

Abstract
ADP-glucose pyrophosphorylase (AGPase), a key enzyme involved in higher plant starch biosynthesis, is composed of pairs of large (LS) and small subunits (SS). Ample evidence has shown that the AGPase catalyzes the rate limiting step in starch biosynthesis in higher plants. In this study, we compiled detailed comparative information about ADP glucose pyrophosphorylase in selected plants by analyzing their structural features e.g. amino acid content, physico-chemical properties, secondary structural features and phylogenetic classification. Functional analysis of these proteins includes identification of important 10 to 20 amino acids long motifs arise because specific residues and regions proved to be important for the biological function of a group of proteins, which are conserved in both structure and sequence during evolution. Phylogenetic analysis depicts two main clusters. Cluster I encompasses large subunits (LS) while cluster II contains small subunits (SS).
Keywords: Computational tools, Aliphatic index, instability index, isoelectric point
Background
Starch is an important carbohydrate and the primary energy source in plants. It has numerous industrial applications as reviewed in Slattery et al. [1, 6]. Starch biosynthesis occurs mainly by the participation of three enzymes: ADP-glucose pyrophosphorylase (AGPase), starch synthase, and branching enzymes [2, 3]. The first enzyme in starch biosynthesis is the AGPase that catalyzes the conversion of Glc-1-P and ATP to ADP-glucose and pyrophosphate (PPi). ADP-glucose is then used by starch synthase for the synthesis of polyglucans. Many researchers have revealed that the AGPase catalyzes the rate limiting step in starch biosynthesis in higher plants [1, 2, 4]. AGPase from higher plants has a heterotetrameric structure (ά2β2) composed of pairs of small (SS) and large (LS) subunits encoded by at least two different genes [5]. The large subunit (LS) plays a major role in allosteric regulation through its interaction with the small catalytic subunit (SS). The LS is encoded by the shrunken-2 (Sh2) and the SS by brittle-2 (Bt2) [7]. Both Sh2 and Bt2 genes show considerable amino acid identity (43.2%) and similarity (61%). Maize (Zea mays) and rice (Oryza sativa), the two major cereals, show 93% identity in the amino acids sequence for the LS of AGPase enzyme. In case of wheat both large and small subunit show only 49 % identity in their amino acid sequences.
Wheat, Rice, maize, barley, and potato are important amongst staple crops as these are primarily consumed by humans otherwise also these are the cheapest source of carbohydrates and proteins used as food by one third population of the world. Economically, wheat is one of the major food crops both in terms of area and production. Wheat is grown in diverse environments, from cool rain-fed to hot dry-land areas around the world. This wide spread cultivation of the crop all along the globe is largely due to high versatility of its genome. The seed number and seed weight are important yield components of wheat for determining wheat production. Particularly starch, which accounts for 65-75% of wheat grain dry weight and composed of glucan chains, amylopectin and amylase, a major determinant of yield. This has rendered the use for comparative analysis of ADP-glucose pyrophosphorylase in selected plants. In addition, phylogenetic analysis was also performed to know the evolutionary relatedness among AGPase.
Methodology
Protein sequence retrieval:
Protein sequences of small subunit (SS) and large subunit (LS) of AGPase from wheat, rice, maize, potato and Arabidopsis were retrieved from protein database of NCBI (National Center for Biotechnology Information, (http://www.ncbi.nlm.nih.gov/protein/) in FASTA format.
Physico-chemical characterization:
The ProtParam tool (http://web.expasy.org/protparam/) of ExPASy was used to compute amino acid composition (%), molecular weight, theoretical isoelectric point (pI), number of positively and negatively charged residues, extinction coefficient, instability and aliphatic index, Grand Average of Hydropathy (GRAVY).
Secondary structural properties:
Secondary structural properties of the protein including alpha helix, 310 helix, Pi helix, beta bridge, extended strand, beta turns, bend region, random coil, ambiguous and other states were computed by using SOPMA (Self Optimized Prediction Method with Alignment, http://npsapbil.ibcp.fr/cgibinnpsa_automat.pl?page=/NPSA/ npsa_sopma.html) tool of NPS (Network Protein Sequence Analysis).
Prediction of functional properties:
The motif prediction analysis was carried out with the help of Expasy's prosite tool. For functional analysis, the motifs of the AGPase protein sequences were identified by using Prosite (http://prosite.expasy.org/). Input data type was in FASTA format and motifs were scanned against prosite patterns.
Identification of Signature Logo using Web tool:
Logo of AGPase was generated using Web Logo tool (http://weblogo.berkeley.edu/). In this overall height of the stack indicates the sequence conservation at that position, while the heights of the symbols within the stack indicate the relative frequency of each amino acid at that position.
Phylogenetic analysis:
Twelve sequences of both large and small subunits of wheat, rice, maize, barley, potato and Arabidopsis were aligned by ClustalW tool and output file of this program was used for generation of phylogenetic tree (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Results & Discussion
For all the AGPase physicochemical characterization, secondary structure properties, motif and phylogenetic analysis was carried out by using various computational tools. In maize additional tyrosine and serine residues increases the seed weight 11-18% without increasing or decreasing the percentage of starch [8]. From ProtParam result, it was observed that for all the residues on average the percentage of serine was higher than the other residues but the percentage of tyrosine was average. The percentage of serine was higher as compare to tyrosine in all subunits analyzed however it was highest in large subunit of Arabidopsis thaliana and lowest in small subunit of maize. In contrast to this the percentage of tyrosine was approximately equal in all the sequences except small subunits of wheat and maize (Figure 1).
Figure 1.
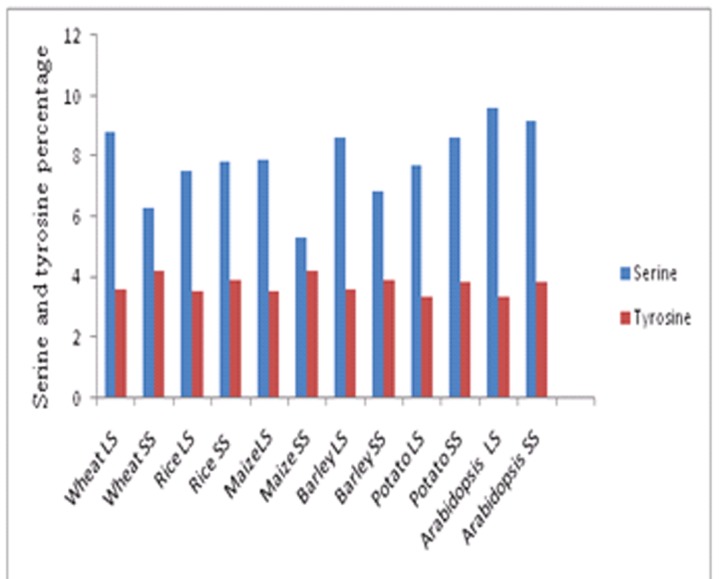
Serine and tyrosine percentage of AGPase in selected plants
The total number of positively (Arg + Lys) and negatively (Asp + Glu) charged residues of AGPase members were observed Table 1 (see supplementary material). For all members, the total number of negatively charged residues exceeded the total number of positive charged residues except large subunit of potato and Arabidopsis thaliana. This possible variation might be due to their isoelectric point in acidic range. For the remaining members, the isoelectric point was within alkaline range. Extinction coefficient for all AGPase was observed higher almost with in a same range. High extinction coefficient means higher concentration of lysine, tryptophan and tyrosine. This prediction is useful to study protein-protein interaction studies. Stability of protein is described in terms of its stability index whether a protein is stable or not, can be described by its instability index. Instability index for large subunit of wheat, barley, Arabidopsis and small subunit of potato and rice is higher than 40 and thus describing these proteins unstable. It is noteworthy that high aliphatic index was observed for small subunit of all plants as compare to large subunit. The higher aliphatic index indicates higher concentration of alanine, valine, isoleucine and leucine occupying the relative volume of a protein [9].
In addition to this higher aliphatic index also provides higher thermo stability. The results obtained in case of instability index and aliphatic index were contradictory while compared for AGPase analysis in rice, potato and Arabidopsis. According to instability index, these proteins are unstable but their aliphatic index is high enough to say that they are stable. These finding are in consistent with earlier research [8]. Grand Average of Hydropathy (GRAVY) was computed for all the members. A range of GRAVY value was observed from -0.253 to -0.131'for AGPase in selected plants. SOPMA analysis was done for all AGPase members and it showed a high value for random coil in all the members Table 2 (see supplementary material) while alpha helix were found approximately equal to extended strands.
High value for random coil bears important significance in the study of protein tertiary structure and related functions. Functional analysis of these proteins includes identification of important motifs Table 3 (see supplementary material). These motifs were 10 to 20 amino acids in length arise because specific residues and regions proved to be important for the biological function of a group of proteins, which are conserved in both structure and sequence during evolution. In this study, a signature logo of ADP glucose pyrophosphorylase was also generated by web logo tool. The overall height of the stack indicates the sequence conservation at that position, while the height of the symbols within the stack indicates the relative frequency of each amino acid at that position (Figure 2). Phylogenetic analysis depicts two main clusters (Figure 3). Cluster I encompasses large subunits (LS) while cluster II contains Small subunits (SS). This study will provide a good foundation for further functional analysis of AGPase of other crops. However, the outcome of this study needs further validation by experimental approach.
Figure 2.

WebLogo representation of motif of ADP Glucose pyrophosphorylase. The amino acid type and position are shown on the x axis. The overall height of the amino acid stacks, plotted on the y axis, indicates the sequence conservation at a given position, while the height of individual symbols within a stack indicates the relative frequency of an amino acid at that position. Amino acids are color coded according to their type as basic (blue), hydrophobic (black), polar/nonpolar (green), and acidic (red).
Figure 3.
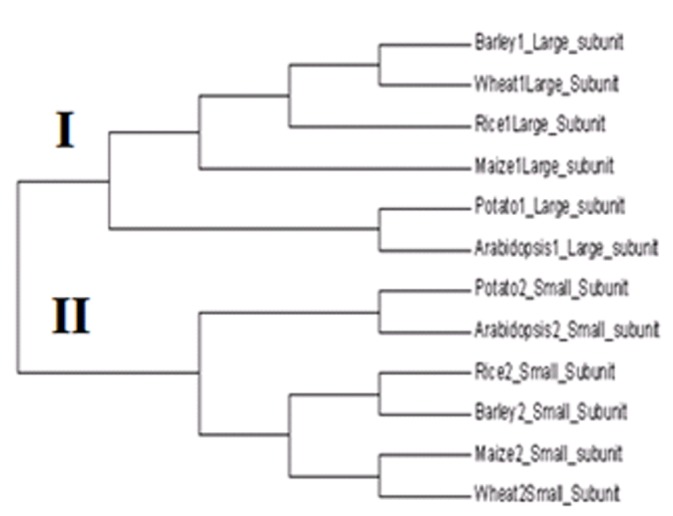
Phylogenetic tree of ADP Glucose Pyrophosphorylase obtained using ClustalW.
Conclusion
In this study, we compiled detailed comparative information about ADP-glucose pyrophosphorylase in selected plants by analyzing their structural features e.g. amino acid content, physico-chemical properties, secondary structural features and phylogenetic classification. Present investigation will provide an insight for the biologists working with ADP-glucose pyrophosphorylase in order to understand the functionality of AGPase.
Supplementary material
Acknowledgments
This work was supported by the UGC sponsored Rajiv Gandhi National Fellowship sponsored by University Grants Commission to SR, Agri Bioinformatics Promotion Program, Department of Information Technology, Ministry of Communications & Information Technology, Government of India, New Delhi to RC and Indian Council of Agricultural research of New Delhi, India to PS. The authors are thankful to Project Director, DWR, Karnal for providing facilities for this work.
Footnotes
Citation:Rani et al, Bioinformation 9(11): 572-576 (2013)
References
- 1.Gasteiger E, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker John M., editor. The Proteomics Protocols Handbook. Humana Press; 2005. p. 571. [Google Scholar]
- 2.Kyte J, Doolottle RF. J Mol Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 3.Martin C, Smith AM. Plant Cell. 1995;7:971. doi: 10.1105/tpc.7.7.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salamone PR, et al. FEBS Lett. 2000;482:113. doi: 10.1016/s0014-5793(00)01985-2. [DOI] [PubMed] [Google Scholar]
- 5.Tuncel A, et al. Biophysical J. 2008;95:3628. doi: 10.1529/biophysj.107.123042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slattery CJ, et al. Trends Plant Sci. 2000;5:291. doi: 10.1016/s1360-1385(00)01657-5. [DOI] [PubMed] [Google Scholar]
- 7.Bhave MR, et al. Plant Cell. 1990;2:581. doi: 10.1105/tpc.2.6.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giroux MJ, et al. Proc Natl Acad Sci U S A. 1996;93:5824. doi: 10.1073/pnas.93.12.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saha A, et al. Bioinformation. 2013;9:42. doi: 10.6026/97320630009042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.