

Abstract
Background and Objectives
Although the pharmacokinetics of dexmedetomidine in healthy volunteers have been studied, there are limited data about the pharmacokinetics of long-term administration of dexmedetomidine in critically ill patients.
Methods
This population pharmacokinetic analysis was performed to quantify the pharmacokinetics of dexmedetomidine in critically ill patients following infusions up to 14 days in duration. The data consisted of three phase III studies (527 patients with sparse blood sampling, for a total of 2,144 samples). Covariates were included in a full random-effects covariate model and the most important covariate relationships were tested separately. The linearity of dexmedetomidine clearance was evaluated by observing steady-state plasma concentrations acquired at various infusion rates.
Results
The data were adequately described with a one-compartment model. The clearance of dexmedetomidine was 39 (95 % CI 37–41) L/h and volume of distribution 104 (95 % CI 93–115) L. Both clearance and volume of distribution were highly variable between patients (coefficients of variation of 62 and 57 %, respectively), which highlights the importance of dose titration by response. Covariate analysis showed a strong correlation between body weight and clearance of dexmedetomidine. The clearance of dexmedetomidine was constant in the dose range 0.2–1.4 μg/kg/h.
Conclusions
The pharmacokinetics of dexmedetomidine are dose-proportional in prolonged infusions when dosing rates of 0.2–1.4 μg/kg/h, recommended by the Dexdor® summary of product characteristics, are used.
Introduction
Dexmedetomidine is a highly selective, lipophilic α2 adrenoceptor agonist [1]. It is used as a sedative agent in intensive care and can be considered an alternative to more traditionally used midazolam and propofol, which act by potentiation of GABAA receptors. Compared with other sedatives, dexmedetomidine does not depress respiration in healthy volunteers [2] and results in better cognitive function than propofol in intensive care unit (ICU) patients [3], allowing better patient arousability and interaction [4, 5], and possibly earlier extubation [5].
The pharmacokinetics of dexmedetomidine have been previously studied in healthy volunteers [6–8], post-operative patients [9], renal disease patients [10] and intensive care patients [11–14]. Dexmedetomidine is mainly metabolised by direct glucuronidation [15], which is a high-capacity pathway and has a high hepatic extraction ratio of 0.71 [6]. We are aware of two studies, which involved a total of 34 patients, concerning the pharmacokinetics of long-term dexmedetomidine in doses higher than 0.7 μg/kg/h [11, 12]. In this paper, pharmacokinetic data from three phase III clinical trials with more than 500 critically ill patients were used to evaluate the impact of a variety of covariates on pharmacokinetics of dexmedetomidine and to confirm the results of the two previous studies in a larger patient group. A further objective was to investigate the dose proportionality of dexmedetomidine pharmacokinetics.
Methods
For this population pharmacokinetic study, the three phase III studies of prolonged dexmedetomidine treatment in critical-care patients sponsored by Orion Pharma were analysed, including MIDEX (Midazolam vs. Dexmedetomidine) and PRODEX (Propofol vs. Dexmedetomidine) studies [5, 16] (ClinicalTrials.gov identifiers: NCT00226785, NCT00481312, NCT00479661). The studies were conducted according to Good Clinical Practice standards and in accordance with the Declaration of Helsinki, subject to ethics committee review and informed consent obtained for all patients according to local regulations. Patients who subsequently withdrew consent were not included in any analyses. No new data were generated during the current study and thus further ethics approval was not required.
Patients
All studies included adult patients who were initially intubated, mechanically ventilated and expected to require light to moderate sedation for at least a further 24 h. The main exclusion criteria were (1) acute severe intracranial or spinal neurological disorder due to vascular causes, infection, intracranial expansion or injury; (2) uncompensated acute circulatory failure at time of randomisation (severe hypotension with mean arterial pressure <55 mmHg despite volume and pressors); (3) severe bradycardia (heart rate <50 beats/min); (4) atrioventricular-conduction block II–III (unless pacemaker installed); (5) severe hepatic impairment (bilirubin >101 μmol/L); (6) burn injuries and other injuries requiring regular anaesthesia or surgery; (7) use of centrally acting α2 agonists or antagonists (e.g. clonidine, titzanidine, apraclonidine and brimonidine) within 24 h prior to randomisation; or (8) investigators’ own judgement.
Treatments
The patients received an initial infusion of 0.7 μg/kg/h for 1 h. Thereafter, the dosing was titrated to clinical effect to maintain patients in the pre-defined target sedation range (Richmond Agitation and Sedation Scale 0 to −3 in all cases) using fixed dose levels ranging between 0.2 and 1.4 μg/kg/h. The maximum duration of treatment was 14 days.
Sampling and Analytical Methods
Blood samples were taken at the following times: baseline, 1 h (±15 min) after starting study treatment and every day at approximately the same time until the end of study treatment. Additionally, two follow-up samples were taken at 24 and 48 h after the end of study treatment.
Concentrations of dexmedetomidine in EDTA plasma samples were determined with high-performance liquid chromatography–tandem spectrometry (HPLC-MS/MS; Shimadzu Prominence HPLC, Kyoto, Japan) and mass spectrometric detection (AB Sciex API4000 mass spectrometer, Toronto, ON, Canada), as previously described [17]. The lower limit of quantification was 0.02 ng/mL. The within- and between-run precision of the assay (coefficient of variation) was within 7.5 % in the relevant concentration range. Deuterated medetomidine was used as the internal standard.
As part of the safety monitoring, the values of aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, creatinine clearance [18] and albumin were measured at baseline and on days 2, 4, 6, 9 and 14 after start of study drug infusion, and at 48 h post-dose. The baseline values of these markers were used for each patient as predictors of dexmedetomidine pharmacokinetics. If no baseline data were available for a patient, the average of all measurements from that patient was used. If no measurements from any time point were available for a patient, the median value of the whole population was substituted for the covariate value of that patient.
Modelling Strategy and Population Pharmacokinetic Model
Data were analysed using the NONMEM® software (version 7.2; ICON Development Solutions, Ellicott City, MA, USA) [19] with Intel Visual Fortran 11 compiler and Perl-speaks-NONMEM [20]. The model was fitted to data using the stochastic approximation expectation/maximisation algorithm [19], with 5,000 burn-phase iterations and 2,000 accumulation-phase iterations. The standard errors were calculated with importance sampling [19], using 20 iterations with 3,000 samples per subject and two degrees of freedom because of the sparseness of the data. One-compartment and two-compartment models with first-order elimination were tested before the inclusion of covariates to describe the time–concentration data.
Between-subject variability was modelled using log-normal distributions of individual parameter values, as shown in Eq. 1:
| 1 |
where P i is the parameter value of the ith subject, θ pop is the typical (median) value of this parameter and η is a random variable with mean of zero and variance of ω 2. Residual error was implemented as a combination of additive and proportional residual errors.
A full random-effects covariate model was used to quantify the relationship between pharmacokinetic parameters and covariates [21]. Briefly, a full random-effects covariate model uses random effects to both quantify the variability in pharmacokinetic parameters and to describe the individual values of observed covariate values. The covariate values are included in the dataset as observations. A full covariance matrix is estimated for the random effects, which means that the correlations between pharmacokinetic parameters, correlations between covariates, and the correlations between pharmacokinetic parameters and covariates are estimated. Advantages of this approach are that (1) it is not sensitive to correlated covariates, which means that all potential covariates can be included in the model; and (2) it may be more stable than a covariate model based on fixed effects. The full random-effects covariate model was considered the most appropriate approach for this project because many of the covariates were correlated. Furthermore, the dexmedetomidine dosing is based on dose titration by response. Because of this, there was considered to be no need to provide dosing guidelines based on covariates, unless dramatically altered pharmacokinetics could be associated with any single covariate. The following covariates were included in the model: body weight, age, creatinine clearance, AST, ALT, bilirubin and albumin. Log-normal distributions were used to describe the between-subject variability in covariate values. Prognostic indicators such as Simplified Acute Physiology Score (SAPS) were not applied in all studies and so could not be used as a covariate.
As an additional verification step, the strongest covariate relationships that were observed in the full covariate model were also tested for significance one at a time with the Likelihood Ratio Test. Briefly, a model without any covariates or covariances between random effects was used as the base model. Candidate covariates were included as predictors with a power model, as shown in Eq. 2:
| 2 |
where COVi is the individual value of the covariate, COVstd is the reference value of covariate, and θ exp is an estimated parameter signifying the relationship between the covariate and the parameter. This way, p-values could be calculated for the significance of the covariates by comparing objective function values. The difference in objective function values between nested models is chi-square distributed. The estimation method used in this step was QRPEM [19] using 80 iterations with 3,000 samples per subject.
Pharmacokinetic Analysis Based on Steady-State Concentrations
A subset of the whole dataset was used for an additional analysis of steady-state concentrations (C ss). Based on previous work [11], samples taken from patients after 15 h (five half-lives) of continuous infusion with a constant infusion rate (R inf) were considered to be at steady state. From these samples, the linearity of dexmedetomidine pharmacokinetics in doses up to 1.4 μg/kg/h was assessed.
Briefly, the analysis consisted of calculating the clearance (CL) of dexmedetomidine based on these single observations, and plotting the calculated CLs against R inf. The C ss of a drug are dependent only on R inf and CL of the drug (Eq. 3) [22].
| 3 |
Therefore, the CL of the drug can be calculated as the drug R inf divided by the C ss of the drug. The observed CL versus R inf was plotted and a linear model was fitted. The main interest was whether the slope of CL, as a function of R inf, is different from zero. If the metabolism of dexmedetomidine became saturated at higher doses, then a negative trend would be visible in the plot of CL versus R inf.
Results
Demographic Data
A total of 527 patients were included in the study. The overwhelming majority (96 %) of patients were Caucasian. The mean age (± standard deviation) was 62 (±15) years and there were more males (65 %) than females in the study population, which is typical of ICU settings [23]. Other relevant demographic factors have been summarised in Table 1. A total of 21 covariate values were missing, and population median values were substituted in their place.
Table 1.
Patient baseline characteristics
| Variable | Value |
|---|---|
| No. of subjects | 527 |
| Age (years) | 62.2 (15) |
| Body weight (kg) | 80.1 (20) |
| Sex [n (%) males] | 340 (65 %) |
| Use of inotropes and vasopressors [n (%)] | 339 (63 %) |
| Reason for ICU admission: surgical [n (%)] | 140 (27 %) |
| Reason for ICU admission: medical [n (%)] | 334 (63 %) |
| Reason for ICU admission: post-operative/trauma [n (%)] | 53 (10 %) |
| SAPS II scorea | 46.8 (15) |
| Overall SOFA scorea | 1.4 (1.5) |
| Creatinine clearance (mL/min) | 58.5 (38) |
| Aspartate aminotransferase baseline (IU/L) | 163.5 (572) |
| Alanine aminotransferase baseline (IU/L) | 101.0 (303) |
| Bilirubin baseline (μmol/L) | 13.1 (13) |
| Albumin baseline (g/L) | 23.4 (6.4) |
Values are expressed as mean (SD) unless specified otherwise
ICU intensive care units, SAPS II Simplified Acute Physiological Score, SD standard deviation, SOFA Sequential Organ Failure Assessment
aThe SOFA score and SAPS II score reflect the overall condition of the patient. Higher score means more severe impairment. SAPS II was not measured in one of the three clinical trials. SOFA scores had some missing information and the overall score is calculated as the average of the existing data
Observed Concentrations
There were a total of 2,144 dexmedetomidine concentrations above the limit of quantification available in the dataset. Figure 1a presents the number of samples taken after different R inf. In total, there were 47 plasma samples taken during dexmedetomidine infusion, which contained dexmedetomidine concentrations below the limit of quantification. Furthermore, 95 % (n = 458) of the samples taken at baseline, and 72 % (n = 634 out of 875) of the samples taken at 24 and 48 h after end of infusion, were below the limit of quantification. Samples below the limit of quantification were excluded from the analysis (M1 method introduced by Beal [24]). The M3 method, which consists of treating the samples below limit of quantification (BLQ) as censored and maximising the likelihood for them being censored, was also tried. The M3 method resulted in similar parameter estimates [less than 1 % difference in population estimates of CL and volume of distribution (V d)], but the NONMEM® software failed in calculating the standard errors of parameters when the M3 method was used. Therefore, the M1 method was considered the most appropriate method for treating the BLQ observations.
Fig. 1.
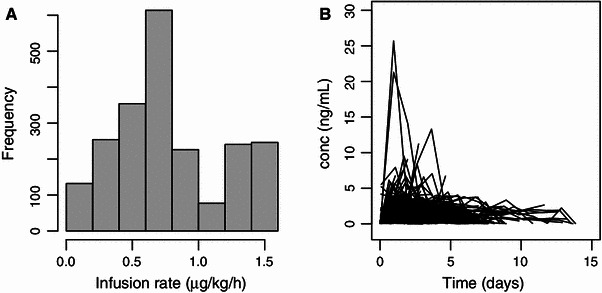
a The number of samples taken after different infusion rates of dexmedetomidine. It should be noted that the time interval between the change in infusion rate and the blood sampling was variable. b Dexmedetomidine concentrations in plasma versus time after the start of study drug treatment. conc concentration
Some atypically high concentrations of dexmedetomidine were encountered. The highest concentration was 383 ng/mL. There were four other unexpectedly high concentrations: 89, 82, 56 and 48 ng/mL. These samples were reanalysed and the same results were observed. These outlier concentrations occurred in four distinct patients, and they were mostly not preceded nor followed by atypically high dexmedetomidine concentrations.
One possible explanation for the high concentrations encountered is that the blood sample could have been taken downstream from the study drug infusion, for example from the same arm. To test this hypothesis, a semi-quantitative metabolite analysis was made for all samples of all the individuals from whom dexmedetomidine concentrations higher than 30 ng/mL were observed. The analysis showed no increase in metabolite concentrations during or after the high dexmedetomidine concentrations (data not shown). Based on this evidence, it seems that these high dexmedetomidine concentrations were not likely to reflect the true venous concentrations in patients and the concentrations were excluded from the model-building process. The final model was run both with and without these concentrations and both results are reported.
Figure 1b presents the time–concentration data for all individuals without the concentrations above 30 ng/mL. The numbers of blood samples per patient are summarised in Fig. 2a. The average duration of treatment was 2 days 14 h. The treatment durations of the patients are summarised in Fig. 2b.
Fig. 2.
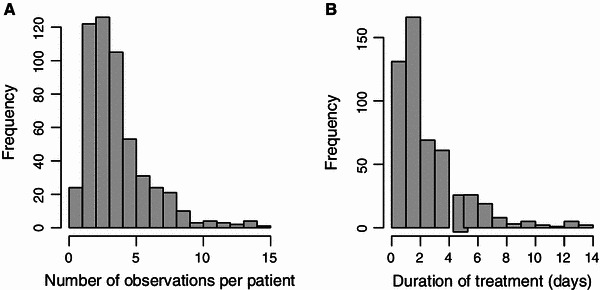
a Number of dexmedetomidine observations per patient. b Histogram of treatment durations
Population Pharmacokinetic Model
Dexmedetomidine time–concentration data were best described with a one-compartment model. Although the two-compartment model resulted in a decreased objective function value (p < 0.001), the parameter values were highly dependent on initial estimates and resulted in implausible results, such as the distribution half-life ranging between 32 s and 1.5 h depending on the run. The previously reported distribution half-life of dexmedetomidine is 6 min [8, 9], and the blood sampling in the current study was sparse. Therefore, the data were not considered adequate to identify the parameters of a two-compartment model, and the one-compartment model was chosen.
The typical CL of dexmedetomidine was 39 L/h and V d 104 L. The final model (with and without outlier concentrations) parameter estimates and standard errors are presented in Table 2. The inclusion of outlier concentrations had little impact on the estimates of CL (38 L/h) or V d (108 L). However, there were increases in between-subject variability and standard errors of estimates when the outlier concentrations were included. The observations versus model predictions are shown in Fig. 3a, b.
Table 2.
Pharmacokinetic parameter estimates and standard errors of the model
| Parameter | Estimate (95 % CI), outlier concentrations excluded | Estimate (95 % CI), outlier concentrations included |
|---|---|---|
| Clearance (L/h) | 39 (37–41) | 38 (36–40) |
| Between-subject variability of clearance (%) | 62 (52–72) | 67.6 (40–95) |
| Volume of distribution (L) | 104 (93–115) | 108 (38–177) |
| Between-subject variability of volume of distribution (%) | 57 (13–100) | 64.7 (0–129) |
| Residual error, additive (ng/mL) | 0.086 (0.067–0.10) | 0.086 (0.054–0.12) |
| Residual error, proportional (%) | 32.6 (32.0–33.2) | 38.7 (−6 to 83) |
Fig. 3.
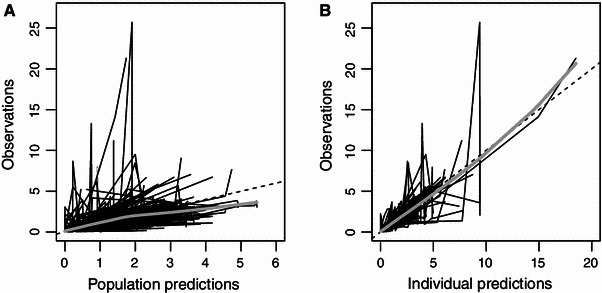
Plots of a population predictions and b individual predictions versus observations
The correlations between pharmacokinetic parameters and covariates are reported in Table 3. The strongest correlation (31 %) was found between body weight and CL; other covariates with higher than 10 % correlation to CL were AST and bilirubin (both with inverse correlation). Only albumin had a correlation to V d that had a relative standard error less than 100 % (inverse correlation of −12.7 %, standard error of 9.41 %).
Table 3.
Correlations between random effects of pharmacokinetic parameters and covariates (from the model without outliers)
| CL | V d | Age | WT | ALT | AST | BIL | ALB | CRCL | |
|---|---|---|---|---|---|---|---|---|---|
| CL | 100 | 15 (−36 to 66) | −7.7 (−22 to 6.3) | 31 (13 to 48) | −8.5 (−22 to 5.4) | −12 (−25 to −0.15) | −11 (−33 to 10) | −8.5 (−26 to 9.4) | 8.2 (−13 to 29) |
| V d | 100 | 9.5 (−44 to 63) | 13 (−20 to 47) | −5.1 (−27 to 17) | −0.13 (−19 to 19) | 12 (−18 to 41) | −13 (−31 to 5.7) | −8.1 (−39 to 23) |
Data are percentage correlation of random effects (95 % CI). Correlations between covariates are not shown. The correlations where the standard errors were less than 100 % of the correlation value are bolded
ALB albumin, ALT alanine aminotransferase, AST aspartate aminotransferase, BIL bilirubin, CL clearance, CRCL creatinine clearance, V d volume of distribution, WT body weight
When body weight was included as a predictor of CL into the base model, there was a significant improvement (p < 0.001) in the model, and the estimate of θ exp was 0.76, indicating an almost linear relationship. AST and bilirubin were also significant predictors of CL (p < 0.01 and p < 0.05) and their respective estimates of θ exp were −0.067 and −0.091. Albumin was a significant predictor of V d (p < 0.001) with an θ exp estimate of −0.49. Table 4 presents the predicted changes in CL and V d for the 2.5th and 97.5th percentiles of the covariate values.
Table 4.
Model-predicted changes in clearance and volume of distribution by change in the covariate values
| Covariate | 95 % percentile interval of the covariate | Relationship | Predicted parameter value range | IIV after covariate inclusion (%) |
|---|---|---|---|---|
| Body weight | 48–130 kg | CL = 36.6 × (WT/70)0.76 | 27–59 L/h | 55.9 |
| AST | 11–1020 IU/L | CL = 40.3 × (AST/41)−0.067 | 32–44 L/h | 58.4 |
| BIL | 2–55 μmol/L | CL = 39.8 × (BIL/9)−0.091 | 34–46 L/h | 58.4 |
| ALB | 11–36 g/L | V d = 91 × (ALB/23)−0.49 | 130–73 L | 52.7 |
IIV before the inclusion of covariates: CL 62 %, V d 57 %. Although the changes in covariates predict large changes in relevant parameters, the decrease in IIV is not clinically significant
ALB albumin, AST aspartate aminotransferase, BIL bilirubin, CL clearance, IIV inter-individual variability, V d volume of distribution, WT body weight
Pharmacokinetic Analysis Based on Steady-State Concentrations
A total of 643 observations (out of the 2,144) at R inf equal to or below 1.4 μg/kg/h were included in the analysis of C ss (Fig. 4a, b). One steady-state sample was considered an outlier and excluded because of being over 30 ng/mL. The calculated CLs versus dexmedetomidine R inf are presented in Fig. 4c. Although the linear model estimated a CL slope significantly different from zero (p < 0.05), the estimate of slope was slightly positive. Based on this, the CL of dexmedetomidine does not decrease at higher doses, and it seems that no saturation of metabolism is evident in continuous dexmedetomidine infusions up to 1.4 μg/kg/h.
Fig. 4.
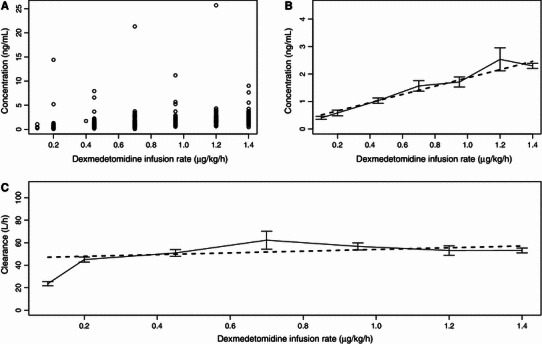
Steady-state plasma concentrations (n = 643) of dexmedetomidine versus infusion rate presented as a raw data and b descriptive plots. c Calculated clearance of dexmedetomidine versus infusion rate presented as a descriptive plot. The descriptive plots consist of means and standard errors of means (error bars) and linear model predictions (bold dashed line)
Discussion
The parameter estimates obtained from this study are similar to previous findings in both healthy volunteers and ICU patients. The CL estimate was 39 L/h in this study. A range of CLs between 31 and 53 L/h has been reported in previous studies in healthy volunteers [6–8, 10], and a range of 28–57 L/h in intensive care patients [11–14]. The lowest CL values (28 L/h) were reported in Chinese intensive care patients. One possible reason for the small CL estimate in that study is that the mean body weight of patients in that study was 60 kg [13].
The V d was estimated to be 104 L in this study, which is slightly lower than the V d at steady state (V ss) values between 121 and 194 L that have been reported in healthy volunteers [7, 8, 10]. In intensive care patients, V ss values between 123 and 389 L have been reported [12–14] and the reason for the lower value in this analysis is not clear.
The strongest covariate relationship was between dexmedetomidine CL and body weight. Some markers of hepatic dysfunction, such as high levels of AST and bilirubin, were associated with decreased CL. This is in agreement with previous knowledge, since hepatic impairment has been reported to result in decreased dexmedetomidine CL [25] and lower initial doses of dexmedetomidine should be considered in patients with hepatic impairment. An inverse association between plasma albumin and V d was also found. This was expected, since dexmedetomidine is 93 % bound to plasma proteins [25]. Therefore, lower concentrations of albumin cannot bind dexmedetomidine into the blood as effectively, which may drive dexmedetomidine into other tissues. However, the inclusion of these covariates as predictors of dexmedetomidine pharmacokinetics resulted in minimal decrease in between-subject variability (Table 4).
No signs of decreasing CL with higher dexmedetomidine concentrations were found by the analysis of C ss. This result is in contrast with recently published work, where dexmedetomidine was found to decrease cardiac output and cardiac output was found to affect the CL of dexmedetomidine [12]. In that study, the dexmedetomidine concentration to produce 50 % of maximum decrease in cardiac output was estimated at 2.4 ng/mL, and R inf of up to 2.5 μg/kg/h were used, which resulted in overall higher dexmedetomidine concentrations than those observed in the current study. It may be that the current study could not quantify a decreased CL resulting from decreased cardiac output because the average dexmedetomidine C ss of 2.3 ng/mL (Fig. 3a, b) after the highest R inf of 1.4 μg/kg/h were lower than the dexmedetomidine concentrations required to produce 50 % of maximum effect on this variable [12]. Although cardiac output data were not collected in these studies, one might speculate that within the usual dose range (0.2–1.4 μg/kg/h) the range of concentrations observed is not sufficient to demonstrate this pharmacodynamic relationship clearly.
A total of five concentrations above 30 ng/mL were observed, and considered as outliers. In the current study, the data were analysed both with and without the outlier concentrations. The presence of outlier concentrations in dexmedetomidine pharmacokinetic studies has been documented and discussed in previous studies [26, 27], and a Bayesian mixture model has been published solely for the handling of outliers [26].
In these data, both intra-individual and inter-individual variabilities were high, which is likely to reflect the highly variable physiological and medical condition of ICU patients. For example, the hepatic blood flow is temporarily reduced after injury [28], which might affect the CL of dexmedetomidine. Decreased cytochrome P450 enzyme activities have been reported in hepatocytes exposed to cytokines [29] but, to our knowledge, no similar experiments have been reported for glucuronidation enzymes, which are in this case more relevant since dexmedetomidine is mostly metabolised by direct glucuronidation [15]. It should also be mentioned that ICU patients are subject to many concomitant medications and the medications may change over time, which could impact pharmacokinetics of dexmedetomidine. For example, 63 % of the patients were given vasopressors or inotropes (Table 1), which could affect hepatic blood flow. Since dexmedetomidine is a high extraction ratio drug [6], changes in hepatic blood flow and cardiac output are more likely to affect CL than are changes in liver enzyme activity.
Since most of these changes in ICU patients are time-dependent, they contribute both to intra-individual and inter-individual variability. For a more extensive discussion of pharmacokinetic alterations in ICU patients, several reviews are available (see, for example [30, 31]).
There was some missing covariate information. The approach taken in this study was to substitute median values for missing information (see Sect. 2.5). This approach is conservative and may increase the risk of false negative findings (type II error) while decreasing the risk of false positive findings (type I error). However, in this case less than 1 % of covariate records had to be substituted with a median value of the covariate.
This population pharmacokinetic study features the largest patient population in a dexmedetomidine pharmacokinetic study to date. Because of the sparse blood sampling, a one-compartment had to be used to describe the pharmacokinetic data, although a two-compartment model would be necessary to describe the concentrations during the first minutes after change of R inf. However, the estimate of CL should be accurate despite the use of a one-compartment model, since samples typically were not taken shortly after change in study drug R inf (data not shown). Despite the simplicity of the structural model, the large number of patients provides a good basis for covariate analysis.
Conclusion
In conclusion, based on C ss analysis, no saturation of dexmedetomidine metabolism occurs at dexmedetomidine doses up to 1.4 μg/kg/h and infusions lasting up to 2 weeks. Body weight is an important, intuitive and easily available predictor of dexmedetomidine CL. Other statistically significant covariates were also identified, but they are not as easily available and did not result in a clinically important decrease in inter-individual variation and so are not suitable for determining appropriate individual patient dose. Therefore, based on the results of this study, dexmedetomidine should continue to be dosed by body weight with titration according to clinical response.
Acknowledgments
We gratefully acknowledge the Dexmedetomidine Investigators for conducting the clinical studies, Lauri Mantere for compiling the datafile and Petri Toivanen for helpful discussions about statistical modelling.
Conflict of interest
This pharmacokinetic analysis is an extension of work done for Orion Pharma during December 2009–December 2010 and February 2011–March 2011, during which time all authors were Orion Pharma employees. The study was sponsored by Orion Pharma; this involved the design of the study, collection of the data and formatting the data into a format suitable for pharmacokinetic analysis. Orion Pharma is the manufacturer and Marketing Authorisation holder of dexmedetomidine in the European Union.
References
- 1.Virtanen R, Savola JM, Saano V, Nyman L. Characterization of the selectivity, specificity and potency of medetomidine as an alpha 2-adrenoceptor agonist. Eur J Pharmacol. 1988;150:9–14. doi: 10.1016/0014-2999(88)90744-3. [DOI] [PubMed] [Google Scholar]
- 2.Hsu YW, Cortinez LI, Robertson KM, Keifer JC, Sum-Ping ST, Moretti EW, et al. Dexmedetomidine pharmacodynamics: part I: crossover comparison of the respiratory effects of dexmedetomidine and remifentanil in healthy volunteers. Anesthesiology. 2004;101:1066–1076. doi: 10.1097/00000542-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Mirski MM, Gill RG, Murakami PM, Thompson CT, Lewin JL. Dexmedetomidine improves attention and recall in agitated critically ill patients. Crit Care. 2011;15:P355. doi: 10.1186/cc9775. [DOI] [Google Scholar]
- 4.Venn M, Newman J, Grounds M. A phase II study to evaluate the efficacy of dexmedetomidine for sedation in the medical intensive care unit. Intensive Care Med. 2003;29:201–207. doi: 10.1007/s00134-002-1579-9. [DOI] [PubMed] [Google Scholar]
- 5.Jakob SM, Ruokonen E, Grounds RM, Sarapohja T, Garratt C, Pocock SJ, et al. Dexmedetomidine vs midazolam or propofol for sedation during prolonged mechanical ventilation: two randomized controlled trials. JAMA. 2012;307:1151–1160. doi: 10.1001/jama.2012.304. [DOI] [PubMed] [Google Scholar]
- 6.Dutta S, Lal R, Karol MD, Cohen T, Ebert T. Influence of cardiac output on dexmedetomidine pharmacokinetics. J Pharm Sci. 2000;89:519–527. doi: 10.1002/(SICI)1520-6017(200004)89:4<519::AID-JPS9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Dyck JB, Maze M, Haack C, Vuorilehto L, Shafer SL. The pharmacokinetics and hemodynamic effects of intravenous and intramuscular dexmedetomidine hydrochloride in adult human volunteers. Anesthesiology. 1993;78:813–820. doi: 10.1097/00000542-199305000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Anttila M, Penttila J, Helminen A, Vuorilehto L, Scheinin H. Bioavailability of dexmedetomidine after extravascular doses in healthy subjects. Br J Clin Pharmacol. 2003;56:691–693. doi: 10.1046/j.1365-2125.2003.01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talke P, Richardson CA, Scheinin M, Fisher DM. Postoperative pharmacokinetics and sympatholytic effects of dexmedetomidine. Anesth Analg. 1997;85:1136–1142. doi: 10.1097/00000539-199711000-00033. [DOI] [PubMed] [Google Scholar]
- 10.De Wolf AM, Fragen RJ, Avram MJ, Fitzgerald PC, Rahimi-Danesh F. The pharmacokinetics of dexmedetomidine in volunteers with severe renal impairment. Anesth Analg. 2001;93:1205–1209. doi: 10.1097/00000539-200111000-00031. [DOI] [PubMed] [Google Scholar]
- 11.Iirola T, Aantaa R, Laitio R, Kentala E, Lahtinen M, Wighton A, et al. Pharmacokinetics of prolonged infusion of high-dose dexmedetomidine in critically ill patients. Crit Care. 2011;15:R257. doi: 10.1186/cc10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iirola T, Ihmsen H, Laitio R, Kentala E, Aantaa R, Kurvinen J-P, et al. Population pharmacokinetics of dexmedetomidine during long-term sedation in intensive care patients. Br J Anaesth. 2012;108:460–468. doi: 10.1093/bja/aer441. [DOI] [PubMed] [Google Scholar]
- 13.Lin L, Guo X, Zhang MZ, Qu CJ, Sun Y, Bai J. Pharmacokinetics of dexmedetomidine in Chinese post-surgical intensive care unit patients. Acta Anaesthesiol Scand. 2011;55:359–367. doi: 10.1111/j.1399-6576.2010.02392.x. [DOI] [PubMed] [Google Scholar]
- 14.Venn RM, Karol MD, Grounds RM. Pharmacokinetics of dexmedetomidine infusions for sedation of postoperative patients requiring intensive care. Br J Anaesth. 2002;88:669–675. doi: 10.1093/bja/88.5.669. [DOI] [PubMed] [Google Scholar]
- 15.Kaivosaari S, Toivonen P, Aitio O, Sipila J, Koskinen M, Salonen JS, et al. Regio- and stereospecific N-glucuronidation of medetomidine: the differences between UDP glucuronosyltransferase (UGT) 1A4 and UGT2B10 account for the complex kinetics of human liver microsomes. Drug Metab Dispos. 2008;36:1529–1537. doi: 10.1124/dmd.108.021709. [DOI] [PubMed] [Google Scholar]
- 16.Ruokonen E, Parviainen I, Jakob SM, Nunes S, Kaukonen M, Shepherd ST, et al. Dexmedetomidine versus propofol/midazolam for long-term sedation during mechanical ventilation. Intensive Care Med. 2009;35:282–290. doi: 10.1007/s00134-008-1296-0. [DOI] [PubMed] [Google Scholar]
- 17.Ji QC, Zhou JY, Gonzales RJ, Gage EM, El-Shourbagy TA. Simultaneous quantitation of dexmedetomidine and glucuronide metabolites (G-Dex-1 and G-Dex-2) in human plasma utilizing liquid chromatography with tandem mass spectrometric detection. Rapid Commun Mass Spectrom. 2004;18:1753–1760. doi: 10.1002/rcm.1548. [DOI] [PubMed] [Google Scholar]
- 18.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 19.Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user’s guides. Ellicott City: ICON Development Solutions; 2009.
- 20.Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson MO. A full model approach based on the covariance matrix of parameters and covariates. PAGE. 2012;Abstr 2455. http://www.page-meeting.org/?abstract=2455. Accessed 20 Jun 2013
- 22.Rowland M, Tozer TN. Clinical pharmacokinetics and pharmacodynamics: concepts and applications. Philadelphia: Wolters Kluwer Health/Lippincott William & Wilkins; 2011. [Google Scholar]
- 23.Dodek P, Kozak JF, Norena M, Wong H. More men than women are admitted to 9 intensive care units in British Columbia. J Crit Care. 2009;24:630.e1–8. [DOI] [PubMed]
- 24.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 25.Karol MD, Maze M. Pharmacokinetics and interaction pharmacodynamics of dexmedetomidine in humans. Best Pract Res Clin Anaesthesiol. 2000;14:261–269. doi: 10.1053/bean.2000.0081. [DOI] [Google Scholar]
- 26.Choi L, Caffo BS, Kohli U, Pandharipande P, Kurnik D, Ely EW, et al. A Bayesian hierarchical nonlinear mixture model in the presence of artifactual outliers in a population pharmacokinetic study. J Pharmacokinet Pharmacodyn. 2011;38:613–636. doi: 10.1007/s10928-011-9211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohli U, Pandharipande P, Muszkat M, Sofowora GG, Friedman EA, Scheinin M, et al. CYP2A6 genetic variation and dexmedetomidine disposition. Eur J Clin Pharmacol. 2012;68:937–942. doi: 10.1007/s00228-011-1208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gottlieb ME, Sarfeh IJ, Stratton H, Goldman ML, Newell JC, Shah DM. Hepatic perfusion and splanchnic oxygen consumption in patients postinjury. J Trauma. 1983;23:836–843. doi: 10.1097/00005373-198309000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Razzak Z, Loyer P, Fautrel A, Gautier JC, Corcos L, Turlin B, et al. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol. 1993;44:707–715. [PubMed] [Google Scholar]
- 30.De Paepe P, Belpaire FM, Buylaert WA. Pharmacokinetic and pharmacodynamic considerations when treating patients with sepsis and septic shock. Clin Pharmacokinet. 2002;41:1135–1151. doi: 10.2165/00003088-200241140-00002. [DOI] [PubMed] [Google Scholar]
- 31.Zagli G, Tarantini F, Bonizzoli M, Di Filippo A, Peris A, De Gaudio AR, et al. Altered pharmacology in the intensive care unit patient. Fundam Clin Pharmacol. 2008;22:493–501. doi: 10.1111/j.1472-8206.2008.00623.x. [DOI] [PubMed] [Google Scholar]