

Abstract
Genome assembly has always been complicated due to the inherent difficulties of sequencing technologies, as well the computational methods used to process sequences. Although many of the problems for the generation of contigs from reads are well known, especially those involving short reads, the orientation and ordination of contigs in the finishing stages is still very challenging and time consuming, as it requires the manual curation of the contigs to guarantee correct identification them and prevent misassembly. Due to the large numbers of sequences that are produced, especially from the reads produced by next generation sequencers, this process demands considerable manual effort, and there are few software options available to facilitate the process. To address this problem, we have developed the Graphic Contig Analyzer for All Sequencing Platforms (G4ALL): a stand-alone multi-user tool that facilitates the editing of the contigs produced in the assembly process. Besides providing information on the gene products contained in each contig, obtained through a search of the available biological databases, G4ALL produces a scaffold of the genome, based on the overlap of the contigs after curation.
Availability
The software is available at: http://www.genoma.ufpa.br/rramos/softwares/g4all.xhtml
Keywords: Genome assembly, Bioinformatic tools, sequence analysis, software
Background
The main advantage of the “high-throughput” sequencing technologies known as “next-generation sequencing” (NGS) is the production of a large amount of data at a low cost, when compared to the Sanger method. They also make it possible to study complete genomes and develop systematic comparative analyses [1]. The NGS platforms, such as Illumina (Genome Analyzer), Sequencing by Oligonucleotide Ligation and Detection - SOLiD (Applied Biosystems), 454 GS FLX Titanium (Roche), Polonator G.007(Azco Biotech) and HeliScope (Helicos Biosciences) are capable of producing millions to billions of short reads, providing a high degree of coverage and accuracy, with reduced run times [1–3]. In the assembly of NGS genomes, the small size of the reads makes it difficult to correctly identify repetitive regions. In addition, problems related to the quality of the bases can provoke assembly errors [4].
The principal programs used to assemble short reads are based on graph theory, although they use quite different approaches: SSAKE [5], SHARCGS [6] and VCAKE [7] use “greedy algorithms”, while Edena [8] and Newbler [9] are part of a software group that implements “overlap-layout-consensus” (OLC), and among the representatives of the De Bruijn graphs we have: EULER-SR [2], Allpaths [10] and Velvet [11]. Nevertheless, the difficulties involved in the production of a complete genome, due to the problems inherent to the NGS technology and the artefacts that appear during assembly [12], has led to the use of hybrid assembly approaches, involving various algorithms and sequencing technologies [13, 14]. At the end of the process, the collection of contiguous DNA sequences (contigs) produced by de novo assembly are put in order and oriented, in a process known as scaffolding [15], which can be simplifier using a reference genome to align the contigs, thus constituting a hybrid approach to the genome assembly.
In the finishing stage it is necessary to analyze the produced sequences in order to correct possible errors in the assembly. As such, it is necessary to use tools that enable viewing and editing sequences [16] as the CLC Genomics Workbench (commercially available from CLC Bio) that despite their extremely useful resource for the curation and editing of sequences, when used for the alignment of contigs (sequences much larger than reads) against a reference are not as efficient in function of their alignment settings being based on percentage of aligned bases and not quantity, such that a contig with 2000bp that has 200bp mapped against a reference would be considered only if the minimum percentage of alignment is less than or equal to 10%, which can result in problems due to the size range of the contigs, which can initiate in a few base pairs up to kilobases. As alternatives to commercial software, Consed and GAP are freely available. However, none of the tools mentioned above allow data sharing among multiple users by storing the information locally in the curatorial files, which limits the number of people involved in the conclusion of the same project, requiring a longer time in the curation of the genomes assembly.
We have developed the Graphic Contig Analyzer for All Sequencing Platforms (G4ALL), multiuser software that allows the visualization and curation of a group of contigs that are aligned locally to a reference genome. This program identifies where the contigs are superimposed, so that the sequences can be extended. In addition, G4ALL can be used to edit and validate contigs through homology searches of public databases, such as the NCBI non-redundant protein bank, and, once curated, to assemble the scaffold.
Methodology
Data:
As a model organism, we used Corynebacterium pseudotuberculosis lineage Cp258, sequenced using SOLiD version 3 and a genome library that produced reads of 50 bp fragments, with a total of 70,521,987 reads which represented 1533× sequence coverage. For the reference genome, we used strain FRC41 (NC_014329) of this same organism.
Data treatment:
The quality of the bases that compose a reads affects the quantity of errors arising during sequence alignment [4]. Consequently, the quality of the raw data obtained from the sequencing was evaluated with the software Quality Assessment [17] to find the best quality filter to apply to the data. The reads which presented mean quality value less than 20 were removed.
De novo assembly:
In order to increase the representation of the sequenced genome in the de novo assembly allowing complementary strategies, given that different approaches or assemblies can produce different set of contigs [13, 18], we have applied the Edena which uses the OLC depending on overlap graph which represents the sequencing reads and their overlaps, and Velvet software which uses the “Eulerian Path” which uses the K-mer graph in which nodes represent all its fixed-length subsequences obtained from larger sequences. Nevertheless, other genome assemblers based on alternative approaches can be used to generate the set of contigs.
After applying the quality filter, the reads were run through both programs with varying parameters, in order to identify the best assembly as a function of N50, the number of bases, and the largest and smallest contigs. In Velvet, the k-mer parameter was varied to include coverage cut-off and expected coverage in Table 1 (see supplementary material).
Elimination of redundant contigs:
The contigs produced by Velvet and Edena can cover the same portions of the genome, so, to reduce the work needed to manually curate the contigs and improve the assembly indicators, such as N50, we used Simplifier (available at https://sourceforge.net/projects/simplifier/) to remove the redundant contigs from a file in multi-FASTA format.
Synteny Analysis between genomes C. pseudotuberculosis:
Currently available in the NCBI, the genome of C. pseudotuberculosis 316 (CP003077) which was sequenced using the platform Ion Torrent [19], whose assembly process was facilitated due to the average size of the readings being 120pb. Thus, since this is an organism of the same biovar of C. pseudotuberculosis 258 and have a very conserved gene order, we conducted an analysis of synteny with the software OSLay [20] between the two genomes.
Search for homology:
We used the Basic Local Alignment Search Tool – BLAST [21] (blastn or megablast), which employs an heuristic algorithm to run a search for homologies with the reference genome, in order to anchor the contigs produced by the de novo assemblies, and provide the basis for the orientation and sorting of the genome assembly. BLAST (blastx) was also used to search for homologies in the available biological databases, that is, the non-redundant (nr) protein data banks of the National Center of Biotechnology Information (NCBI), in order to identify the gene products contained in each contig and provide G4ALL with this information. The standard BLAST parameters were used, the low complexity filter was turned off, and this process was run locally because of the large amount of data.
G4ALL:
G4ALL has a graphic edition and validation interface (Figure 1) for de novo -assembled contigs, which allows the orientation and sorting of these contigs in relation to a reference genome based on the results of the alignment generated by the appropriate software in table format, with 11 columns (query, reference, alignment length, mismatches, gaps, query start, query end, source start, source end, evaluate, bitscore and identity). The last three columns are used only for the BLAST result. G4ALL is able to convert the alignment results of BLAST (table format) and Nucmer (Mummer [22] package) to the G4ALL format through the Convert alignment results option in the utility menu. This software allows the contigs to be extended with the aid of the information on the gene products provided by the homology search (see above). To install a project in G4ALL, the name of the project is entered, followed by the reference genome (fasta file), sequences produced by the genome assembly (multifasta file), and the results (table format) of the alignment of the contigs in relation to the reference genome. This information is entered into a MySQL database, so that it can be accessed by the sequence curating interface.
Figure 1.

The interface lists the contigs that were curated, showing their position in the reference genome and in relation to the other contigs. After selecting one of these contigs, it is possible to move it to the left or to the right to correct the alignment in relation to the others, copy, save or edit each sequence, and trim both ends. In addition, the BLAST results can be observed through the menu “Blast Result”, in order to identify the existing gene products.
To visualize the project information registered in G4ALL, the following filters can be used: minimum identity of the BLAST hits, minimum alignment extension, contigs with multiple alignments in the genome, contigs selected during curation (marked as “used”), along with the option “all alignments”, in which all previously-defined filters are ignored. Because of the considerable computing power needed to load all of the project data, render and position all of the sequences in the curation window, threads were implemented in some processes, in order to improve the performance of the program.
During curation, the following actions can be taken for each contig: save in a FASTA format file, copy, edit, and visualize the results produced by the alignment against the non-redundant protein database, previously inserted through G4ALL, in addition to generating a report of the list of gene products for each of the contigs, with their respective e-values and identity values, when available. In order to facilitate navigation through the sequences produced by the assembly software, the screen “Contig Details” presents navigation options. Within this screen, the sequence ends can be trimmed, the sequences can be moved in relation to the other sequences to allow alignment, and the original sequence can be recovered using the resource “Recover Original Sequence”. In order to produce the scaffold of the genome, the button “Scaffold” is activated. The curated sequences are evaluated as a function of the overlap (size defined by the user) of each sequence in relation to its neighbours, based on the alignment results inserted previously, in order to represent possible rearrangements in the genome, for the construction of the consensus sequence (Figure 2).
Figure 2.

Assembly of the consensus sequence. When the contigs are aligned with each other by similarity, they are extended to produce the consensus sequence
Implementation:
To develop G4ALL, we used the JAVA language, with the Swing library (visual resources), and java.util.concurrent (http://java.sun.com/docs/books/tutorial/essential/concurrency/pools.html) to implement the threads. We used the Java Persistence API (http://java. sun.com/developer/technicalArticles/J2EE/jpa/) to communicate with the MySQL database. The system reports were developed using the iReport software (http://jasperforge.org/projects/ireport). G4ALL is available at: http://www.genoma.ufpa.br/rramos/softwares/g4all.xhtml
Results & Discussion
The sequencing of strain Cp258 generated 70,521,987 reads, which were submitted to the phred >=20 quality filter, considering the mean, using the Quality Assessment function, with left 40,589,132 reads representing 882× sequence coverage. In order to assemble the genome using Velvet and Edena, we ran 378 and 336 assemblies, respectively, with variations in the parameters. Considering the largest N50, the number of bases included, and the largest and smallest contig, 1269 contigs were obtained with Velvet, with an N50 of 3.2Kb and 2,343,443 bases, using the parameters k-mer, coverage cut-off and expected coverage, of 31, 15 and 260, respectively. In Edena, the best run produced 6,735 contigs, an N50 of 0.5Kb and 2,590,667 bases, with: MinOverlap set at 36, OverlapCutoff at 11 and DephLimit at 14.
The contigs in Velvet and Edena were saved in a single multifasta file with 8,004 sequences and submitted to Simplifier, which returned only 5,271 contigs (65% of the sequences that were entered). These were used for the BLAST run (blastn) against the reference genome FRC41 (CP002097). The contigs were divided into four groups: (a) single alignments, (b) more than one alignment with the reference genome, (c) no significant alignment and (d) no alignment with the reference, with the criterion of alignment equal to or greater than 40 nucleotides in Table 2(see supplementary material). Contigs with significant alignments (extension greater than or equal to 40 bps) in more than one region of the domain, generally represent repetitions such as ribosomal RNAs, but they can also involve the deletion (Figure 3A) or insertion (Figure 3B) of regions in the contig when compared to the reference sequence, or even assembly errors, where they do not represent the gene product correctly (Figure 3C).
Figure 3.
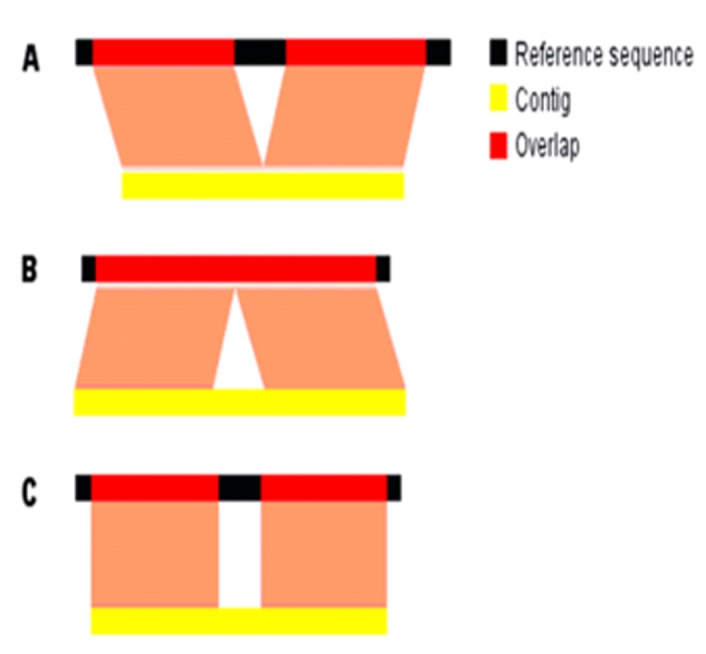
Possible alignments of the contigs against the reference sequence provoke multiple Blast alignments; A) Two regions of the contig align with two parts of the reference due to the deletion of a region in the contig; B) A region was inserted into the contig that is not in the reference; C). The central region of the contig does not align with the reference, which could indicate an error in assembly or sequence insertion.
The lack of significant alignments or alignment with the reference sequence may be the result of regions that are not present in FRC41 or because of assembly errors. In either case, G4ALL can use the gene product reports for each contig to identify the cause. After curation in G4ALL, of the 5,118 contigs that aligned significantly with the reference, 2,084 were selected for the production of the scaffold, resulting in 655 contigs (2,263,398 bases) oriented and ordered with respect to the reference, representing 96.81% of the reference genome (2,337,913 bases), when compared to the size of FRC41.
In the synteny analysis using the genome of C. pseudotuberculosis 316 and 258 (Figure 4), we observed a highly conserved gene order between genomes, which was expected since these are organisms of the same species and biovar.
Figure 4.
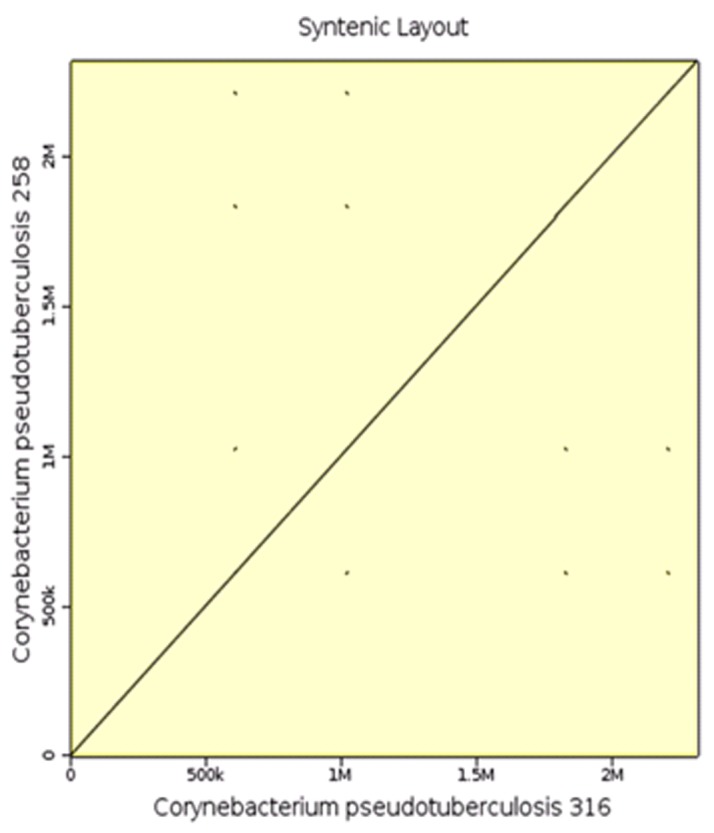
Analysis of synteny using the genome of C. pseudotuberculosis 316 and 258. We observed a highly conserved gene order between genomes.
Conclusion
G4ALL is a computational tool integrated with a database that allows the curation and extension of contigs produced by de novo assemblers, and the production of a scaffold, even when there is little overlap between the sequences (which can be validated by searching for homologies in biological databases). In addition, the tool allows various users to work on the project simultaneously, reducing the time needed for curation. This software has been successfully used in a number of projects involving organisms such as Archaea and viruses.
Supplementary material
Acknowledgments
This work was part of the Rede Paraense de Genômica e Proteômica supported by Fundação de Amparo a Pesquisa do Estado do Pará. A.S., M.P.S., A.R.C., P.H.C.G.S. and V.A. were supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). R.T.J.R. acknowledges support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Footnotes
Citation:Ramos et al, Bioinformation 9(11): 599-604 (2013)
References
- 1.Metzker ML, et al. Nature Reviews Genetics. 2009;11:31. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 2.Chaisson MJ, Pevzner PA. Genome Res. 2008;18:324. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster SC. Nat Methods. 2008;5:16. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Homer N. Brief Bioinform. 2010;11:473. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warren RL, et al. Bioinformatics. 2007;23:500. doi: 10.1093/bioinformatics/btl629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dohm JC, et al. Genome Res. 2007;17:1697. doi: 10.1101/gr.6435207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeck WR, et al. Bioinformatics. 2007;23:2942. doi: 10.1093/bioinformatics/btm451. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez D, et al. Genome Res. 2008;18:802. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margulies M, et al. Nature. 2005;437:376. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler J, et al. Genome Res. 2008;18:810. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zerbino DR, Birney E. Genome Res. 2008;8:821. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birney E. Nat Methods. 2011;8:59. doi: 10.1038/nmeth0111-59. [DOI] [PubMed] [Google Scholar]
- 13.Cerdeira LT. J Microbiol Methods. 2011;86:218. doi: 10.1016/j.mimet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Nijkamp J, et al. Bioinformatics. 2010;26:i433. doi: 10.1093/bioinformatics/btq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roach JC, et al. Genomics. 1995;26:345. doi: 10.1016/0888-7543(95)80219-c. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen CB, et al. Nat Methods. 2010;7:S5. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- 17.Ramos RT, et al. BMC Res Notes. 2011;18:130. doi: 10.1186/1756-0500-4-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JR, et al. Genomics. 2010;95:315. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos RT, et al. Microb Biotechnol. 2013;6:150. doi: 10.1111/1751-7915.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richter DC, et al. Bioinformatics. 2007;23:1573. doi: 10.1093/bioinformatics/btm153. [DOI] [PubMed] [Google Scholar]
- 21.Altschul SF, et al. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 22.Kurtz S, et al. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.