuses on the diverse role and mechanism of ceramide, a prime sphingolipid signaling molecule, in the pathogenesis of type 1 and type 2 diabetes and its complications. Studies using cultured cells, animal models, and human subjects demonstrate that ceramide is a key player in the induction of β-cell apoptosis, insulin resistance, and reduction of insulin gene expression. Ceramide induces β-cell apoptosis by multiple mechanisms namely; activation of extrinsic apoptotic pathway, increasing cytochrome c release, free radical generation, induction of endoplasmic reticulum stress and inhibition of Akt. Ceramide also modulates many of the insulin signaling intermediates such as insulin receptor substrate, Akt, Glut-4, and it causes insulin resistance. Ceramide reduces the synthesis of insulin hormone by attenuation of insulin gene expression. Better understanding of this area will increase our understanding of the contribution of ceramide to the pathogenesis of diabetes, and further help in identifying potential therapeutic targets for the management of diabetes mellitus and its complications.
Keywords: Diabetes, Sphingolipid, Ceramide, Insulin resistance, Pancreatic apoptosis
Introduction
Diabetes mellitus is one of the leading causes of death worldwide, ranking amongst cardiovascular diseases and cancer. Diabetes may be classified into two groups based on its pathophysiology. One is caused by autoimmune destruction of β-cells (Type 1 or Insulin dependent diabetes), while the other is due to the progressive decline of pancreatic β-cell function in the context of insulin resistance (Type 2 or Non-insulin dependent diabetes) [1]. The major morbidity threats of diabetes are often microvascular (retinopathy, nephropathy and neuropathy) and macrovascular (cardiovascular diseases) complications [1,2]. According to a recent World Health Organization report, 3.2 million deaths worldwide are attributable to diabetes every year [2]. The number of people with diabetes will be more than double over the next 20 years, reaching a total of 366 million by 2030 [2]. Therefore, a better understanding of the disease and its pathophysiological mechanism is of paramount importance.
Sphingolipids, one of the major classes of lipid within the mammalian lipidome, are characterized by the presence of sphingoid base in their structure [3]. They are ubiquitous constituents of eukaryotic membranes that play a key role in the regulation of signal transduction pathways [4,5]. Deregulation of sphingolipids is implicated in numerous diseases including cancer [6], cardiovascular diseases [7], and neurodenerative disorders [8]. Over the past two decades, accumulating evidence has shown a clear indication that sphingolipids such as ceramide, sphingosine and glycosphingolipids (GSL), have important role in the pathogenesis of both type 1 and 2 diabetes and its associated complications.
The aim of this review is to shed light on the current understanding of the role and mechanism of ceramide in diabetes, and to highlight areas requiring further study. Understanding the complex mechanism of ceramide action in diabetes requires adequate knowledge of insulin signal transduction and sphingolipid biosynthesis. Therefore, the first two sections of this review provide an overview of insulin signaling and sphingolipid metabolic pathways. This review also summarizes the role, and potential cellular signaling mechanisms of ceramide in β-cell apoptosis, insulin resistance, and attenuation of insulin gene expression. This review will also very briefly discuss the role of ceramide in some of the diabetic complications.
Insulin signaling pathway
Insulin, an endocrine hormone produced from the pancreatic β-cells, plays a key role in glucose homeostasis [9,10]. Insulin action is mediated through insulin receptor (IR) that propagates its activity via three different pathways: phosphatidylinositol-3 kinase (PI3K) pathway, mitogen-activated protein kinase (MAPK) pathway, and Cbl-associated protein (CAP) pathway [11,12]. The PI3K pathway is the major pathway involved in glucose transport, and is significantly distorted by ceramide. The present review specifically focuses on this particular pathway of glucose regulation.
Insulin receptor is a tyrosine kinase receptor which has two extracellular α-subunits, and two transmembrane β-subunits [11]. Upon insulin binding to the α-subunit, IR undergoes autophosphorylation of tyrosine residues in the intracellular β-domain. Insulin receptor substrate (IRS) has a phosphotyrosine binding domain that recognizes the activated IR, and in turn leads to tyrosine phosphorylation and activation of IRS (Figure 1) [11]. Once activated, IRS allows the binding and activation of PI3K which phosphorylates the membrane lipid phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-triphosphate (PIP3). Protein kinase B (PKB, also known as Akt) is recruited to the plasma membrane and activated at PIP3 site in the presence of phosphoinositide-dependant protein kinase-1 (PDK1) [10]. The activation of Akt allows its relocalization to the cytosol, where it causes the translocation of the glucose transporter-4 (Glut-4) to the plasma membrane, thereby promoting glucose uptake (Figure 1) [13]. Akt also phosphorylates and inactivates glycogen synthase kinase-3 (GSK3), an enzyme involved in glycogen synthase phosphorylation and inactivation. This results in an increase in glucose storage as glycogen [12]. Glycogen synthase kinase-3 is also an endogenous inhibitor of guanine nucleotide exchange factor eIF2B, an essential participant in the initiation of protein translation. Hence, the inactivation of GSK3 by Akt promotes protein synthesis making the amino acids less available for gluconeogenesis [12]. Akt also excludes transcription factor Forkhead box O1 (FOXO1) from the nucleus, abolishing FOXO1 mediated expression of gluconeogenic enzymes such as glucose-6-phosphatase (G6P), fructose-1,6-biphosphatase (F16BP) and phosphoenolpyruvate carboxykinase (PEPCK) (Figure 1) [12,14]. Akt activates mammalian target of rapamycin (mTOR) which promotes protein synthesis through activation of p70 ribosomal S6 kinase (p70S6K), and the inhibition of eIF4E binding protein (eIF4EBP) [15,16]. The activation of mTOR by Akt also promotes fatty acid (FA) uptake and synthesis by activating transcription factor sterol regulatory binding protein 1C (SREBP-1c) which is a protein that enhances the transcription of FA and triglyceride biosynthetic enzymes (Figure 1) [12,14].
Figure 1.

Major pathways in insulin receptor signaling and mechanism of ceramide in insulin resistance. Details of these processes and abbreviations are described in the text. Briefly, activation of insulin receptor leads to Akt activation. Once activated, Akt reduce blood glucose level by inducing glucose uptake, glycogen synthesis, protein synthesis and fatty acid synthesis. Akt also act by inhibiting gluconeogenesis. Some of the metabolic effects of insulin may also be mediated via other signaling branches than those depicted. Ceramide causes insulin resistance by PP2A and PKCζ mediated inhibition of Akt and MLK-3 mediated inhibition of IRS. Ceramide may also decrease Glut-4 gene transcription. Cer, Ceramide.
Sphingolipid biosynthesis: regulation and compartmentalization
The biosynthetic pathway of sphingolipids consists of a complex network of synthetic and degradative reactions. The de novo biosynthesis of sphingolipids occurs at the cytosolic leaflet of the endoplasmic reticulum (ER). During this, 3-ketosphinganine is formed by the condensation of L-serine and palmitoyl CoA by the action of an enzyme serine palmitoyl transferase (SPT). The newly formed 3-ketosphinganine first undergoes rapid reduction to dihydrosphingosine by the action of 3-ketosphinganine reductase, which is then acetylated to form dihydroceramide (dh-Cer) by the action of dh-Cer synthase. The enzyme dh-Cer desaturase reduces dh-Cer to ceramide (Figure 2) [4,5,17]. Ceramide serves as a ‘metabolic hub’ in the sphingolipid metabolic pathway as a substrate for subsequent production of other sphingolipid signaling intermediates [18-20]. Neutral ceramidase in the ER, alkaline ceramidase in the plasma membrane, and acid ceramidase in the lysosome may hydrolyze ceramide to generate sphingosine. Sphingosine may be phosphorylated to sphingosine-1-phosphate (S1P) by sphingosine kinase. The formation of sphingosine from ceramide and S1P from sphingosine can be reversed by enzymes ceramide synthase and S1P phosphatase respectively (Figure 2).
Figure 2.

Regulation of sphingolipid biosynthesis. Details of these processes and abbreviations are described in the text. Briefly, ceramide is formed from L-serine and palmitoyl CoA via de novo pathway. Ceramide can also be formed from SM hydrolysis by SMase, glucosyl ceramide hydrolysis by Glucocerebrosidase and from sphingosine by ceramide synthase. Sphingolipid metabolites are depicted in box and enzymes are shown outside the box. Some pharmacological inhibitors of sphingolipid biosynthetic enzymes are also shown.
Ceramide once formed in ER, is transported to the Golgi with the help of a transfer protein CERT [20]. The Golgi is the site for the synthesis of sphingomyelin (SM) and glucosylceramide, with the latter serving as the precursor for complex GSLs. An enzyme SM synthase-1 at Golgi, and SM synthase-2 at plasma membrane transfer a phosphorylcholine head group to ceramide and form SM with the release of diacylglycerol (DAG) [4,20]. Alkaline and neutral sphingomyelinase (SMase) at the plasma membrane, and acid SMase at the lysosome can reverse this reaction to generate ceramide back from SM. Glucosylceramide is formed from ceramide by action of membrane bound glucosylceramide synthase (GCS), which can be further converted to complex GSLs by different sialyl transferases. In the lysosome, complex GSLs are degraded back to glucosylceramide, and then to ceramide by the enzymes glycosidases and glucocerebrosidase, respectively [20]. In an alternative pathway, ceramide is phosphorylated at the plasma membrane by ceramide kinase. Its product, ceramide-1-phosphate (C1P), can be hydrolyzed back by C1P phosphatase to generate ceramide (Figure 2) [21].
Ceramide in pancreatic beta-cell apoptosis
Role of ceramide in beta-cell apoptosis
Apoptosis is a programmed death of cells that plays an important role in the maintenance of tissue homeostasis by eliminating harmful or unwanted cells. The mechanism of apoptosis involves complex signaling pathways. The three currently recognized apoptotic signaling pathways are: the extrinsic death receptor, the intrinsic mitochondrial, and the intrinsic ER pathways [22,23]. Literature suggests that the excessive apoptosis of pancreatic β-cell contributes significantly in the pathogenesis of both type 1 [24] and type 2 [25] diabetes. The role of ceramide in β-cell apoptosis in both type 1 and type 2 diabetes is also well established.
Cytokines such as tumor necrosis factor-alpha (TNF-α), Interleukin-1 beta (IL-1β) and Interferon-gamma (IFN-γ) are known to exert cytotoxic effects on pancreatic β-cells [26]. Emerging evidence suggest that ceramide has a role in β-cell apoptosis induced by these cytokines [27]. Exposure of insulin-producing MIN6 cells and RINm5F cells to TNF-α and IL-1β, respectively, results in an increase in ceramide production. In addition, ceramide, either delivered exogenously or generated endogenously, mimicked the cytotoxic effect of TNF-α and IL-1β in these cell lines [28-30]. On the other hand, another β-cell model, β-TC3 cells did not show an increase in ceramide when treated with cytokines [31]. This disparity may be due to an immediate activation of ceramidase in β-TC3 cells as reported with INS-1 β-cells [32]. Ceramidase is capable of converting the ceramide to sphingosine which can be further converted to S1P by sphingosine kinase. This is further evidenced with the presence of enhanced S1P level in INS-1 cells following treatment with IL-1β and TNF-α [33]. Such a possibility has to be tested in β-TC3 cells employing sensitive assay methods.
Long-standing evidence implicates that prolonged exposure to free fatty acid (FFA) has detrimental effects on pancreatic β-cells. Ceramide has been proposed to be a mediator of FFA-induced β-cell toxicity. Palmitate, a precursor of ceramide has been found to induce islet cell apoptosis in diabetic rat models, healthy rats, and human β-cells [34-36]. However, unsaturated FA failed to induce this effect [35,37,38]. Similarly, studies conducted in Zucker diabetic fatty (ZDF) rats demonstrated that ceramide formed via the de novo pathway is responsible for the FFA-induced β-cell apoptosis. This is evidenced with the appearance of marked increase in [3H]-ceramide up on culturing islet cells in the presence of either [3H]-serine or [3H]-palmitate [34,39]. In addition, inhibition of ceramide synthesis using SPT inhibitor (L-cycloserine) or ceramide synthase inhibitor (fumonisin-B1), has been reported to attenuate FFA-induced cytotoxicity in both rodent [34,35,39] and human [37,40] β-cells. In contrast, some studies with β-cell apoptosis have reported only modest increase in ceramide upon treatment with FFA. However, inhibition of de novo ceramide synthesis reduced the apoptosis significantly [41]. There could be three possible explanations for this contradictory result. First, an increase in ceramide at particular cellular location, without altering the total ceramide mass may be sufficient to induce apoptosis. This is supported by the fact that ceramide synthesized in the ER can be transported to other organelles including mitochondria, and moreover, mitochondria itself contains the enzymatic machinery necessary for ceramide synthesis [42]. A second possible explanation is conversion of ceramide to another sphingolipid metabolite after apoptotic induction. To support this, Boslem et al. identified an increase in glucosyl ceramide, a metabolite of ceramide up on treatment of MIN β-cells with FFA. They also found that the overexpression of glucosyl ceramide synthase, an enzyme that converts ceramide to glucosyl ceramide, did not exacerbate, but partially protected from apoptosis [43]. Finally, apoptotic potential of ceramide may vary with different isoforms. Veret et al. demonstrated that FFA, at low glucose concentration, increased all ceramide species, but at elevated glucose concentration increased the more toxic C18:0, C22:0 and C24:1 isoforms of ceramide specifically to enhance apoptosis [44]. Conclusive data from these studies demonstrate that ceramide generated via the de novo pathway is the key player in the execution of FFA-induced β-cell apoptosis.
Mechanism of ceramide induced beta-cell apoptosis
Activation of extrinsic apoptotic pathway
It has been generally accepted over the past few decades that activation of the extrinsic pathway of apoptosis plays a significant role in β-cell loss. The extrinsic pathway begins outside the cell with the binding of death ligands, such as TNF-α and Fas ligand, to their respective cell-surface death receptors such as TNF receptor and Fas (CD95). This binding of the receptor by its cognate ligand results in the recruitment and activation of initiator caspase (mostly caspase-8), which then propagates apoptosis by cleaving and activating downstream effector caspases such as caspase-3 [45,46]. Agent that induces ceramide accumulation en route to β-cell apoptosis, such as cytokines and saturated FAs, were also reported to activate the extrinsic pathway of apoptosis [27,35] (Figure 3). Normal pancreatic β-cells exhibit only minimal expression of Fas, but when exposed to cytokines, such as IL-1, an increase in Fas expression was observed [47]. Pancreatic β-cell that lacks caspase-8 has shown protection against both Fas and ceramide-induced cell death, suggesting the role of ceramide in it [48]. Moreover, Liadis et al. demonstrated that caspase-3 knockout mice protected from developing diabetes [49], signifying the role of caspase cascade in β-cell apoptosis.
Figure 3.

Schematic representation of mechanism of beta-cell apoptosis caused by ceramide. Details of the processes and abbreviations are described in the text. Briefly, cellular ceramide can be formed by de novo biosynthesis from precursor palmitate and by the activation of extrinsic pathway of apoptosis by FasL or TNF-α binding to the Fas and TNF receptor (TNFR) respectively. Ceramide acts on the mitochondria and causes the release of ROS/RNS and cytochrome c and activates intrinsic apoptotic pathway. Increased ROS can further increase ceramide generation. Ceramide can act on the ER and cause ER stress mediated apoptosis. Ceramide also inhibits insulin gene expression.
Alteration of mitochondrial membrane permeation
Ceramide induces β-cell apoptosis by increasing the mitochondrial membrane permeability, leading to the activation of the intrinsic pathway of apoptosis. The intrinsic mitochondrial pathway is initiated within the cell in response to a wide range of death stimuli. Regardless of the stimuli, pro-apoptotic molecule, such as cytochrome c, is released into the cytosol which together with another pro-apoptotic protein, Apoptotic protease activating factor 1 (Apaf-1) activates the initiator caspase (mostly caspase-9). The initiator caspase then activates an effector caspase such as caspase-3 that propagates the apoptotic signal [50]. The intrinsic pathway is regulated by a group of proteins belonging to the B-cell lymphoma-2 (Bcl-2) family. There are two main groups of the Bcl-2 proteins, namely the pro-apoptotic proteins (e.g. Bad, Bid, Bax, Bak, Bcl-Xs, Bim, etc) and the anti-apoptotic proteins (e.g. Bcl-2, Bcl-xL, Mcl-1, etc). The anti-apoptotic proteins reduce apoptosis by blocking the mitochondrial release of cytochrome-c, while the pro-apoptotic proteins act by promoting it. The fate of the cell is determined by the tilt in their ratio towards one or the other [46,50].
Ceramide either delivered exogenously or generated endogenously when targeted to the mitochondria, an increase in its permeability to cytochrome c and apoptosis is observed [51,52] (Figure 3). The apoptotic action of ceramide is mediated by the recruitment and activation of pro-apoptotic Bax at the mitochondria. Birbes et al. [53] and Kashkar et al. [54], demonstrated that induction of SMase, an enzyme that converts SM to ceramide, promotes the translocation and activation of Bax at the mitochondria which in turn leads to the release of cytochrome c [55]. In agreement with this, depleting the Bax or antagonizing its activity by overexpressing Bcl-2, prevented ceramide-induced apoptosis [56,57]. The intrinsic mitochondrial pathway is also involved in the FFA-induced β-cell apoptosis. It has been reported that exposure of β-cells to saturated FAs results in enhanced mitochondrial membrane permeability and cytochrome c release [58]. Furthermore, reduction in Bcl-2 expression [36] and Bax up-regulation [58] was also observed when β-cells were exposed to FFAs. Jointly, these studies suggest that ceramide generated from FFA may very well be involved in the activation of the intrinsic apoptosis pathway. Yet, another mechanism of ceramide in mitochondrial membrane alteration has also been proposed. Siskind et al. proposed that ceramide is directly capable of forming channels in the mitochondrial membrane, thus, increasing its permeability to cytochrome c [59]. This action of ceramide has not been extensively studied yet. Ceramide also interferes with Akt activation, and thereby, promotes the mitochondrial membrane permeability, and cytochrome c release by preventing the Akt-induced inactivation of pro-apoptotic Bcl-2 members [60].
Free radical generation
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are some of the key integrating mediators in the development of β-cell apoptosis. Although, relevant amount of ROS and RNS are essential for maintaining normal physiological signaling and metabolic functions, excessive amount of these radicals can lead to cell death and tissue damage [61]. Ceramide plays an important role in ROS and RNS generation through activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [62], initiation of mitochondrial dysfunction [63], induction of inducible nitric oxide synthase (iNOS) gene expression [64], and down-regulation of anti-apoptotic Bcl-2 proteins [65]. Conversely, a growing body of evidence also suggests that ROS and RNS enhance ceramide generation by inducing SMase [66], or inhibiting ceramidase [67] enzymes (Figure 3). Although, these effects are not extensively studied in the pancreatic β-cells, the bidirectional interaction of ceramide and ROS/RNS may also be involved in the apoptotic cascades in β-cells.
The major source of ROS in most cells is the leakage of electrons from complex I and complex III of the mitochondrial respiratory chain [14]. Inhibition or reduction of activities of these complexes results in electron leakage, and elevated ROS level which in turn can lead to β-cell apoptosis. Ceramide has been shown to disrupt electron transport at complex I [68] and complex III [69], resulting in an enhanced ROS generation which facilitates cytochrome c release and caspase activation [63]. Similarly, TNF-α, a known inducer of ceramide is also found to inhibit complex III activity [14]. As described earlier, activation of NADPH oxidase by ceramide also leads to ROS generation and apoptosis if it is not rapidly removed by the antioxidant defense [62]. The exact mechanism of ceramide-mediated NADPH oxidase activation is not known, but it is believed that ceramide activates protein kinase C zeta (PKCζ), which facilitates binding of p67phox, a catalytic subunit of NADPH oxidase, to its holo enzyme complex [70,71]. Another mechanism of ceramide-induced free radical generation is via regulation of anti-apoptotic Bcl-2 protein. Bcl-2 prevents ROS production, increases antioxidant GSH pool and redistributes it [65]. In islet cells, ceramide is reported to decrease Bcl-2 mRNA expression [72]. Yet, another mechanism of ceramide-induced free radical generation is via increased expression of iNOS, which codes for iNOS enzyme involved in the synthesis of nitric oxide (NO) in response to inflammatory stimuli such as cytokines and lipotoxicity. Ceramide, either generated via the de novo pathway [73] or SM hydrolysis [74], is reported to induce nuclear factor kappa B (NFκB), a transcription factor involved in the expression of iNOS gene activation. When islets of ZDF rats were supplied with FAs, an increase in apoptosis along with increased iNOS expression and nitric oxide formation was observed [34]. These effects were blocked by fumonisin B1 and aminoguanidine, a NOS inhibitor [34], suggesting the role of ceramide in it. Finally, ROS also acts by inducing ER stress that further induces β-cell apoptosis [75]. In conclusion, all these studies suggest that ROS/RNS generation triggered by ceramide may be one of the crucial factors in promoting apoptosis in many mammalian cells and animal models, but further studies are required to understand the complete involvement of these free radicals in β-cell apoptosis.
Induction of ER stress
Accumulating evidence suggests that ER stress plays an important role in β-cell apoptosis [76,77]. During diabetes there will be high secretory demand for insulin placed on the pancreatic β-cells to counteract hyperglycemia. The ER, being the site of synthesis and folding of secreted proteins, is highly susceptible to stress when the demand for insulin folding and secretion exceeds its capacity [78,79]. This increased ER stress leads to accumulation of misfolded and unfolded insulin in the ER, which may activate Unfolded Protein Response (UPR) to restore the normal ER function. When restoration fails, UPR switches in to an alternative mode and turns on the apoptotic signaling pathway in the β-cells. Oyadomari et al. and Socha et al. demonstrated ER stress-induced β-cell apoptosis in diabetic animal models [80,81]. Similarly, elevated levels of ER stress markers were observed in islets of diabetic human subjects [82]. Fatty acids, particularly saturated FAs have been shown to promote ER stress to induce β-cell apoptosis [82,83]. This specific toxic effect of saturated FAs may be related to the formation of ceramide from it [41]. Collectively, observations from several studies demonstrate that ceramide synthesis via the de novo pathway is involved in ER stress-induced β cell apoptosis [43,44]. In line with this, ceramide generated via SM hydrolysis has also been reported to cause ER stress-induced β-cell apoptosis. Lei et al demonstrated that activation of Ca2 + -independent phospholipase A2 (iPLA2β), an inducer of β-cell apoptosis in response to strong ER stress, promotes ceramide accumulation secondary to the activation of neutral SMase, an enzyme that hydrolyzes SM to generate ceramide [84]. Inhibition of neutral SMase protected β-cell from ER stress induced apoptosis, demonstrating the importance of ceramide in ER stress-induced cell death. Ceramide generated by ER stress also activates intrinsic mitochondrial pathway of apoptosis in β-cells by altering the mitochondrial membrane permeability and release of cytochrome c [84]. ER stress and induction of UPR can also result in ROS generation, and the role of ROS, both as upstream and downstream, to ceramide activation is well known [85].
Inhibition of Akt
A major mechanism through which ceramide induces β-cell apoptosis is by its inhibitory action on Akt, a serine/threonine kinase which regulates several biological processes including cellular growth, proliferation and survival in multiple organs (Figure 3). Akt mediates its proliferative and anti-apoptotic action on the pancreatic β-cell through several mechanisms. First, Akt phosphorylates and induces cytosolic retention of cyclin-dependent kinase inhibitors (CKI) such as p21Cip1 and p27Kip1. This enhances the proteosomal degradation of these CKIs [86]. Second, Akt negatively regulates the transcriptional activity of FOXO1, which is known to upregulate p27kip1 [86]. Third, Akt directly phosphorylates and inactivates pro-apoptotic Bcl-2 members such as Bad, Bax and Bid [87]. Finally, Akt activates mTOR/p70S6K mediated cell growth and proliferation [15,16]. Through all these mechanisms, Akt promotes the cell cycle, proliferation and inhibition of apoptosis.
An inverse correlation between ceramide and Akt activation has been reported in cultured cells when exposed to ceramide [12,15,16,88-90]. Akt inactivation along with ceramide accumulation is also observed in rats treated with glucocorticoids and saturated fat [91]. Inhibition of ceramide biosynthesis using myriocin, cycloserine, or fumonisin B1 restored the Akt activity [15,91,92]. As an alternative strategy to manipulate endogenous ceramide, cultured cells when treated with PDMP, an inhibitor of ceramide glucosylation, exacerbated palmitate-induced Akt inactivation [93]. This palmitate effect was reversed by over expressing acid ceramidase [92,94]. These studies support the hypothesis that Akt inactivation by ceramide is one of the contributing mechanisms by which ceramide causes β-cell apoptosis. The mechanism through which ceramide inhibits Akt activity will be discussed later in this review.
Ceramide in insulin resistance
Role of ceramide in insulin signaling and action
It is becoming increasing apparent that ceramide plays significant role in insulin resistance, a metabolic state in which cells fail to respond to the normal hormonal actions of insulin. Evidence from various studies revealed that excess FFA intake, glucocorticoid administration, obesity, and lack of physical exercise are some of the important causations leading to insulin resistance. Plethora of studies using wide variety of cultured cells, animal models and human subjects demonstrated ceramide as the key intermediate linking all these conditions to insulin resistance [95].
Preliminary evidence underpinning the role of ceramide in insulin resistance came from the direct application of ceramide to isolated skeletal muscles and cultured adipocytes. These studies indicate that ceramide inhibits insulin-stimulated glucose uptake and glycogen synthesis [96,97]. Increased delivery of saturated FA in excess of a tissue’s oxidative or storage capacity is one of the main reasons for insulin resistance. Prolonged exposure of palmitate to cultured myotubes [15], L6 skeletal muscle cells [16], 3 T3-L1 adipocytes [98] and cardiac myocytes [99] increases ceramide accumulation with simultaneous inhibition of Akt. Consistent with this, palmitate exposure also caused a reduction in glucose uptake and glycogen synthesis [92]. Subsequent studies using pharmacological inhibitors or small interfering RNA (siRNA) to block the enzymes involved in ceramide biosynthesis, have demonstrated that ceramide is an obligate intermediate in saturated FA-induced insulin resistance [94,100,101]. In agreement with this, overexpression of acid ceramidase as an alternative approach to reduce the ceramide level, negated the palmitate-induced ceramide accumulation and improved insulin signaling [93]. Similarly, acute application of ceramide analogue to 3 T3-adipocytes mimicked the palmitate induced insulin desensitizing effect [102].
As mentioned earlier, some groups also studied the role of saturated FA in insulin resistance using animal models, including high fat-fed mice, ob/ob mice, lipid-infused rats, dexamethasone-treated rats and ZDF rats [103-105]. Recently, Frangioudakis et al. demonstrated that mice fed with high-fat diet shows increased expression of ceramide synthase [103]. Infusion of lipid emulsion in animal model has shown to increase muscle ceramide content and decrease peripheral insulin sensitivity [105]. These effects were blocked by using SPT inhibitors, indicating the role of ceramide [105]. Recently, Holland et al. compared the effect of lard oil (high in saturated FAs) and soy oil (high in unsaturated FAs) on insulin sensitivity in rats. They found that both of these treatments decreased glucose uptake and Akt activation, but ceramide increase was observed only with lard-oil infusion [105]. Similarly, several other studies also could not find considerable increase in ceramide in response to lipid supplementation, but an elevated DAG level was observed [106,107]. When carefully examined, some of these studies were found to use lipid rich in unsaturated FA, indicating that saturated fats and unsaturated fats have different mechanisms of promoting insulin resistance, and ceramide plays a role only in insulin resistance induced by saturated fats [105]. As mentioned earlier, the experiments conducted to study the effect of FA on ceramide generation and insulin resistance, were not only restricted to cell lines and animal models. Studies conducted in human subjects also provided precious information. Studies conducted using insulin-resistant human subjects demonstrated almost two fold increase in ceramide accumulation compared to normal subjects [108]. In another study, lipid infusion in humans is found to increase skeletal muscle ceramide and decrease insulin sensitivity [109].
Apart from inducing ceramide generation, chronic exposure to elevated concentrations of FAs have been reported to modulate several other effectors and signaling pathways, to induce beta-cell dysfunction and apoptosis. Some of those effectors include endocannabinoids, eicosanoids, cytokines, and transcription factors. Endocannabinoids are FA derivatives implicated in the regulation of energy balance, hepatic lipogenesis, and glucose homeostasis [110,111]. Elevated levels of endocannabinoids, such as 2-arachidonoylglycerol or anandamide, have been implicated in hyperglycemia and decreased insulin sensitivity in high fat fed mice and obese human subjects [111]. Inhibition of endocannabinoid activity, using endocannabinoid receptor antagonist, resulted in increased glucose uptake in diabetic mice models [111]. Eicosanoids are another class of FA derivatives that play vital role in the control of pancreatic β-cell function and survival [112,113]. Cyclooxygenase derived eicosanoids, such as prostaglandin E2 and eicosanoids of 12-Lipoxygenases, have been found to attenuate glucose-stimulated insulin secretion and increase beta-cell destruction in the pancreas [113]. NF-κB is a transcription factor which is best known for its immune and inflammatory responses. Key role of the NF-kB pathway in the induction of inflammatory responses, that underlie type 2 diabetes, has been highlighted in several studies [114,115]. Fatty acids, in particular saturated FAs induce the expression of NF-κB through Toll-like receptor 4 (TLR4) signaling [115]. An improvement in insulin sensitivity was observed in animal model of FFA-induced insulin resistance when the gene encoding for TLR4 was mutated [115].
Glucocorticoids are most commonly used therapeutic agents, although contraindicated in diabetes as they can cause insulin resistance [116]. Previously, it has been demonstrated that glucocorticoids induce ceramide generation which may be responsible for its induction of insulin resistance [105]. Dexamethasone, a widely used glucocorticoid was shown to increase ceramide level in broad range of cell types, and animal subjects through stimulation of enzymes such as SPT, SMase and ceramide synthase [105,117,118]. Some of this effect was completely prevented by pre-treating the subjects with myriocin [105]. It has been reported that glucocorticoid-induced ceramide accumulation and associated insulin resistance requires activation of peroxisome proliferator activated receptor (PPAR) alpha. As an evidence to this, genetic ablation of PPAR alpha, or disruption of hepatic vagal nerves (which decreases hepatic PPAR alpha expression) prevented dexamethasone-induced insulin resistance [119,120]. Thiazolidinediones (TZDs) are most commonly used anti-diabetic agent which functions as insulin sensitizers [121]. It is found that TZDs like pioglitazone [122], troglitazone [123], and rosiglitazone [124] decrease ceramide accumulation in muscles of rat or mice. Fenretinide, a chemotherapeutic agent is found to improve insulin sensitivity in high fat-fed mice, was recently identified as an inhibitor of dihydroceramide desaturase. Thus, insulin-sensitizing actions of some of these drugs may result from its inhibitory effects on ceramide synthesis [125].
Physical exercise is widely perceived to be beneficial for glycemic control in type 2 diabetes, as it improves insulin sensitivity and glucose homeostasis [126]. Routine exercise training is found to decrease ceramide content in skeletal muscles of rat, mice [127,128] and human subjects [129,130]. In contrast, some studies could not report significant decrease in muscle ceramide level even after exercise training in rats and humans [131,132]. The reason for this discrepancy remains unclear. Obesity and associated increase in pro-inflammatory cytokines is another key player of insulin resistance [133]. Patients with obesity have elevated level of the inflammatory cytokine such as TNF-α [134]. The role of TNF-α in ceramide generation has already been discussed in this review. Finally, insulin resistance increases with age. Wu et al. demonstrated that adipocytes from older mice contained higher ceramide compared to younger one [135]. Conclusive evidence from all these studies suggests that inhibitors of ceramide synthesis or activators of ceramide degradation may prove efficacious as therapeutics to combat insulin resistance.
Mechanism of ceramide-induced insulin resistance
The precise molecular mechanisms by which changes in ceramide might cause insulin resistance are not entirely clear. Although, it seems likely that ceramide influences several distinct intermediates in the insulin signaling pathway. Some of these potential mechanisms by which ceramide may impair insulin signaling and action are discussed below.
Ceramide on IRS
Kanety et al. reported that ceramide inhibits insulin stimulated tyrosine phosphorylation of IRS-1 [136]. This is confirmed by some other studies which demonstrated that ceramide phosphorylates inhibitory serine residues on IRS-1 [136,137]. The probable mechanism is that ceramide activates mixed lineage kinase-3 [138] (Figure 1), which in turn activates stress-activated protein kinases such as p38 and c-Jun N-terminal kinase (JNK) [139,140]. These enzymes have been implicated in phosphorylation of serine-307 on IRS-1 [141,142]. This in turn inhibits the necessary tyrosine phosphorylation needed for insulin signal transduction [143]. In agreement to this, Hirosumi et al. demonstrated an increase in JNK activity and serine-307 phosphorylation, and a decrease in tyrosine phosphorylation of IRS-1 in tissues of obese mice [142]. In another study, mutations in the gene coding for JNK-binding protein (an inhibitor of JNK activity) in humans caused type 2 diabetes [10,144]. Even though these studies demonstrated ceramide-induced inhibition of IRS-1, other studies did not find any correlation [16,92].
Ceramide on PI3K, PDK1, Phosphoinositides (PIP2 and PIP3) and Glut-4
Several studies evaluated the role of sphingolipids on PI3K [16,91,92], PDK1 [145], phosphoinositide (PIP2 and PIP3) [146,147] and Glut-4 [98]. However, majority of these studies failed to see any direct effect. Although Zundel et al. reported that ceramide inhibits PI3K activity [148], yet, its relevance in the regulation of glucose homeostasis remains unclear.
Ceramide on Akt/PKB
The role of ceramide in regulating the Akt to induce β-cell apoptosis has been discussed in the preceding section of this review. Indeed, the involvement of Akt in β-cell physiology might go beyond the induction of apoptosis and include the regulation of insulin secretion. Therefore, the inhibition of Akt by ceramide might have a negative impact on insulin sensitivity as well by abrogating all Akt mediated insulin activities (Figure 1).
Inhibition of Akt activation by ceramide is thought to be accomplished by at least two mechanisms. First, ceramide activates protein phosphatase 2A [92,149] (Figure 3) which catalyses the dephosphorylation of Akt by removing activating phosphates [92,145,146,150]. The Akt inhibitory effect of ceramide in cell lines like PC12 cells [145], C2C12 myotubes [92], human glioblastoma cell [150], and brown adipocytes [149], was negated by the PP2A inhibitor, Okadaic acid. When PP2A activity was impaired by over expressing SV40 small T antigen (which displaces the regulatory subunits that target PP2A to its substrates), the effect of ceramide on Akt was blocked [99]. Second, ceramide blocks insulin-stimulated Akt translocation to the PIP3-PDK1 complex at the plasma membrane [16,147]. Powell et al. and Bourbon et al demonstrated that ceramide binds to cysteine rich ceramide binding domain on PKCζ and activates it [94,151,152] (Figure 3). The activated PKCζ in turn phosphorylates inhibitory the serine or threonine residue (depending on the Akt isoforms) at the site 34 in the pleckstrin homology (PH) domain of Akt [152]. This prevents its interactions with PIP3, may be by forming more stable Akt-PKCζ complex [94,152]. In agreement with this mechanism, PKCζ inhibitors were found to increase insulin sensitivity and prevent ceramide-induced loss of Akt activation [152,153].
Ceramide in lipid raft and diabetes
Lipid rafts are specialized micro-domains of plasma membrane that contain high concentrations of lipid derived molecules such as cholesterol, sphingolipids and a subset of phospholipids [154]. Sphingomyelin is the most prevalent sphingolipid present in the outer leaflet of the plasma membrane and hence in the lipid raft. The SM within the raft can be hydrolyzed by SMase and lead to the generation of ceramide [155] (Figure 4). Previously discussed key players of diabetes induction such as TNF receptor activation, Fas activation and ROS generation were reported to activate SMase and hence generate ceramide in the rafts [156]. Ceramide molecules associate with each other and immediately form small ceramide-enriched membrane microdomains. The generation of ceramide and its tight packing within rafts dramatically alters the structure and composition of these domains. This results in spatial re-organization and clustering of several cytokine and death receptors, including Fas and TNF receptors [155,156]. This receptor clustering perhaps could promote β-cell apoptosis through proximity induced caspase activation (Figure 4). In line with this, cells that lack SMase activity failed to release ceramide and induce apoptosis upon Fas and TNF receptor stimulation [157]. Consistent with this, neutralization of ceramide in ceramide-enriched membrane domains using anti-ceramide antibodies inhibited Fas-induced apoptosis [158]. However, additional experimental evidence is required to elucidate the entire role of receptor clustering in diabetes.
Figure 4.
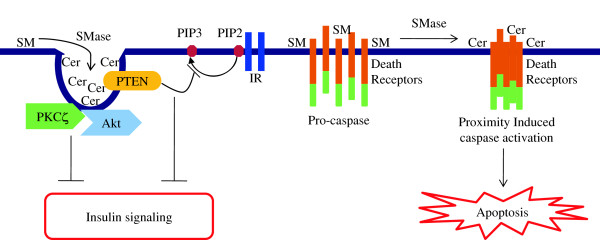
Schematic representation of role of ceramide in lipid raft and diabetes. Details of the processes and abbreviations are described in the text. Briefly, the SM within the raft can be hydrolyzed by SMase to form ceramide which immediately self-associate and form ceramide enriched membrane domains. This raft coalescence facilitates apoptosis by initiating proximity induced caspase activation. Ceramide recruits both PKCζ and Akt to the caveolae. By doing so, ceramide can not only activate PKCζ, but also causes co-localization of activated PKCζ with Akt, there by suppressing Akt activity. Ceramide also recruits PTEN in the caveolae which dephosphorylate PIP3 and impair insulin signaling. Cer, Ceramide.
Caveolae are a subset of lipid rafts seen as flask like invaginations on the plasma membrane [159]. Apart from ceramide, these structures are characterized by the presence of proteins termed caveolin, a family of scaffolding proteins which play significant role in numerous signaling pathways by compartmentalizing and concentrating signaling molecules [159]. Generally, optimal level of caveolae is important for the proper insulin signaling. However, excessive caveolae is reported to impair insulin signaling and action, basically via two mechanisms. First, ceramide extensively recruits and elevates phosphatase and tensin homolog deleted on chromosome 10 (PTEN) in caveolae. The anti-proliferative PTEN specifically catalyses the dephosporylation of the 3′ phosphate in PIP3 and convert it to PIP2 (Figure 4). This decrease in PIP3 in turn, impairs insulin signaling because it is essential for the activation of Akt in response to the IR activation by insulin [160,161]. Second, ceramide recruits both PKCζ and Akt to the caveolae which will have synergetic repressive effect on insulin signaling. Briefly, recruitment of both PKCζ and Akt by ceramide leads to the collective presence of ceramide, PKCζ and Akt in the caveolae [153,161] (Figure 4). By doing so, PKCζ would not only be exposed to ceramide rendering it active, but at the same time would be co-localized with Akt, there by suppressing its activity. Both of these mechanisms together create a highly repressive environment for Akt, and hence, diminish insulin mediated signaling response.
Ceramide and insulin synthesis
Thus far, we have mainly considered the role for ceramide in inducing β-cell apoptosis and insulin resistance. However, several studies reveal that ceramide also diminishes insulin synthesis by decreasing the insulin mRNA levels in islets. In pancreatic β cells, saturated FA has been shown to impair insulin gene expression with associated increase in ceramide [162,163]. This effect was largely prevented by inhibitors of de novo ceramide synthesis suggesting the role for ceramide [163]. Furthermore, addition of exogenous ceramide or elevation of endogenous ceramide, using ceramidase inhibitor, has been demonstrated to reduce insulin mRNA levels [163,164]. Two postulations have been made to explain the mechanisms of inhibition of insulin gene transcription by ceramide. First, ceramide activates JNK which inhibits insulin gene transcription both via c-jun-dependent [165], and oxidative stress-dependent [166] pathways. Second, ceramide directly activates PKCζ [167] which phosphorylates and inactivates Pancreatic and duodenal homeobox gene-1 (PDX-1), a transcription factor which regulates insulin gene expression [168]. Apart from down-regulating insulin gene transcription, ceramide has also been shown to decrease transcription of the Glut-4 gene [169] (Figure 1). In conclusion, these results suggest that ceramide modulates signaling pathways implicated in the transcriptional regulation of the insulin gene. However, further investigation is required to understand the exact mechanism of inhibition of insulin gene expression by ceramide.
Ceramide in diabetic complications
The major threats of diabetes are its serious, sometimes life-threatening complications. A growing body of evidence has identified the role of sphingolipids, particularly ceramide, in the pathogenesis of both microvascular and macrovascular diabetic complications. Cardiovascular diseases such as atherosclerosis, myocardial infarction and stroke, are major cause of mortality in diabetic patients. Diabetic cardiomyopathy is characterized by the apoptosis of cardiomyocytes [170]. Increased myocardial ceramide content was observed in various rodent models of lipotoxic cardiomyopathy [171,172]. De novo ceramide biosynthesis, when inhibited either pharmacologically by myriocin, or genetically by heterozygous deletion of SPT subunit, an improvement in the cardiac function was observed [171,172]. Apart from this, Gorska et al. demonstrated an elevated acid SMase level in plasma of type 2 diabetic patients [173]. Ceramide has also been implicated in atherosclerosis in both human subjects and in animal models [174,175], and regression of atherosclerotic plaques was observed when treated with myriocin [176]. Conclusively, these studies suggest that ceramide generated via de novo pathway and SM hydrolysis may be involved in the development of diabetic cardiovascular complications.
Nephropathy represents another major threat in diabetic patients which is characterized by the apoptosis of renal mesangial cells. Several studies reported the role of ceramide in mesangial cell apoptosis. Increased expression of SPT was seen in renal tubular epithelial and microvascular endothelial cells, which are the main sites of apoptosis observed in diabetic patients [177]. When ceramide generation was inhibited using SPT inhibitors and ceramide synthase inhibitors, a reduction in tubular epithelial cell death was observed [178,179]. Another important complication associated with diabetes is development of neuropathy. Except for the gangliosides, role of sphingolipids in diabetic neuropathy is not extensively studied. When mouse Schwann cells were cultured with palmitate, an enhancement in apoptosis was observed. This effect was significantly suppressed by myriocin and fumonisin B1, suggesting a role for ceramide [180]. Another important complication of diabetes is retinopathy, the second leading cause of blindness in the developed countries [181]. When cultured retinal pericytes were incubated with palmitate, an increase in cellular ceramide content with subsequent increase in apoptosis was observed [182]. This effect was reversed by overexpression of ceramidase [182]. In another study where ceramide accumulation was induced by using Advanced Glycation End products, Fumonisin B1 did not reverse the retinal pericyte apoptosis, suggesting that de novo biosynthesis is not involved in the ceramide generation [183]. Here, desipramine, an acid SMase inhibitor, almost completely abolished ceramide generation and apoptosis, demonstrating that ceramide is formed from SM hydrolysis [183].
These observations underline the potential involvement of ceramide in the pathogenesis of diabetic complications. Moreover, some of the studies also demonstrated the role of other sphingolipids such as GSL in diabetic nephropathy [184] and retinopathy [185]. However, whether it is ceramide or GSL that play a more important role in these diabetic complications is not known. Further research employing more advanced lipidomic screening will definitely bring the answer to this question.
Conclusion
There has been considerable progress over the last few years in unraveling the role and mechanism of ceramide action in diabetes and its complications. Taken together, evidence from a plethora of studies indicates that ceramide plays a significant role in diabetes by at least three different mechanisms: inducing pancreatic β-cell apoptosis, increasing insulin resistance, and reducing insulin gene expression. Translated to the clinical level, all of these ceramide mediated pathways remain candidates for their putative contributions to the pathogenesis of diabetes. However, there are still several unanswered questions. First, it is not known what enables ceramide to mediate so many diverse signaling events in the cell. Though different species of ceramide exist, the role and critical importance of each species in the pathophysiology of diabetes is not as yet completely known. May be this species diversity is responsible for the multiple cellular events mediated by ceramide. Secondly, Ceramide generated in the ER can be transported to different organelles, and moreover, many organelles contain the machinery that enables them to synthesize ceramide. Hence, the quantitative and qualitative aspects of ceramide in different sub-cellular compartments must be extensively studied. Third, sphingolipids have highly interconnected metabolic networks, so alteration of ceramide might have deleterious impact on other sphingolipid metabolites. The impact of such modifications on the normal functioning of cells must be studied. Finally, the factors that regulate sphingolipid biosynthetic enzymes to generate ceramide during diabetes induction have to be investigated extensively. Further elucidation of these molecular details will be essential to develop better understanding of the validity of ceramide modulation as a strategy for treating diabetes. Advances in analytical lipidomics such as tandem mass spectrometry and lipid imaging could provide more crucial information regarding the role of ceramide in the etiology and pathogenesis of diabetes in the coming years. Such an understanding undoubtedly will have a direct impact on future therapies for diabetes.
Abbreviations
Apaf-1: Apoptotic protease activating factor 1; Bcl-2: B-cell lymphoma-2; C1P: Ceramide-1-phosphate; CAP: Cbl-associated protein; CKI: Cyclin-dependent kinase inhibitors; DAG: Diacylglycerol; dh-Cer: Dihydroceramide; eIF4EBP: elF4E binding protein; ER: Endoplasmic reticulum; F16BP: Fructose-1,6-biphosphatase; FA: Fatty acid; FFA: Free fatty acid; FOXO1: Forkhead box O1; G6P: Glucose-6-phosphatase; GCS: Glucosylceramide synthase; Glut4: Glucose transporter-4; GSK3: Glycogen synthase kinase-3; GSL: Glycosphingolipid; IFN-γ: Interferon-gamma; IL-1β: Interleukin-1 beta; iNOS: inducible nitric oxide synthase; iPLA2β: Ca2 + -independent phospholipase A2 beta; IR: Insulin receptor; IRS: Insulin receptor substrate; JNK: c-Jun N-terminal kinase; MAPK: Mitogen-activated protein kinase; mTOR: mammalian target of rapamycin; NADPH: Nicotinamide adenine dinucleotide phosphate; NFκB: Nuclear factor kappa B; NO: Nitric oxide; p70S6K: p70 ribosomal S6 kinase; PDK1: Phosphoinositide dependant protein kinase-1; PDX-1: Pancreatic and duodenal homeobox gene-1; PEPCK: Phosphoenolpyruvate carboxykinase; PH: Pleckstrin homology; PI3K: Phosphatidylinositol-3 kinase; PIP2: Phosphatidylinositol (4,5)-bisphosphate; PIP3: Phosphatidylinositol (3,4,5)-triphosphate; PKB: Protein kinase B; PKCζ: Protein kinase C zeta; PPAR: Peroxisome proliferator activated receptor; PTEN: Phosphatase and tensin homolog deleted on chromosome 10; RNS: Reactive nitrogen species; ROS: Reactive oxygen species; S1P: Sphingosine-1-phosphate; SM: Sphingomyelin; SMase: Sphingomyelinase; SPT: Serine palmitoyl transferase; SREBP-1c: Sterol regulatory binding protein 1C; TLR4: Toll-like receptor 4; TNF-α: Tumor necrosis factor-alpha; TZD: Thiazolidinediones; UPR: Unfolded protein response; ZDF: Zucker diabetic fatty.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SG and AR drafted the first version of the manuscript. SP, AG and FT participated in researching the literature and critical discussion of the work. All authors read and approved the final manuscript.
Contributor Information
Sehamuddin Galadari, Email: sehamuddin@uaeu.ac.ae.
Anees Rahman, Email: anees@uaeu.ac.ae.
Siraj Pallichankandy, Email: sirajpk@uaeu.ac.ae.
Alaa Galadari, Email: alaa.galadari@gmail.com.
Faisal Thayyullathil, Email: t.faisal@uaeu.ac.ae.
Acknowledgements
This work was supported by grant from The Sheikh Hamdan Award for Medical Sciences.
References
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- World health organization. Definition and diagnosis of diabetes mellitus and intermediate hyperglycemia: report of a WHO/IDF consultation. Geneva: WHO press; 2006. [Google Scholar]
- Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res. 2009;50:S91–S96. doi: 10.1194/jlr.R800080-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanian J, Ohanian V. Sphingolipids in mammalian cell signaling. Cell Mol Life Sci. 2001;58:2053–2068. doi: 10.1007/PL00000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhakkayil J, Thayyullathil F, Chathoth S, Hago A, Patel M, Galadari S. Glutathione regulates caspase-dependent ceramide production and curcumin-induced apoptosis in human leukemic cells. Free Radic Biol Med. 2012;52:1854–1864. doi: 10.1016/j.freeradbiomed.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Alewijnse AE, Peters SL. Sphingolipid signaling in the cardiovascular system: good, bad or both? Eur J Pharmacol. 2008;585:292–302. doi: 10.1016/j.ejphar.2008.02.089. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- Keller SR, Lienhard GE. Insulin signalling: the role of insulin receptor substrate 1. Trends Cell Biol. 1994;4:115–119. doi: 10.1016/0962-8924(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Langeveld M, Aerts JM. Glycosphingolipids and insulin resistance. Prog Lipid Res. 2009;48:196–205. doi: 10.1016/j.plipres.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Che. 1999;274:24202–24210. doi: 10.1074/jbc.274.34.24202. [DOI] [PubMed] [Google Scholar]
- Hajduch E, Balendran A, Batty IH, Litherland GJ, Blair AS, Downes CP, Hundal HS. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia. 2001;44:173–183. doi: 10.1007/s001250051596. [DOI] [PubMed] [Google Scholar]
- Merrill AH Jr. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem. 2002;277:25843–25846. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010–1018. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Wennekes T, Van den Berg RJ, Boot RG, Van der Marel GA, Overkleeft HS, Aerts JM. Glycosphingolipids–nature, function, and pharmacological modulation. Angew Chem Int Ed Engl. 2009;48:8848–8869. doi: 10.1002/anie.200902620. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta. 2006;1758:2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- Mathis D, Vence L, Benoist C. Beta-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- Chandra J, Zhivotovsky B, Zaitsev V, Juntti-Berggren L, Berggren PO, Orrenius S. Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes. 2001;50:S44–S47. doi: 10.2337/diabetes.50.2007.S44. [DOI] [PubMed] [Google Scholar]
- Kim KA, Lee MS. Recent progress in research on beta-cell apoptosis by cytokines. Front Biosci. 2009;14:657–664. doi: 10.2741/3271. [DOI] [PubMed] [Google Scholar]
- Lang F, Ullrich S, Gulbins E. Ceramide formation as a target in beta-cell survival and function. Expert Opin Ther Targets. 2011;15:1061–1071. doi: 10.1517/14728222.2011.588209. [DOI] [PubMed] [Google Scholar]
- Ishizuka N, Yagui K, Tokuyama Y, Yamada K, Suzuki Y, Miyazaki J, Hashimoto N, Makino H, Saito Y, Kanatsuka A. Tumor necrosis factor alpha signaling pathway and apoptosis in pancreatic beta cells. Metabolism. 1999;48:1485–1492. doi: 10.1016/S0026-0495(99)90234-2. [DOI] [PubMed] [Google Scholar]
- Sjoholm A. Ceramide inhibits pancreatic beta-cell insulin production and mitogenesis and mimics the actions of interleukin-1 beta. FEBS Lett. 1995;367:283–286. doi: 10.1016/0014-5793(95)00470-T. [DOI] [PubMed] [Google Scholar]
- Welsh N. Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J Biol Chem. 1996;271:8307–8312. doi: 10.1074/jbc.271.14.8307. [DOI] [PubMed] [Google Scholar]
- Major CD, Gao ZY, Wolf BA. Activation of the sphingomyelinase/ceramide signal transduction pathway in insulin-secreting beta-cells: role in cytokine-induced beta-cell death. Diabetes. 1999;48:1372–1380. doi: 10.2337/diabetes.48.7.1372. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Shan X, Miao H, Lu Y, Xu J, You N, Liu C, Liao DF, Jin J. Acute activation of acid ceramidase affects cytokine-induced cytotoxicity in rat islet beta-cells. FEBS Lett. 2009;583:2136–2141. doi: 10.1016/j.febslet.2009.05.047. [DOI] [PubMed] [Google Scholar]
- Mastrandrea LD, Sessanna SM, Laychock SG. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: response to cytokines. Diabetes. 2005;54:1429–1436. doi: 10.2337/diabetes.54.5.1429. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 1998;95:2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes. 2001;50:69–76. doi: 10.2337/diabetes.50.1.69. [DOI] [PubMed] [Google Scholar]
- Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patané G, Boggi U, Piro S, Anello M, Bergamini E, Mosca F, Di Mario U, Del Prato S, Marchetti P. Prolonged exposure to free fatty acids has cytostatic and proapoptotic effects on human pancreatic islets: evidence that beta cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52:726–733. doi: 10.2337/diabetes.52.3.726. [DOI] [PubMed] [Google Scholar]
- de Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, Van der Vusse GJ, Van Bilsen M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384–1394. [PubMed] [Google Scholar]
- Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- Lupi R, Del Guerra S, Fierabracci V, Marselli L, Novelli M, Patanè G, Boggi U, Mosca F, Piro S, Del Prato S, Marchetti P. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes. 2002;51:S134–S137. doi: 10.2337/diabetes.51.2007.S134. [DOI] [PubMed] [Google Scholar]
- Boslem E, Meikle PJ, Biden TJ. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets. 2012;4:177–187. doi: 10.4161/isl.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M, Laybutt DR, Meikle PJ, Biden TJ. A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J. 2011;435:267–276. doi: 10.1042/BJ20101867. [DOI] [PubMed] [Google Scholar]
- Veret J, Coant N, Berdyshev EV, Skobeleva A, Therville N, Bailbé D, Gorshkova I, Natarajan V, Portha B, Le Stunff H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem J. 2011;438:177–189. doi: 10.1042/BJ20101386. [DOI] [PubMed] [Google Scholar]
- Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009;14:1389–1404. doi: 10.1007/s10495-009-0339-5. [DOI] [PubMed] [Google Scholar]
- Liadis N, Salmena L, Kwan E, Tajmir P, Schroer SA, Radziszewska A, Li X, Sheu L, Eweida M, Xu S, Gaisano HY, Hakem R, Woo M. Distinct in vivo roles of caspase-8 in beta-cells in physiological and diabetes models. Diabetes. 2007;56:2302–2311. doi: 10.2337/db06-1771. [DOI] [PubMed] [Google Scholar]
- Liadis N, Murakami K, Eweida M, Elford AR, Sheu L, Gaisano HY, Hakem R, Ohashi PS, Woo M. Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005;25:3620–3629. doi: 10.1128/MCB.25.9.3620-3629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Novgorodov SA, Szulc ZM, Luberto C, Jones JA, Bielawski J, Bielawska A, Hannun YA, Obeid LM. Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J Biol Chem. 2005;280:16096–16105. doi: 10.1074/jbc.M411707200. [DOI] [PubMed] [Google Scholar]
- Birbes H, El Bawab S, Hannun YA, Obeid LM. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J. 2001;15:2669–2679. doi: 10.1096/fj.01-0539com. [DOI] [PubMed] [Google Scholar]
- Birbes H, Luberto C, Hsu YT, El Bawab S, Hannun YA, Obeid LM. A mitochondrial pool of sphingomyelin is involved in TNFalpha-induced Bax translocation to mitochondria. Biochem J. 2005;386:445–451. doi: 10.1042/BJ20041627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkar H, Wiegmann V, Yazdanpanah B, Haubert D, Kronke M. Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J Biol Chem. 2005;280:20804–20813. doi: 10.1074/jbc.M410869200. [DOI] [PubMed] [Google Scholar]
- Birbes H, El Bawab S, Obeid LM, Hannun YA. Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv Enzyme Regul. 2002;42:113–129. doi: 10.1016/s0065-2571(01)00026-7. [DOI] [PubMed] [Google Scholar]
- Allison J, Thomas H, Beck D, Brady JL, Lew AM, Elefanty A, Kosaka H, Kay TW, Huang DC, Strasser A. Transgenic overexpression of human Bcl-2 in islet beta cells inhibits apoptosis but does not prevent autoimmune destruction. Int Immunol. 2000;12:9–17. doi: 10.1093/intimm/12.1.9. [DOI] [PubMed] [Google Scholar]
- Von Haefen C, Wieder T, Gillissen B, Starck L, Graupner V, Dorken B, Daniel PT. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–4019. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- Maestre I, Jordan J, Calvo S, Reig JA, Cena V, Soria B, Prentki M, Roche E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology. 2003;144:335–345. doi: 10.1210/en.2001-211282. [DOI] [PubMed] [Google Scholar]
- Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6:118–125. doi: 10.1016/j.mito.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Valverde AM, Rohn JL, Benito M, Lorenzo M. Akt mediates insulin rescue from apoptosis in brown adipocytes: effect of ceramide. Growth Horm IGF Res. 2000;10:256–266. doi: 10.1054/ghir.2000.0165. [DOI] [PubMed] [Google Scholar]
- Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci. 2011;32:82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang AY, Teggatz EG, Zou AP, Campbell WB, Li PL. Endostatin uncouples NO and Ca2+ response to bradykinin through enhanced O2*- production in the intact coronary endothelium. Am J Physiol Heart Circ Physiol. 2005;288:H686–H694. doi: 10.1152/ajpheart.00174.2004. [DOI] [PubMed] [Google Scholar]
- García-Ruiz C, Colell A, Marí M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Fujii J, Fukutomi T, Watanabe T, Che W, Sanada Y, Igarashi Y, Taniguchi N. Reactive oxygen species enhances the induction of inducible nitric oxide synthase by sphingomyelinase in RAW264.7 cells. Biochim Biophys Acta. 1998;1393:203–210. doi: 10.1016/S0005-2760(98)00066-6. [DOI] [PubMed] [Google Scholar]
- Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc Natl Acad Sci U S A. 1998;95:2956–2960. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilane CM, LaBelle EF. NO induced apoptosis of vascular smooth muscle cells accompanied by ceramide increase. J Cell Physiol. 2004;199:310–315. doi: 10.1002/jcp.10464. [DOI] [PubMed] [Google Scholar]
- Franzen R, Fabbro D, Aschrafi A, Pfeilschifter J, Huwiler A. Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J Biol Chem. 2002;277:46184–46190. doi: 10.1074/jbc.M204034200. [DOI] [PubMed] [Google Scholar]
- Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry. 2000;39:6660–6668. doi: 10.1021/bi9924415. [DOI] [PubMed] [Google Scholar]
- Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- Reinehr R, Becker S, Eberle A, Grether-Beck S, Haussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem. 2005;280:27179–27194. doi: 10.1074/jbc.M414361200. [DOI] [PubMed] [Google Scholar]
- Morgan D, Rebelato E, Abdulkader F, Graciano MF, Oliveira-Emilio HR, Hirata AE, Rocha MS, Bordin S, Curi R, Carpinelli AR. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology. 2009;150:2197–2201. doi: 10.1210/en.2008-1149. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Wang MY, Zhou YT, Newgard CB, Unger RH. Protection against lipoapoptosis of beta cells through leptin-dependent maintenance of Bcl-2 expression. Proc Natl Acad Sci U S A. 1998;95:9558–9561. doi: 10.1073/pnas.95.16.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. 2002;1585:202–212. doi: 10.1016/S1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- Won JS, Im YB, Khan M, Singh AK, Singh I. The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem. 2004;88:583–593. doi: 10.1046/j.1471-4159.2003.02165.x. [DOI] [PubMed] [Google Scholar]
- Leibowitz G, Bachar E, Shaked M, Sinai A, Ketzinel-Gilad M, Cerasi E, Kaiser N. Glucose regulation of β-cell stress in type 2 diabetes. Diabetes Obes Metab. 2010;12:66–75. doi: 10.1111/j.1463-1326.2010.01280.x. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- Fonseca SG, Burcin M, Gromada J, Urano F. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr Opin Pharmacol. 2009;9:763–770. doi: 10.1016/j.coph.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/A:1016175429877. [DOI] [PubMed] [Google Scholar]
- Socha L, Silva D, Lesage S, Goodnow C, Petrovsky N. The role of endoplasmic reticulum stress in nonimmune diabetes: NOD.k iHEL, a novel model of beta cell death. Ann N Y Acad Sci. 2003;1005:178–183. doi: 10.1196/annals.1288.022. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lai E, Bikopoulos G, Wheeler MB, Rozakis-Adcock M, Volchuk A. Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2008;294:E540–E550. doi: 10.1152/ajpendo.00478.2007. [DOI] [PubMed] [Google Scholar]
- Lei X, Barbour SE, Ramanadham S. Group VIA Ca2 + -independent phospholipase A2 (iPLA2beta) and its role in beta-cell programmed cell death. Biochimie. 2010;92:627–637. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer J, Bulleid N, Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/S0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Kellerer M, Mushack J, Seffer E, Mischak H, Ullrich A, Haring HU. Protein kinase C isoforms alpha, delta and theta require insulin receptor substrate-1 to inhibit the tyrosine kinase activity of the insulin receptor in human kidney embryonic cells (HEK 293 cells) Diabetologia. 1998;41:833–838. doi: 10.1007/s001250050995. [DOI] [PubMed] [Google Scholar]
- Pickersgill L, Litherland GJ, Greenberg AS, Walker M, Yeaman SJ. Key role for ceramides in mediating insulin resistance in human muscle cells. J Biol Chem. 2007;282:12583–12589. doi: 10.1074/jbc.M611157200. [DOI] [PubMed] [Google Scholar]
- Sabin MA, Stewart CE, Crowne EC, Turner SJ, Hunt LP, Welsh GI, Grohmann MJ, Holly JM, Shield JP. Fatty acid-induced defects in insulin signalling, in myotubes derived from children, are related to ceramide production from palmitate rather than the accumulation of intramyocellular lipid. J Cell Physiol. 2007;211:244–252. doi: 10.1002/jcp.20922. [DOI] [PubMed] [Google Scholar]
- Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, Loviscach M, Stumvoll M, Claussen CD, Schick F, Haring HU, Jacob S. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. 2001;50:2579–2584. doi: 10.2337/diabetes.50.11.2579. [DOI] [PubMed] [Google Scholar]
- Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280:20148–20153. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. 2004;382:619–629. doi: 10.1042/BJ20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing’s syndrome. Diabetes. 2005;54:591–602. doi: 10.2337/diabetes.54.3.591. [DOI] [PubMed] [Google Scholar]
- Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Bio. 1998;18:5457–5464. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CN, O’Brien L, Brindley DN. Effects of cell-permeable ceramides and tumor necrosis factor-alpha on insulin signaling and glucose uptake in 3T3-L1 adipocytes. Diabetes. 1998;47:24–31. doi: 10.2337/diabetes.47.1.24. [DOI] [PubMed] [Google Scholar]
- Van Epps-Fung M, Williford J, Wells A, Hardy RW. Fatty acid-induced insulin resistance in adipocytes. Endocrinology. 1997;138:4338–4345. doi: 10.1210/en.138.10.4338. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2000;279:H2124–H2132. doi: 10.1152/ajpheart.2000.279.5.H2124. [DOI] [PubMed] [Google Scholar]
- Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem J. 2009;417:791–801. doi: 10.1042/BJ20081149. [DOI] [PubMed] [Google Scholar]
- Hu W, Ross J, Geng T, Brice SE, Cowart LA. Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids: implications for insulin resistance. J Biol Chem. 2011;286:16596–16605. doi: 10.1074/jbc.M110.186916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J, Wang CN, O’Brien L, Brindley DN. Cell-permeable ceramides increase basal glucose incorporation into triacylglycerols but decrease the stimulation by insulin in 3T3-L1 adipocytes. Int J Obes Relat Metab Disord. 2003;27:31–39. doi: 10.1038/sj.ijo.0802183. [DOI] [PubMed] [Google Scholar]
- Frangioudakis G, Garrard J, Raddatz K, Nadler JL, Mitchell TW, Schmitz-Peiffer C. Saturated- and n-6 polyunsaturated-fat diets each induce ceramide accumulation in mouse skeletal muscle: reversal and improvement of glucose tolerance by lipid metabolism inhibitors. Endocrinology. 2010;151:4187–4196. doi: 10.1210/en.2010-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2009;297:E211–E224. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen WE, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53:1215–1221. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- Dain A, Repossi G, Das UN, Eynard AR. Role of PUFAs, the precursors of endocannabinoids, in human obesity and type 2 diabetes. Front Biosci (Elite Ed) 2010;2:1432–1447. doi: 10.2741/e203. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. The endocannabinoid system in obesity and type 2 diabetes. Diabetologia. 2008;51:1356–1367. doi: 10.1007/s00125-008-1048-2. [DOI] [PubMed] [Google Scholar]
- Comba A, Lin YH, Eynard AR, Valentich MA, Fernandez-Zapico ME, Pasqualini ME. Basic aspects of tumor cell fatty acid-regulated signaling and transcription factors. Cancer Metastasis Rev. 2011;30:325–342. doi: 10.1007/s10555-011-9308-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo P, Wang MH. Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011;95:1–4. doi: 10.1016/j.prostaglandins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 2012;22:557–566. doi: 10.1016/j.tcb.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Solinas G, Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24:2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- Quintans J, Kilkus J, McShan CL, Gottschalk AR, Dawson G. Ceramide mediates the apoptotic response of WEHI 231 cells to anti-immunoglobulin, corticosteroids and irradiation. Biochem Biophys Res Commun. 1994;202:710–714. doi: 10.1006/bbrc.1994.1988. [DOI] [PubMed] [Google Scholar]
- Linn SC, Kim HS, Keane EM, Andras LM, Wang E, Merrill AH Jr. Regulation of de novo sphingolipid biosynthesis and the toxic consequences of its disruption. Biochem Soc Trans. 2001;29:831–835. doi: 10.1042/BST0290831. [DOI] [PubMed] [Google Scholar]
- Lepine S, Lakatos B, Maziere P, Courageot MP, Sulpice JC, Giraud F. Involvement of sphingosine in dexamethasone-induced thymocyte apoptosis. Ann N Y Acad Sci. 2002;973:190–193. doi: 10.1111/j.1749-6632.2002.tb04631.x. [DOI] [PubMed] [Google Scholar]
- Baranowski M, Blachnio A, Zabielski P, Gorski J. Pioglitazone induces de novo ceramide synthesis in the rat heart. Prostaglandins Lipid Mediat. 2007;83:99–111. doi: 10.1016/j.prostaglandins.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev. 2002;18:S10–S15. doi: 10.1002/dmrr.249. [DOI] [PubMed] [Google Scholar]
- Zendzian-Piotrowska M, Baranowski M, Zabielski P, Gorski J. Effects of pioglitazone and high-fat diet on ceramide metabolism in rat skeletal muscles. J Physiol Pharmacol. 2006;57:101–114. [PubMed] [Google Scholar]
- Planavila A, Alegret M, Sanchez RM, Rodriguez-Calvo R, Laguna JC, Vazquez-Carrera M. Increased Akt protein expression is associated with decreased ceramide content in skeletal muscle of troglitazone-treated mice. Biochem Pharmacol. 2005;69:1195–1204. doi: 10.1016/j.bcp.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lessard SJ, Lo Giudice SL, Lau W, Reid JJ, Turner N, Febbraio MA, Hawley JA, Watt MJ. Rosiglitazone enhances glucose tolerance by mechanisms other than reduction of fatty acid accumulation within skeletal muscle. Endocrinology. 2004;145:5665–5670. doi: 10.1210/en.2004-0659. [DOI] [PubMed] [Google Scholar]
- Wolf G. Serum retinol-binding protein: a link between obesity, insulin resistance, and type 2 diabetes. Nutr Rev. 2007;65:251–256. doi: 10.1111/j.1753-4887.2007.tb00302.x. [DOI] [PubMed] [Google Scholar]
- Thomas DE, Elliott EJ, Naughton GA. Exercise for type 2 diabetes mellitus. Cochrane Database Syst Rev. 2006;3 doi: 10.1002/14651858.CD002968.pub2. CD002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzyn A, Zendzian-Piotrowska M, Gorski J. Effect of endurance training on the sphingomyelin-signalling pathway activity in the skeletal muscles of the rat. J Physiol Pharmacol. 2004;55:305–313. [PubMed] [Google Scholar]
- Smith AC, Mullen KL, Junkin KA, Nickerson J, Chabowski A, Bonen A, Dyck DJ. Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia. Am J Physiol Endocrinol Metab. 2007;293:E172–E181. doi: 10.1152/ajpendo.00677.2006. [DOI] [PubMed] [Google Scholar]
- Dube JJ, Amati F, Stefanovic-Racic M, Toledo FG, Sauers SE, Goodpaster BH. Exercise-induced alterations in intramyocellular lipids and insulin resistance: the athlete’s paradox revisited. Am J Physiol Endocrinol Metab. 2008;294:E882–E888. doi: 10.1152/ajpendo.00769.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- Lessard SJ, Rivas DA, Chen ZP, Bonen A, Febbraio MA, Reeder DW, Kemp BE, Yaspelkis BB 3rd, Hawley JA. Tissue-specific effects of rosiglitazone and exercise in the treatment of lipid-induced insulin resistance. Diabetes. 2007;56:1856–1864. doi: 10.2337/db06-1065. [DOI] [PubMed] [Google Scholar]
- Helge JW, Dobrzyn A, Saltin B, Gorski J. Exercise and training effects on ceramide metabolism in human skeletal muscle. Exp Physiol. 2004;89:119–127. doi: 10.1113/expphysiol.2003.002605. [DOI] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25:4–7. doi: 10.1016/j.it.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Moon YS, Kim DH, Song DK. Serum tumor necrosis factor-alpha levels and components of the metabolic syndrome in obese adolescents. Metabolism. 2004;53:863–867. doi: 10.1016/j.metabol.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Wu D, Ren Z, Pae M, Guo W, Cui X, Merrill AH, Meydani SN. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J Immunol. 2007;179:4829–4839. doi: 10.4049/jimmunol.179.7.4829. [DOI] [PubMed] [Google Scholar]
- Kanety H, Hemi R, Papa MZ, Karasik A. Sphingomyelinase and ceramide suppress insulin-induced tyrosine phosphorylation of the insulin receptor substrate-1. J Biol Chem. 1996;271:9895–9897. doi: 10.1074/jbc.271.17.9895. [DOI] [PubMed] [Google Scholar]
- Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibitstheir binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997;272:29911–29918. doi: 10.1074/jbc.272.47.29911. [DOI] [PubMed] [Google Scholar]
- Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, Bergmann A, Tzivion G, Rana A. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527–1533. doi: 10.1016/S1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Kim BC, Xu Z, Kim SJ. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-beta-induced apoptosis in hepatoma cells. J Biol Chem. 2004;279:29478–29484. doi: 10.1074/jbc.M313947200. [DOI] [PubMed] [Google Scholar]
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Gual P, Le Y. Marchand-Brustel, J.F. Tanti, Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet. 2000;24:291–295. doi: 10.1038/73523. [DOI] [PubMed] [Google Scholar]
- Salinas M, Lopez-Valdaliso R, Martin D, Alvarez A, Cuadrado A. Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells. Mol Cell Neurosci. 2000;15:156–169. doi: 10.1006/mcne.1999.0813. [DOI] [PubMed] [Google Scholar]
- Schubert KM, Scheid MP, Duronio V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem. 2000;275:13330–13335. doi: 10.1074/jbc.275.18.13330. [DOI] [PubMed] [Google Scholar]
- Stratford S, DeWald DB, Summers SA. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem J. 2001;354:359–368. doi: 10.1042/0264-6021:3540359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel W, Giaccia A. Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev. 1998;12:1941–1946. doi: 10.1101/gad.12.13.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- Zinda MJ, Vlahos CJ, Lai MT. Ceramide induces the dephosphorylation and inhibition of constitutively activated Akt in PTEN negative U87MG cells. Biochem Biophys Res Commun. 2001;280:1107–1115. doi: 10.1006/bbrc.2000.4248. [DOI] [PubMed] [Google Scholar]
- Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem. 2002;277:3286–3292. doi: 10.1074/jbc.M110541200. [DOI] [PubMed] [Google Scholar]
- Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduch E, Turban S, Le Liepvre X, Le Lay S, Lipina C, Dimopoulos N, Dugail I, Hundal HS. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem J. 2008;410:369–379. doi: 10.1042/BJ20070936. [DOI] [PubMed] [Google Scholar]
- George KS, Wu S. Lipid raft: a floating island of death or survival. Toxicol Appl Pharmacol. 2012;259:311–319. doi: 10.1016/j.taap.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li X, Becker KA, Gulbins E. Ceramide-enriched membrane domains–structure and function. Biochim Biophys Acta. 2009;1788:178–183. doi: 10.1016/j.bbamem.2008.07.030. [DOI] [PubMed] [Google Scholar]
- Grassme H, Riethmuller J, Gulbins E. Biological aspects of ceramide-enriched membrane domains. Prog Lipid Res. 2007;46:161–170. doi: 10.1016/j.plipres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, Gulbins E, Kolesnick R. Ceramide enables fas to cap and kill. J Biol Chem. 2001;276:23954–23961. doi: 10.1074/jbc.M101866200. [DOI] [PubMed] [Google Scholar]
- Hueber AO, Bernard AM, Herincs Z, Couzinet A, He HT. An essential role for membrane rafts in the initiation of Fas/CD95-triggered cell death in mouse thymocytes. EMBO Rep. 2002;3:190–196. doi: 10.1093/embo-reports/kvf022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stralfors P. Caveolins and caveolae, roles in insulin signalling and diabetes. Adv Exp Med Biol. 2012;729:111–126. doi: 10.1007/978-1-4614-1222-9_8. [DOI] [PubMed] [Google Scholar]
- Goswami R, Singh D, Phillips G, Kilkus J, Dawson G. Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J Neurosci Res. 2005;81:541–550. doi: 10.1002/jnr.20550. [DOI] [PubMed] [Google Scholar]
- Lipina C, Hundal HS. Sphingolipids: agents provocateurs in the pathogenesis of insulin resistance. Diabetologia. 2011;54:1596–1607. doi: 10.1007/s00125-011-2127-3. [DOI] [PubMed] [Google Scholar]
- Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–30021. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- Guo J, Qian Y, Xi X, Hu X, Zhu J, Han X. Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol Cell Biochem. 2010;338:283–290. doi: 10.1007/s11010-009-0362-4. [DOI] [PubMed] [Google Scholar]
- Henderson E, Stein R. c-jun inhibits transcriptional activation by the insulin enhancer, and the insulin control element is the target of control. Mol Cell Biol. 1994;14:655–662. doi: 10.1128/mcb.14.1.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem. 2002;277:30010–30018. doi: 10.1074/jbc.M202066200. [DOI] [PubMed] [Google Scholar]
- Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- Furukawa N, Shirotani T, Araki E, Kaneko K, Todaka M, Matsumoto K, Tsuruzoe K, Motoshima H, Yoshizato K, Kishikawa H, Shichiri M. Possible involvement of atypical protein kinase C (PKC) in glucose-sensitive expression of the human insulingene: DNA binding activity and transcriptional activity of pancreatic and duodenal homeobox gene-1 (PDX-1) areenhanced via calphostin C-sensitive but phorbol 12-myristate 13-acetate (PMA) and Go 6976-insensitive pathway. Endocr J. 1999;46:43–58. doi: 10.1507/endocrj.46.43. [DOI] [PubMed] [Google Scholar]
- Long SD, Pekala PH. Lipid mediators of insulin resistance: ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem J. 1996;319:179–184. doi: 10.1042/bj3190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol. 2009;297:H2096–H2108. doi: 10.1152/ajpheart.00452.2009. [DOI] [PubMed] [Google Scholar]
- Gorska M, Baranczuk E, Dobrzyn A. Secretory Zn2 + -dependent sphingomyelinase activity in the serum of patients with type 2 diabetes is elevated. Horm Metab Res. 2003;35:506–507. doi: 10.1055/s-2003-41810. [DOI] [PubMed] [Google Scholar]
- Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. 1996;98:1455–1464. doi: 10.1172/JCI118934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin CM, Leventhal AR, Kuriakose G, Schuchman EH, Williams KJ, Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28:1723–1730. doi: 10.1161/ATVBAHA.108.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS, Robertson AW, Kindt EK, Homan R, Karathanasis SK, Rekhter MD. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2004;110:3465–3471. doi: 10.1161/01.CIR.0000148370.60535.22. [DOI] [PubMed] [Google Scholar]
- Liu G, Han F, Yang Y, Xie Y, Jiang H, Mao Y, Wang H, Wang M, Chen R, Yang J, Chen J. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effectsof rapamycin. Nephrol Dial Transplant. 2011;26:1493–1502. doi: 10.1093/ndt/gfq633. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Yano T, Sendo T, Sueyasu M, Hirano K, Kanaide H, Oishi R. Involvement of de novo ceramide synthesis in radiocontrast-induced renal tubular cell injury. Kidney Int. 2006;69:288–297. doi: 10.1038/sj.ki.5000057. [DOI] [PubMed] [Google Scholar]
- Basnakian AG, Ueda N, Hong X, Galitovsky VE, Yin X, Shah SV. Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am J Physiol Renal Physiol. 2005;288:F308–F314. doi: 10.1152/ajprenal.00204.2004. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Akahane K, Nakamura J, Naruse K, Kamiya H, Himeno T, Nakamura N, Shibata T, Kondo M, Nagasaki H, Fujiya A, Oiso Y, Hamada Y. Palmitate induces apoptosis in Schwann cells via both ceramide-dependent and independent pathways. Neuroscience. 2011;176:188–198. doi: 10.1016/j.neuroscience.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, Klein R. Diabetic retinopathy. Diabetes Care. 2003;26:226–229. doi: 10.2337/diacare.26.1.226. [DOI] [PubMed] [Google Scholar]
- Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes. 2005;54:1838–1845. doi: 10.2337/diabetes.54.6.1838. [DOI] [PubMed] [Google Scholar]
- Denis U, Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M. Advanced glycation endproducts induce apoptosis of bovine retinal pericytes in culture: involvement ofdiacylglycerol/ceramide production and oxidative stress induction. Free Radic Biol Med. 2002;33:236–247. doi: 10.1016/S0891-5849(02)00879-1. [DOI] [PubMed] [Google Scholar]
- Masson E, Troncy L, Ruggiero D, Wiernsperger N, Lagarde M, El Bawab S. A series a series gangliosides mediate the effects of advanced glycation end products on pericyte and mesangial cell proliferation: a common mediator for retinal and renal microangiopathy? Diabetes. 2005;54:220–227. doi: 10.2337/diabetes.54.1.220. [DOI] [PubMed] [Google Scholar]
- Fox TE, Han X, Kelly S, Merrill AH 2nd, Martin RE, Anderson RE, Gardner TW, Kester M. Diabetes alters sphingolipid metabolism in the retina: a potential mechanism of cell death in diabetic retinopathy. Diabetes. 2006;55:3573–3580. doi: 10.2337/db06-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]