

Abstract
Striatal fast spiking interneurons (FSIs) modulate output of the striatum by synchronizing medium-sized spiny neurons (MSNs). Recent studies have broadened our understanding of FSIs, showing that they are implicated in severe motor disorders such as parkinsonism, dystonia and Tourette syndrome. FSIs are the only striatal neurons to express the calcium-binding protein parvalbumin (PV). This selective expression of PV raises questions about the functional role of this Ca2+ buffer in controlling FSI Ca2+ dynamics and, consequently, FSI spiking mode and neurotransmission. To study the functional involvement of FSIs in striatal microcircuit activity and the role of PV in FSI function, we performed perforated patch recordings on enhanced green fluorescent protein-expressing FSIs in brain slices from control and PV−/− mice. Our results revealed that PV−/− FSIs fired more regularly and were more excitable than control FSIs by a mechanism in which Ca2+ buffering is linked to spiking activity as a result of the activation of small conductance Ca2+-dependent K+ channels. A modelling approach of striatal FSIs supports our experimental results. Furthermore, PV deletion modified frequency-specific short-term plasticity at inhibitory FSI to MSN synapses. Our results therefore reinforce the hypothesis that in FSIs, PV is crucial for fine-tuning of the temporal responses of the FSI network and for the orchestration of MSN populations. This, in turn, may play a direct role in the generation and pathology-related worsening of motor rhythms.
Key points
Fast spiking interneurons (FSIs) modulate output of the striatum and are implicated in severe motor disorders.
Selective expression of the calcium-binding protein parvalbumin (PV) in FSIs raises questions about how PV controls FSI Ca2+ dynamics.
Here we report a novel mechanism linking PV-Ca2+ buffering and FSI spiking as a result of the activation of small conductance (SK) Ca2+-dependent K+ channels.
We also show that, at the presynaptic terminals, PV prevents synaptic facilitation at narrow frequencies at FSI to striatal output neuron synapses.
Our data establish that PV is a key element in providing rhythm generation in FSIs as well as filtering striatal output. Thus, FSI neuromodulation via PV and/or SK channels is an interesting target for controlling the establishment of oscillatory frequencies related to the induction or worsening of pathology-related motor rhythms.
Introduction
In vivo, striatal fast spiking interneuron (FSI) feed-forward inhibition controls spike timing and filters cortical input to the striatal output neurons, which are the medium-sized spiny neurons (MSNs) (Mallet et al. 2005). The fact that FSIs receive inputs from both motor and somatosensory cortices (Ramanathan et al. 2002), as well as from globus pallidus (Bevan et al. 1998), strongly supports their important role in sensorimotor integration. Shunting inhibition of the MSN cortical afferences by FSI activity renders MSNs silent during sensory stimulation (Pidoux et al. 2011) and epileptic spike-and-wave discharges (Slaght et al. 2004). FSIs possess unique characteristics in the striatal network such as deep spike after-hyperpolarization (AHP), non-accommodation discharge, narrow action potentials (APs), smooth dendrites (Kawaguchi, 1993; Plenz & Kitai, 1998; Koós & Tepper, 1999; Bracci et al. 2002) and a somatic control on MSN (Kita et al. 1990). Given this last property and despite being rather few in numbers, FSIs are able to potently suppress MSN activity across the striatum in both D1- and D2-MSN subpopulations (Planert et al. 2010), although apparently targeting preferentially the D1-population (Gittis et al. 2010). In dopamine depletion models, FSI influence seems to contribute to the imbalance of MSN spiking by inhibiting D2-MSNs more (Gittis et al. 2011) through a mechanism based on the generation of new synapses, instead of an increase in their discharge frequency. Moreover, a reduction in FSI number has been reported in Tourette syndrome (Kalanithi et al. 2005) and paroxysmal dystonia (Gernert et al. 2000), underlining the importance of FSI inhibition. Therefore, restoring and/or modulating FSI function might be viewed as a potential therapeutic target (Wiltschko et al. 2010).
FSIs selectively express parvalbumin (PV), which is a calcium-binding protein (CBP) of the EF-hand family. CBPs have long been considered as useful markers to identify neuronal populations in specific circuits in the brain (Baimbridge et al. 1992; Schwaller, 2009). In the last decade, experimental evidence demonstrating the functional role of the three most broadly distributed CBPs (PV, calbindin D-28k and calretinin) has grown considerably (Schwaller et al. 2002; Schwaller, 2010). Here, we took advantage of the perforated patch configuration of the patch-clamp technique to record striatal FSIs from wild-type (WT) and PV knockout mice (PV−/−) in coronal slices, without disturbing their endogenous Ca2+ buffering capacity. Our experimental and theoretical results provide evidence that PV in FSIs affects the precision of spike-timing and efferent short-term plasticity.
Methods
Ethical approval
The experiments and procedures conformed to the regulations of the Institutional Ethical Committee of the School of Medicine of the Université Libre de Bruxelles.
Animals
We used the PV–enhanced green fluorescent protein (EGFP) BAC transgenic mouse line (Meyer et al. 2002; kind gift of Dr H. Monyer, University of Heidelberg) that expresses EGFP under control of the PV promoter (mouse gene Pvalb), which facilitates the identification of striatal FSIs. PV-EGFP PV−/− mice were obtained by crossing these PV-EGFP mice with the constitutive knockout mice for PV (PV−/−; Schwaller et al. 1999), both lines having the same C57Bl/6J background. In the latter line, EGFP is expressed in the population of ‘PV-immunoreactive’ neurons, although the cells are devoid of PV expression. For each set of experiments presented in this work, recordings were obtained from at least 4–7 different animals per genotype and from 3–4 different litters. Thus, a set of experiments with n= 5 embraces at least four different animals per genotype.
Slices and identification of FSIs by their intrinsic fluorescence
Mice aged 18–24 days (P18–P24) were deeply anaesthetized with halothane and killed by decapitation. Careful dissection was performed and brains were rapidly immersed in ice-cold bicarbonate-buffered saline solution (BBS) at 4°C with the following composition (in mm): 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 26 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose equilibrated with a 95% O2/5% CO2 mixture (pH 7.3). Cortico-striatal coronal slices of 250 μm thickness were prepared with a Leica VT1000S vibratome and incubated in the same BBS at 32°C for at least 1 h before recordings and protected from light. At the end of this period, slices were placed in a recording chamber and submerged in a continuously flowing BBS at 22–24°C with a 1.5 ml min−1 flux rate. FSIs were visualized in the striatum with a 63× water immersion objective of a Zeiss upright microscope (Axioskop 2FS Plus; Zeiss, Oberkochen, Germany) and the intrinsic EGFP fluorescence was detected by using short pulses of blue light from a source composed of an LED array (OptoLED, Cairn Research, Faversham, UK) emitting at 488 nm and coupled to the microscope that was equipped with a dichroic mirror and a high-pass emission filter centred at 505 and 507 nm, respectively. FSIs were visualized with a back-illuminated CCD camera (iXon +; Andor Technology, Belfast, UK). Minimal illumination of the slices was used to reduce bleaching and phototoxicity. Once FSIs were identified, both FSIs and MSNs were targeted with the help of infrared differential interference contrast microscopy.
Electrophysiology
All recordings were made with a Dual EPC-10 operational amplifier and data were acquired using the software Patchmaster (HEKA, Lambrecht-Pfalz, Germany). Using the built-in EPC-10 filter, current-clamp recordings were filtered at 2.9 kHz and digitized at 10 kHz; voltage-clamp recordings were filtered at 2.9 kHz and digitized at 20 kHz. Patch pipettes were made from borosilicate glass capillaries (Hilgenberg GmbH, Malsfeld, Germany) with a two-stage vertical puller (PIP 5, HEKA). Pipette input resistances were in the 5–7 MΩ range.
FSI perforated patch recordings
Perforated patch recordings on FSIs were made with a pipette solution containing the following (in mm): 80 K2SO4, 10 NaCl, 15 glucose, 5 Hepes, 225–230 mOsm l−1 and 400 μg ml−1 amphotericin B (pH 7.2 corrected with KOH). Fresh stocks of the ionophore were prepared every 2 h (4 mg amphotericin B in 50 μl DMSO). This solution was vortexed for 10 min, immediately diluted in the perforated patch solution and protected from light. Leak current values were similar between WT and PV−/− groups (−40.65 ± 26.27 and −40.18 ± 16.54 pA, respectively, P= 0.98). Series resistance was monitored to ensure that voltage attenuation in current-clamp mode was always <10%. Due to the fact that FSIs presented spontaneous spiking in current clamp at zero current injected, the resting potential value was calculated in voltage clamp, as the voltage value at which leak current was zero. Other parameters were measured as indicated for whole-cell recordings. The rhythmicity of spontaneous FSI firing was evaluated by the coefficient of variation for inter-spike intervals (CVISI) as the square root of the ratio of ISI standard deviation to mean ISI.
FSI and MSN whole-cell recordings
When whole-cell recordings were performed on FSIs, pipettes were filled with a solution containing the following (in mm): 150 potassium gluconate, 4.6 MgCl2, 10 K-Hepes, 1 K-EGTA, 0.4 Na-GTP, 4 Na-ATP and 0.1 CaCl2 (pH 7.2). For some experiments, PV−/− FSIs were recorded also with a lower EGTA concentration (100 μm). For FSI–MSN pairs, MSNs were recorded in whole-cell configuration with a chloride-enriched internal solution to maximize the signal-to-noise ratio of postsynaptic events with the following composition (in mm): 150 CsCl, 4.6 MgCl2, 10 Hepes, 4 Na-ATP, 0.4 Na-GTP, 1 EGTA and 0.1 CaCl2 (pH 7.2). Tight giga seals (>2 GΩ) were obtained before rupture of the membrane patch. Holding potential was maintained at −70 and −90 mV for FSIs and MSNs, respectively. As in perforated patch, the resting potential value was calculated in voltage clamp, as the voltage value at which leak current was zero. Recordings were started only after a waiting period of 2 min. Subsequent hyperpolarizing pulses of −10 mV were applied to extract the passive cellular parameters by analysing the elicited current transients (D’Angelo et al. 1995). Thus, series resistance, membrane capacitance and input resistance were monitored during the entire experiment. Experiments with series resistance values >30 MΩ were discarded. All the experiments were performed at 20–23°C.
FSI intrinsic excitability
After 30-s recordings of spontaneous activity in current clamp without current injection, the resting membrane potential was set at −70 mV and steps of depolarizing current from 20 to 380 pA (20 pA increments and either 200 ms or 1 s duration) at 0.25 Hz were injected. AP frequency was measured by dividing the number of spikes by the pulse duration and the results were analysed in frequency vs. injected current plots. Once the spike threshold was reached, a regular spiking was evoked. Averages of sustained spike trains were used to dissect the AP morphology and to calculate: (1) the AP height as the difference between the positive peak and the minimal potential after the fast hyperpolarization; (2) the spike threshold as the sudden increase observed in a plot of the time derivative of the AP (dV/dt) during the depolarizing ramp of the AP, with the chosen value being the voltage at which the derivative experienced a threefold increase; (3) the AP duration as the full-width at half maximum (FWHM) from a Gaussian fit of the depolarized face of the AP immediately after the threshold; and (4) the amplitude of fast hyperpolarization as the difference between threshold and the peak of the fast hyperpolarization. The kinetics of the recovery phase following the action potential AHP was determined during the first 6 ms following the time point at which the membrane potential reached its minimum. This short period was well fitted with mono-exponential functions and avoided the interference of depolarizing currents that follow the AHP and trigger a new AP.
Paired recordings
To test functionally connected FSI–MSN pairs, we first evoked 20 presynaptic depolarizing steps from the holding potential to −10 mV over 1 ms at 0.25 Hz. The postsynaptic MSN was maintained at the holding potential of −90 mV. This protocol evokes a presynaptic transient inward current or ‘active current’ that represents the activation of sodium currents. Previous evidence demonstrated that those currents propagate along the axon and evoke Ca2+ transients in axon terminals of cerebellar interneurons (Tan & Llano, 1999) and Purkinje cell recurrent collaterals (Orduz & Llano, 2007). Moreover, the protocol permits precise timing of single presynaptic stimulation in FSIs. Averages of those traces revealed in successful pairs the presence of corresponding inhibitory postsynaptic currents (IPSCs) in the postsynaptic MSNs. If no IPSCs were recorded in the patched MSNs, other nearby postsynaptic MSNs were patched. Identified connected pairs were continually recorded at 0.25 Hz to study unitary properties of IPSCs, paired-pulse ratio (PPR) and trains. The amplitude of IPSCs during PPR and trains was calculated by subtracting a baseline value from the measured peaks. Amplitudes for the second IPSC were obtained from the exponential fit to the decay of the preceding event. Soma to soma distances between recorded FSI–MSN connected pairs were similar for WT (34.1 ± 3.47 μm, n= 9) and PV−/− slices (36.13 ± 3.99 μm, n= 10; P= 0.7).
Miniature current analysis
For the detection of miniature currents in voltage clamp at a holding potential of −70 mV in our perforated recordings of FSIs, we first determined the threshold detection for positive events as twice the standard deviation of the baseline noise in a period of 50 ms. Once this value was calculated, we introduced it into a template procedure of the package Neuromatic (Jason Rothman, UCL, London, UK) that permitted us to quantify frequencies and amplitudes of those miniature synaptic events. Analysed recordings lasted from 30 s to 2 min for each FSI.
Immunocytochemistry and biocytin labelling
For morphological reconstructions, whole-cell solutions for FSIs and MSNs were complemented with 0.4% biocytin. Two conditions were maintained to improve labelling: a period of 20 min after whole-cell break-in and a high-resistance outside-out patch obtained when the pipette was withdrawn. Slices were fixed by overnight immersion in 4% paraformaldehyde at 4°C, rinsed in PBS at 0.1 mm and, later, immersed for 2 h periods in a mixture of PBS/Triton X-100 (0.1 mm and 0.1%, respectively). Biocytin was revealed in red with streptavidin-conjugated NL557 (R&D systems, Minneapolis, MN, USA), diluted 1:5000, always in PBS/Triton X-100. After a final rinse in TBS, slices were mounted on coverslips with FlourSave reagent (EMD Millipore Corporation, Billerica, MA, USA) and secured with nail polish.
Histology and immunostaining
PV immunostainings were performed on 35 μm coronal cortico-striatal sections prepared from fixed brains of PV-EGFP mice, previously perfused with paraformaldehyde (4%). Primary antibody incubations were performed at 4°C for 48 h, using rabbit anti-PV 25 (1:500, Swant, Marly, Switzerland). Secondary antibody incubations were performed for 1 h at room temperature with donkey anti-rabbit 549 (1:100, Jackson ImmunoResearch, West Grove, PA, USA). Co-localization of endogenous EGFP (green) and PV (red fluorescence) was evaluated on confocal images obtained with a laser scanning confocal microscope (see below). Image J software (http://imagej.nih.gov/ij/) was used to quantify the number of EGFP+-expressing PV neurons (and vice versa) and to confirm, by a line analysis on single neurons, the somatic co-localization of both green and red fluorescent signals.
Confocal microscopy
Confocal images were acquired using an Axiovert 200M-LSM 510 META microscope (Zeiss, Oberkochen, Germany) equipped with two objectives: a Plan-Neofluar 10×/0.3 W or a C-Apochromat 40×/1.2, and 488 nm argon and 543 nm helium–neon lasers. Band-pass emission filters were used for selective detection of the endogenous EGFP (500–550 nm) and red-biocytin labelling (565–615 nm). Single FSI and FSI–MSN pair images were acquired as 50–70 μm-thick Z-stacks composed of 2048 × 2048 pixel images (pixel size 0.22 μm) with a Z-step of 0.62 μm. Cortico-striatal region images were acquired as 30 μm-thick Z-stacks composed of 1300 × 1300 pixel images (pixel size 0.63 μm) with a Z-step of 5 μm.
Data analysis and statistics
Data analyses were performed using the Neuromatic software package (http://www.neuromatic.thinkrandom.com/) and custom routines within the IgorPro environment (Wavemetrics, Lake Oswego, OR, USA). All values are expressed as mean ± SEM. Statistical tests were performed using Student's t test within Excel software package (Microsoft, Redmond, WA, USA) and repeated-measures analyses of variance (ANOVA) were performed using Prism 4.0 (GraphPad Software, Inc., USA). The significance level was established at P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) and when P > 0.05, not significant (NS).
Drugs and reagents
All drugs were dissolved in the bath solution and were applied to the preparation by superfusion. The drug solution reached a steady-state concentration in the experimental chamber within 2 min. After reaching this steady-state period, the response to the drug was measured after a prolonged application (up to 5 min). Apamin and iberiotoxin were purchased from Tocris Bioscience (Bristol, UK) and other reagents and drugs from Sigma-Aldrich (St Louis, MO, USA). Stocks of apamin, gabazine, iberiotoxin and NBQX (2,3-dihydroxy- 6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione) were prepared in water and stored at −20°C. They were diluted in BBS before bath application.
Computational model of striatal FSIs
We have used a single compartment model of FSIs (Erisir et al. 1999), as modified by Bischop et al. (2012). The ionic currents of the Bischop model consist of a fast transient sodium current (INa), a fast delayed rectifier potassium current of the Kv3.1 type (IKv3), a slow delayed rectifier potassium current of the Kv1.3 type (IKv1), a high-voltage-activated Ca2+ current (ICa), a small conductance (SK) potassium current (ISK) and a passive leak current (Ileak). The model includes the presence of a Ca2+ buffer similar to PV. The equation governing the membrane potential dynamics is given by:
where Cm and V are, respectively, the membrane capacitance and the membrane potential of the FSI and Iapp is an external applied current. The ionic currents are given by:
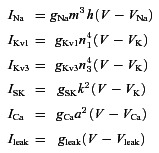 |
The equations governing the gating variables and all the parameters are the same as in Bischop et al. (2012) except for the SK conductance for which we used gSK= 4 nS (instead of 2 nS). Coupling of the Ca2+ and buffer dynamics is described by the following equations:
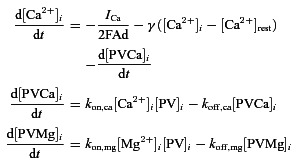 |
[Ca2+]i and [PV]i represent, respectively, the free intracellular Ca2+ concentration and the concentration of free PV. [PVCa2+]i and [PVMg2+]i are the concentrations of PV bound to Ca2+ and Mg2+. We assume that [Mg2+]i is constant, as in Lee et al. (2000). kon,ca, koff,ca, kon,mg and koff,mg are the association and dissociation constants for PV with Ca2+ and Mg2+. We consider Ca2+ fluxes across a shell of thickness d= 0.2 μm beneath the cell surface (area A= 3000 μm2). The inward flux is ICa/2FAd (F is the Faraday constant). The term γ([Ca2+]i−[Ca2+]rest) is the clearance mechanism associated with the Ca2+ fluxes across the plasma membrane or storage organelles. All the parameters are as in Bischop et al. (2012) except for the resting (basal) Ca2+ level, for which we use [Ca2+]rest= 0.93 μm. Both spontaneous spiking and membrane oscillations resulted by injecting current pulses and/or by decreasing the leak reversal potential. To better mimic electrophysiological recordings, in the case of irregular stuttering, we added a Gaussian white noise to the applied current (zero mean, unit variance). The equations of the model are numerically solved using a fourth-order Runge–Kutta integration method (Press et al. 1992). The bifurcation diagrams were obtained with the software XPPAUT 6.10 (Free Software Foundation Inc., Cambridge, MA, USA) using the applied current (Iapp) as a bifurcation parameter. In our model we have simulated three experimental conditions: (1) PV = 1000 μm, gSK= 4 nS (WT phenotype, Fig. 4D); (2) PV = 0 μm, gSK= 4 nS (PV−/− phenotype, Fig. 4E); and (3) PV = 1000 μm, gSK= 0 nS (WT phenotype after apamin application, Fig. 4F). For a full mathematical analysis of our FSI computational model, we refer readers to Bischop et al. (2012) and Ermentrout and Wechselberger (2009).
Figure 4. The absence of parvalbumin regularizes spontaneous spiking of striatal FSIs.
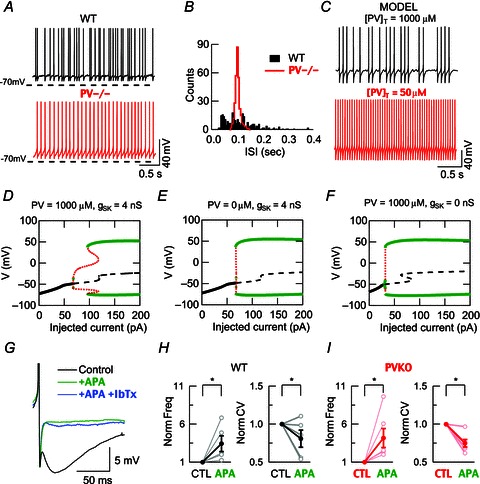
A–C, comparison of FSIs from WT and PV−/− mice recorded using perforated patch recordings and a computational model for FSIs with high and low PV concentrations. A, FSI spontaneous spiking (3 s traces). B, ISI histograms from 30 s recordings from cells recorded in A (5 ms bins). The ISI distribution is much narrower in PV−/− FSIs than in WT neurons. C, spontaneous spiking is obtained in a computational model of FSIs by using Iapp= 60 pA. At a PV concentration of 1000 μm, the spiking appears irregular (top), whereas a reduction to 50 μm converts it to a regular spiking. D–F, bifurcation diagrams for three different conditions: D, high PV buffering with SK conductance; E, low PV buffering with SK conductance; and F, low PV buffering without SK conductance. The absence of PV as well as the absence of SK conductance leads to the disappearance of a stuttering firing in 60–90 pA current range. G, example of spontaneous APs in a WT FSI during control conditions (black trace) and during the consecutive bath addition of apamin (250 nm, green trace) and iberiotoxin (200 nm, blue trace). An apamin-sensitive current is evident without any additional modification after the BK blocker. H and I, similar changes in normalized values of spontaneous spiking frequency and calculated CVs were observed during apamin application in WT FSIs as well as in PV-deficient (PV−/−) FSIs.
Results
PV expression identifies FSIs in rodent striatum
PV-expressing cells constitute only 1–5% of total striatal neurons (Kita et al. 1990; Luk & Sadikot, 2001), which reduces considerably the probability to target these neurons by recording blindly in slices. For this reason we took advantage of the PV-EGFP BAC transgenic mouse strain (Meyer et al. 2002) that expresses EGFP under control of the PV (Pvalb gene) promoter. We determined that 92.7% of the total PV-immunoreactive (PV-ir) striatal neurons co-localized also with EGFP (Fig. 1) and that 93.4% of the EGFP-expressing neurons also stained positive for PV. Co-labelling was easily identified at both somatic (Fig. 1B) and dendritic levels (Fig. 1C). These mice were used as control animals. We then produced PV-EGFP PV−/− mice by crossing the PV-EGFP line with constitutive knockout mice for PV (PV−/−; Schwaller et al. 1999) making it possible to identify both PV-containing and PV-deficient neurons within the FSI population (by their EGFP expression).
Figure 1. Identification of striatal FSIs in coronal slices from PV-EGFP mice.
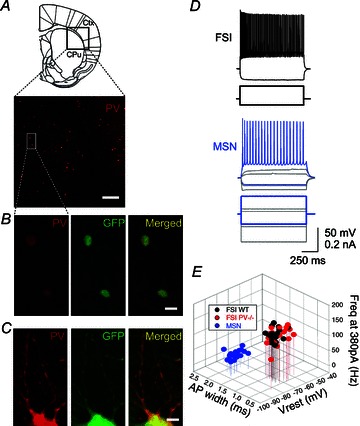
A, image of a cortico-striatal region from a coronal slice labelled with anti-PV antibodies revealing PV-ir cells as well-defined red spots (10× magnification; scale bar 200 μm); the schema of a coronal slice (top) shows the region of interest containing the striatum (CPu) and the adjacent cortex (Ctx). B, two PV-ir cells also express EGFP (green) in their somata, resulting in a yellow colour in the merged image (40×, scale bar 20 μm). C, PV and EGFP co-expression is also observed at the level of dendritic processes (scale bar 5 μm). D, a typical electrophysiological response to hyper- and depolarizing pulses demonstrating that the visually identified EGFP-ir neurons show the well-defined firing pattern of striatal FSIs (top). EGFP-negative MSNs are also clearly recognized by their lower frequency spiking to similar supra-threshold current depolarizations, their sub-threshold depolarization ramps and an inward rectification during hyperpolarizations (bottom). E, 3-D plot of maximal frequency rates at 380 pA, AP widths and resting membrane potentials (Vrest) to distinguish the firing properties of FSIs vs. MSNs. Two different clusters appeared with no difference between FSIs from WT and PV−/−.
Visually identified EGFP-positive cells were subjected to perforated patch recordings and neurons from both genotypes showed the typical electrophysiological signature of striatal FSIs (Table 1). Three parameters were considered as prototypical traces of FSIs compared to other neurons in the striatal circuit (Kawaguchi et al. 1993; Gittis et al. 2010), i.e. narrow APs, sustained non-adaptive spiking at high frequencies during current injection steps and relatively depolarized resting potentials compared to MSNs (Table 1). A typical FSI electrophysiological response to hyper- and depolarizing pulses shows no hyperpolarization-activated currents and a train of sustained APs, respectively (Fig. 1D, upper trace). In the same slice, unlike FSIs, an MSN presented hyperpolarization-activated currents and slower AP frequencies (bottom trace). PV-EGFP PV−/− FSIs showed the same main characteristics without any statistically significant differences with respect to neurons of WT controls (regarding PV expression) concerning their electrophysiological FSI profile (Fig. 1E, Table 1).
Table 1.
Intrinsic properties of FSIs from PV-EGFP WT and PV-EGFP PV−/− mice
| n | WT 19 | PV−/− 20 |
|---|---|---|
| Vrest (mV) | −68.22 ± 1.38 | −69.78 ± 0.73 |
| Rin (GΩ) | 0.38 ± 0.03 | 0.38 ± 0.02 |
| Cm (pF) | 29.02 ± 1.45 | 29.45 ± 0.73 |
| FWHM of AP (ms) | 0.739 ± 0.03 | 0.814 ± 0.03 |
| AP threshold (mV) | −42.71 ± 1.59 | −44.09 ± 1.99 |
| Frequency at 380 pA step (Hz) | 119.26 ± 6.03 | 119.75 ± 5.47 |
| Rheobase (pA) | 47.5 ± 2.23 | 40 ± 5.16 |
All parameters were measured with perforated patch recordings and no significant differences were observed between the two genotypes by Student's t tests. Resting potential (Vrest), input resistance (Rin) and membrane capacitance (Cm) were measured in voltage clamp; full-width at half maximum (FWHM), AP threshold, rheobase and the frequency of AP were measured in current clamp. Data are presented as mean ± SEM.
Absence of PV enhances electroresponsiveness of striatal FSIs
Next we investigated whether PV is able to interfere with FSI firing patterns under conditions of sustained depolarizations. In the same recording mode (perforated patch), the relationship between spiking frequencies and injected current was determined at a membrane potential of −70 mV and by injecting 200 ms steps of depolarizing current from 0 to 380 pA, in steps of 20 pA. Once the injected current reached the threshold for AP firing, higher frequencies at a given injected current were observed in PV−/− neurons compared to control neurons. At the same supra-threshold pulse (80 pA) at which PV−/− neurons fired at 51.74 ± 9.34 Hz (n= 6, from six mice), WT FSIs fired only at 25.9 ± 4.5 Hz (n= 5, from four mice, P < 0.05, Fig. 2A). Note that at maximal injected currents (380 pA), the average spiking frequency was not statistically different for the two groups (see Table 1). In fact, in some FSIs, accommodation or small pauses appeared at the last segment of those maximal pulses. We therefore centred our analysis of FSI excitability on the first injection pulses that presented a nearly linear relationship between AP frequency and injected current. Analysis of the first five pulses permitted us to calculate the excitability slope (see line fits in Fig. 2B). The linear part of the current–frequency plot was steeper for PV−/− (0.91 ± 0.13 Hz pA−1) than for WT FSIs (0.55 ± 0.03 Hz pA−1; P < 0.05), i.e. an increase of 65%. We hypothesized that those changes in excitability slopes were related to altered Ca2+ buffering. To confirm this hypothesis, we performed numerical simulations in the FSI mathematical model. In response to a 200-ms step current injection of 140 pA, we observed an increase in excitability (higher frequency) as the total PV concentration dropped from 1000 to 0 μm. The mean spiking frequency rose from 35 to 55 Hz (Fig. 2F), similar to the effect observed in perforated patch recordings in current-clamp mode (cf. Fig. 2A). Additionally, in the absence of PV, the model predicted an increase in the slope of 58% for the same injected currents, in good agreement with the experimental data.
Figure 2. Parvalbumin deletion enhances FSI electroresponsiveness.
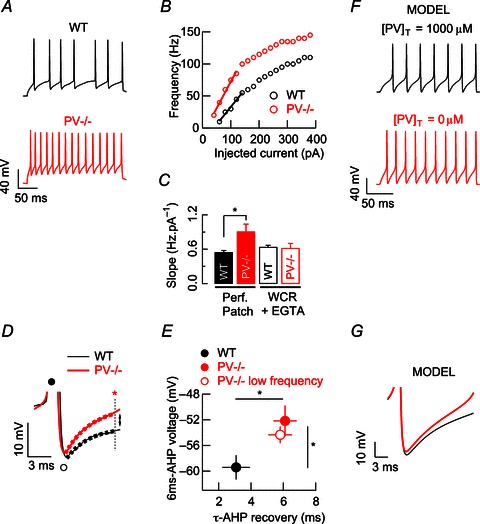
A, examples of action potentials (AP) evoked by a 200 ms suprathreshold pulse during perforated-patch recordings of WT and PV−/− FSIs. B, spiking frequency vs. injected current plots for the two FSIs displayed in A. C, pooled data for FSI excitability slopes during both perforated-patch recordings and whole-cell recordings (WCR, 1 mm EGTA). In PV−/− FSIs, restoration of somatic Ca2+ buffering by EGTA via WCR-patch pipette re-established slope values to those observed in WT FSI perforated-patch recordings. D, superimposed AP averages from A illustrating the afterhyperpolarization (AHP) following AP. Filled and open circles are placed at the positive and negative peaks of the averaged AP, respectively. At 6 ms from the negative peak of the AP (red star) PV−/− FSIs are less hyperpolarized than WT FSIs. The same colour code applies in A to D. E, pooled data for voltage values at 6 ms of AHP recovery (red star in D) and its time constant during the same suprathreshold pulse for WT and PV−/− (filled circles). A rate-matched analysis of AP demonstrates that in AP from PV−/− obtained at lower frequencies (red open circle), both parameters remain different from those recorded in WT FSIs (filled circle). F, in our mathematical model of FSI, the absence of PV enhances spiking frequency, as observed with the experimental data. G, in the model, averages of AP show less hyperpolarization of the AHP when PV is not present (red trace). *P < 0.05, Student's t test.
Moreover, to determine experimentally whether those changes were related to altered Ca2+ buffering we performed whole-cell recordings to dialyse both groups of FSIs with the same exogenous Ca2+ buffer concentration. Restoration of the excitability profile of neurons has been previously demonstrated for calretinin-deficient cerebellar granule cells by adding the fast buffer BAPTA (Gall et al. 2003). Thus, we added an exogenous buffer to the pipette solution with reportedly similar Ca2+ binding kinetics to PV (EGTA, 1 mm). Equal intracellular Ca2+ buffering resulted in almost identical excitability slopes for WT (0.62 ± 0.08 Hz pA−1, n= 6) and PV−/− (0.64 ± 0.03 Hz pA−1, n= 10) FSIs, obliterating any significant difference (P= 0.77) and thus converting the excitability of PV−/− FSIs to the one seen in WT neurons (Fig. 2C). Likewise, the fact that WT FSI excitability slope values from whole-cell recordings were similar to those from perforated patch recordings indicated that the deletion of PV rendered FSIs more excitable through a modification of a Ca2+ buffering-dependent mechanism.
A more detailed analysis of AP shape was performed to better understand the altered electroresponsiveness of PV−/− FSIs. For this, we analysed evoked APs from trains recorded in response to the same injected current (Fig. 2A). Averaged APs from those trains obtained in PV−/− neurons showed a slower AHP recovery, but reached a more depolarized potential compared to WT FSI (Fig. 2D and E). At 6 ms after reaching the minimal voltage during the AHP, the potential was clearly more depolarized in PV−/− FSIs (−52.16 ± 2.41 mV, n= 6) than in WT neurons (−59.42 ± 1.88 mV, n= 5; P < 0.05; red star and dotted line in Fig. 2D). AHP recovery could be fitted by a mono-exponential function and time constants (τ) were clearly larger for PV−/− FSIs (6.1 ± 0.71 ms, n= 6) than for WT neurons (3.08 ± 0.85 ms, n= 5; P < 0.05). As shown in Fig. 2G, our FSI mathematical model allowed us to link the observed AHP alterations to the absence of PV, as it predicts a slower AHP recovery and a more depolarized potential in the absence of PV compared to high (1000 μm) PV concentration. This more depolarized potential due to a decreased SK activation in PV−/− FSI favours the generation of a new spike and consequently results in an increase in the firing frequency.
Given that faster spike rates are associated with shorter AHP, we performed a rate-matched AP analysis to confirm that the observed differences in the AHP recovery were not a frequency artifact. First, we looked at pulses of currents, at which PV−/− neurons discharged at similar frequencies to those of WT. The selected pulses in PV−/− evoked trains of 23.33 ± 2.78 Hz (n= 6), which were not different from WT neurons: 25.9 ± 4.5 Hz (n= 5; P= 0.62). Evaluation of AP averages of those selected pulses in PV−/− cells (Fig. 2E) revealed that (1) the voltage at 6 ms after reaching the minimal voltage during the AHP was more depolarized than in WT neurons (−54.33 ± 1.3 mV, n= 6; P < 0.05) and (2) the time constant (τ) of the AHP recovery also remained larger (5.8 ± 0.73 ms, n= 6; P < 0.05) when PV−/− spikes were obtained at lower frequencies.
Next we investigated how Ca2+ buffering might affect excitability in FSIs. Because AHP is sustained by Ca2+-dependent currents in most neurons (Stocker et al. 2004), we explored the probable modulation of Ca2+-dependent potassium channels in WT FSIs (Gall et al. 2003). Thus, whole-cell recordings using the same Ca2+ buffering conditions (1 mm EGTA in the pipette solution) were performed in both WT and PV−/− FSIs in the presence of two inhibitors: the SK-channel blocker apamin (250 nm) and the large conductance (BK)-channel blocker iberiotoxin (200 nm). Trains of repetitive APs during 40 pA depolarizing supra-threshold current pulses from −70 mV (Fig. 3A) revealed that apamin strongly increased AP frequency in WT FSIs (52.33 ± 8 Hz, n= 6) compared to untreated control FSIs (28.66 ± 4.15 Hz; P < 0.05). The subsequent addition of iberiotoxin did not induce a further effect on AP frequency (51.66 ± 3.13 Hz; P= 0.93). In some experiments, we tested iberiotoxin alone and no effect was observed, either on frequencies (27.33 ± 7.51 vs. 27.66 ± 6.74 Hz, n= 3; P= 0.97) or on excitability slopes (0.58 ± 0.02 vs. 0.64 ± 0.07, n= 3; P= 0.51). These results are in agreement with a recent study in the rat, in which BK blockers do not influence AP duration in striatal FSIs (Sciamanna & Wilson, 2011).
Figure 3. SK channels are involved in FSI excitability control.
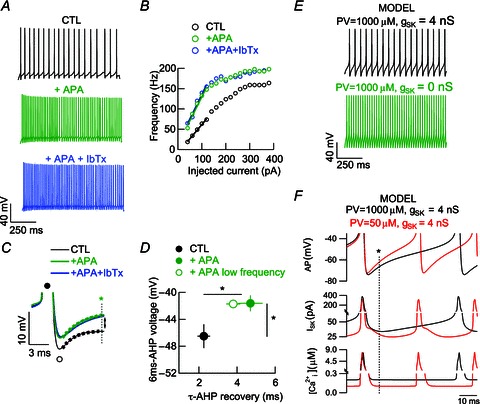
A, examples of evoked AP by a 1 s suprathreshold pulse in WT FSIs in whole-cell recordings in the presence of the SK blocker apamin and BK blocker iberiotoxin. B, spiking frequency vs. injected current plots for the FSI displayed in A. C, superimposed AP averages from A. The traces are displayed similarly as in Fig. 2D. At 6 ms from the negative peak of the AP (green star) apamin (green trace) caused a faster recovery from AHP in FSIs in comparison to untreated control FSIs (black trace). Consecutive addition of iberiotoxin did not exert a further visible effect. The same colour code applies in A to C. D, pooled data for voltage values at 6 ms of AHP recovery (green star in C) and its time constant during the same suprathreshold pulse for control and apamin groups (filled circles). Analysis of AP from apamin series at lower frequencies (similar to controls) revealed no frequency-dependent differences (green open circle). E, in our mathematical model of FSIs, suppression of SK conductance mimics the apamin-mediated enhancement of frequency in the presence of PV. F, time-course of SK conductance (middle lane) and [Ca2+]i (lower lane) in comparison to the AP shape (upper lane) in the FSI model. Note that a strong reduction of buffering (low PV, red trace) leads to a more depolarized AHP (black star and dotted line) that is related to a decrease in the SK current and a decrease in [Ca2+]i.
In WT FSIs, blockage of SK channels by apamin significantly enhanced FSI excitability (slope values 0.41 ± 0.04 Hz pA−1 in controls vs. 0.81 ± 0.08 Hz pA−1 in the presence of apamin, n= 6; P < 0.01; Fig. 3B) with no further alterations after iberiotoxin administration (0.7 ± 0.03 Hz pA−1, n= 6; P= 0.24). In PV−/− FSIs, slopes were also steeper after the addition of apamin (0.54 ± 0.06 in untreated controls vs. 0.71 ± 0.01 after apamin application, n= 5; P < 0.05).
Analysis of AP shape in WT neurons demonstrated that the changes in the spiking frequency induced by the blockade of the SK channels were accompanied by (1) a more depolarized state 6 ms after the AHP minimum (−41.61 ± 1.17 mV, n= 6) when compared to untreated control FSIs (−46.49 ± 1.77 mV, n= 6; P < 0.05; Fig. 3C), and (2) a slower AHP recovery with τ-values being larger in the presence of apamin (4.07 ± 0.64 ms, n= 6) than in controls (2.25 ± 0.33 ms, n= 6; P < 0.01). Also in these experiments, iberiotoxin did not lead to any additional changes, either in AHP recovery (−43.58 ± 0.96 mV; P= 0.22) or in τ (5.91 ± 1.24 ms; P= 0.4).
To confirm that the apamin effect on the AHP recovery was not only due to a higher frequency of spiking (because faster spike rates are associated with shorter AHP), we performed a rate-matched AP analysis on AP trains during apamin application, which presented similar frequencies to those from controls (Fig. 3D, similar to the analysis in Fig. 2E). At those frequencies (30.75 ± 8.98 Hz, n= 6; P= 0.84), apamin-averaged AP continued to present stronger voltage depolarization at 6 ms after the AHP minimum (−41.71 ± 0.47 mV, n= 6; P < 0.05) and larger τ values upon AHP recovery (3.8 ± 0.37 ms, n= 6; P < 0.05).
To further explore how Ca2+ buffering might affect excitability in FSIs, we performed computer simulations mimicking the action of the SK channel blocker apamin in the FSI mathematical model. Decreasing the SK conductance gSK from 4 to 0 nS resulted in an increase in the firing frequency of FSIs, as observed in whole-cell recording experiments after the addition of apamin (cf. Fig. 3A). This change in frequency is similar to that observed by decreasing the PV concentration (Fig. 2F) and is thus suggestive of a possible modulation of SK channels by PV. Results from the mathematical model are presented and include the time course of the intracellular Ca2+ concentration, the SK currents during the AP train and the AHP phase between APs (Fig. 3F). Between two APs, the residual Ca2+ concentration is higher in the presence of 1000 μm PV than at a low (50 μm) PV concentration. This results in stronger activation of SK channels between APs and thus a longer AHP phase. As shown previously (Bischop et al. 2012), PV acts as a Ca2+ source between two APs, thereby prolonging the duration of the AHP. In conclusion, the SK blocker apamin causes alterations in FSI excitability resembling those observed in PV−/− FSIs and strongly supports a crucial modulating role of PV on SK channels during FSI spiking.
PV is involved in the regularity of spontaneous FSI firing
During perforated patch recordings in current-clamp mode, FSIs presented spontaneous APs (Fig. 4A) at relatively low frequencies that were similar in WT (n= 5, from four mice) and PV−/− neurons (n= 6, from six mice): 8.88 ± 2.18 and 12.05 ± 2.31 Hz, respectively (P= 0.34). However, a more regular, rhythmic spiking pattern was observed in PV−/− cells (Fig. 4A). To quantify the extent of this spiking rhythmicity, we determined the inter-spike interval (ISI) distribution for 30 s recordings of spiking FSIs. Histograms for ISI values in PV−/− neurons revealed a much narrower distribution with a maximum around 100 ms, while WT neurons had a much broader ISI distribution (Fig. 4B). In addition, CVISI was significantly smaller for PV−/− FSI (0.55 ± 0.08) than for WT neurons (0.79 ± 0.05; P < 0.05).
Similar results were obtained by simulating spontaneous firing using the FSI mathematical model. At high PV concentration (1000 μm), neurons exhibit stuttering behaviour consisting of brief bursts of APs separated by quiescent periods, which are characterized by sub-threshold membrane potential oscillations (Fig. 4C). Lowering the PV concentration to 50 μm, FSIs exhibit a regular firing pattern (Fig. 4C). To further investigate the role of PV on the regularity of the firing pattern of FSIs, we analysed the dynamic behaviour of the FSI computational model. We have plotted bifurcation diagrams for three conditions (see Methods) representing the WT phenotype (Fig. 4D), the PV−/− phenotype (Fig. 4E) and the WT phenotype after apamin application (Fig. 4F). In the WT, when Iapp > 100 pA the FSIs exhibit a regular firing pattern. In contrast, for Iapp between 70 and 100 pA, the cells exhibit stuttering behaviour (Fig. 4D). In the second and third conditions the intermediate range of applied currents responsible for the stuttering behaviours shrinks to a few picoamps in the PV−/− and WT + apamin conditions (see zone of red traces in Fig. 4E and F). This might therefore explain the more regular spiking pattern observed in PV−/− FSIs during perforated-patch recordings in current-clamp mode. Interestingly, when frequency-matched analyses of CVISI were performed for depolarizing pulses at four representative points of the excitability slope during perforated patch recordings (25, 50, 75 and 100 Hz), WT FSIs revealed higher CVISI values than PV−/− FSIs: 0.55 ± 0.06 (n= 5) vs. 0.31 ± 0.05 (n= 6), respectively (P= 0.022), only at the lowest frequency tested (25 Hz). This difference was not observed for the other three higher frequencies: 50 Hz (0.41 ± 0.06 vs. 0.38 ± 0.04, P= 0.72), 75 Hz (0.45 ± 0.07 vs. 0.37 ± 0.02, P= 0.25) and 100 Hz (0.35 ± 0.04 vs. 0.33 ± 0.02, P= 0.77). These results are in good agreement with our modelling prediction: in spontaneously spiking FSIs or during slightly depolarized states (Iapp 70–100 pA), both PV and functional SK channels cause a more arrhythmic spike-time pattern, which is correlated with the instability region observed in the bifurcation analyses (compare Fig. 4D and Fig. 4E, F).
Consistent with our results on the induced firing pattern, spontaneous spiking was sensitive to apamin in both groups (WT and PV−/−) and insensitive to iberiotoxin (Fig. 4G). An apamin-induced increase in FSI frequency to 343 ± 104% (P < 0.05) and 418 ± 124% (P < 0.05) for WT and PV−/−, respectively (Fig. 4H left and Fig. 4I left), was accompanied by a reduction in the CV, which decreased to 80.9 ± 11.31% (P < 0.05) and 74.87 ± 4.9% (P < 0.05), respectively (Fig. 4H right and Fig. 4I right). This demonstrates that SK channels are functional in both genotypes and that they exert similar effects on the frequency of spiking.
To exclude that a change of firing patterns between WT and PV−/− FSIs resulted from a change in the background synaptic noise, we analysed the spontaneous miniature currents at a holding potential of −70 mV during perforated patch recordings. At this voltage, the recorded miniature currents were mainly due to the activation of AMPA-dependent receptors (Allene et al. 2012), disappearing totally after NBQX application. Miniature excitatory postsynaptic currents displayed similar frequencies (5.08 ± 0.46 vs. 4.6 ± 1.09 Hz; P= 0.7) and amplitudes (−13.68 ± 1.76 vs. −16.91 ± 1.88 pA; P= 0.28) for WT (n= 6; from five mice) and PV−/− (n= 7; from four mice), respectively. These results essentially exclude a synaptic influence on FSI spontaneous spiking and rather reinforce the hypothesis that PV constitutes a key regulator of rhythmicity in striatal FSIs.
Identification of FSI–MSN pairs
Reconstructions of a series of biocytin-loaded connected FSI–MSN pairs subjected to dual whole-cell recordings were carried out to show the identity of the EGFP-ir cells as FSIs (Fig. 5B). A positive correlation between the absence of dendritic spines and EGFP expression (besides the typical electrophysiological profile of presynaptic FSIs) was observed when compared to postsynaptic MSNs. Only the biocityn-loaded presynaptic aspiny FSIs were EGFP-ir (Fig. 5C), while the second biocytin-loaded EGFP-negative soma corresponded to a postsynaptic MSN (Fig. 5B, inset on the right and Fig. 5D). As reported by others (Plenz & Kitai, 1998), somata diameters were larger for FSIs than for MSNs in connected pairs of both genotypes (13.5 ± 0.79 vs. 10.32 ± 0.52 μm for WT FSIs vs. MSNs, respectively, n= 9; P < 0.01; from five mice) and for PV−/− neurons (12.69 ± 0.73 vs. 9.82 ± 0.38 μm, n= 10; P < 0.01; from six mice).
Figure 5. PV-induced modifications of FSIs to MSN inhibitory short-term plasticity are restricted to a narrow time interval.
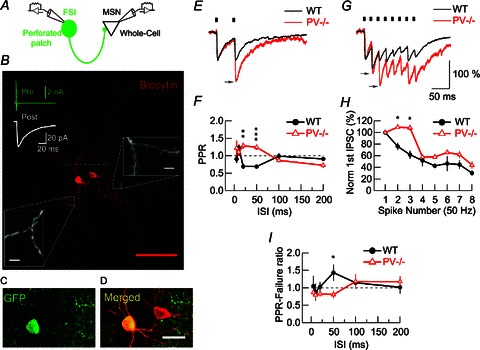
A, recording configuration used to study synaptically connected FSI–MSN pairs. B, confocal image of a biocytin (red)-loaded FSI–MSN connected pair. Seventy-nine images taken at incremental z-axis coordinates were superimposed. Red scale bar: 100 μm. In the top image, averages of 50 tests of action currents generated in the presynaptic FSI by depolarizing steps (green trace) and the corresponding averaged evoked IPSC in the connected MSN (white trace). A higher magnification of the dendrites of both cells allows us to validate the aspiny nature of FSI dendrites (left inset) compared to the spiny MSN dendrites (right inset). White scale bars: 2 μm. C and D, higher magnification of the boxed region (dotted white lines) in B: EGFP (C) and biocytin (D) co-localize selectively in the presynaptic FSI neuron (yellow), while biocytin alone (red) is present in the MSN. Scale bar: 25 μm. E, averaged IPSC from a PPR at 50 ms intervals. Small black bars at the top of the traces denote the time of presynaptic stimulation. The grey arrow indicates a pronounced increase of the second IPSC amplitude in the PV−/− trace. F, pooled data for all PPR protocols. WT, n= 5 pairs; PV−/−, n= 6 pairs. G, averaged IPSC evoked by a 50 Hz train. Grey arrows indicate the increase of the second and third IPSC in the PV−/− trace. H, pooled data for IPSC amplitudes during 50 Hz trains. WT, n= 5 pairs; PV−/−, n= 5 pairs. The second and third IPSCs are significantly larger in the PV−/− MSNs compared to WT MSNs. A two-way ANOVA test demonstrated a significant difference (P < 0.001). I, pooled data for PPR failure ratios. At an ISI of 50 ms, synaptic failures of the second compared to the first pulses were significantly fewer in PV−/− pairs than in WT. Amplitudes were normalized to the first IPSC (E, G and H). Averages derived from 20 tests recorded at 4 s intervals in an MSN (E, G) have identical time scale bars. Dotted lines in F and I represent unity. All values are presented as mean ± SEM, bars denoting the SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t test.
Presynaptic role of PV on short-term plasticity at synapses between FSIs and MSNs
To test the impact of presynaptic PV on FSI neurotransmission, FSIs synaptically connected to MSNs were recorded using the perforated patch technique. Whole-cell recordings were performed on postsynaptic MSNs (Fig. 5A). Based on the unequivocal identification of EGFP-ir FSIs and postsynaptic MSNs, we confirmed that IPSCs in MSNs evoked by FSI somatic depolarizations (to −10 mV over 1 ms) were reversibly blocked by 10 μm gabazine (n= 2 and 3, for WT and PV−/−, respectively; data not shown). This indicated that currents were GABAA receptor mediated (Koós and Tepper, 1999). First, we determined the kinetics of single IPSCs (n= 100 presynaptic stimuli). Here, two parameters relating to presynaptic function and release probability were determined: (1) the failure rate and (2) quantal size defined as the variance to mean ratio of IPSC amplitudes. No differences were observed, either in IPSC kinetics or in failure rate or quantal size, indicating that the general properties of neurotransmission were not changed by PV during single pulse protocols (Table 2).
Table 2.
Properties of FSI–MSN synapses from WT and PV−/− mice
| n | WT 9 | PV−/− 10 |
|---|---|---|
| IPSC amplitude (pA) | 30.59 ± 4.61 | 36.75 ± 7.76 |
| Synaptic delay (ms) | 2.84 ± 0.27 | 2.88 ± 0.14 |
| Rise time 10–90% (ms) | 2.68 ± 0.32 | 2.38 ± 0.23 |
| Decay time, τ (ms) | 23.05 ± 1.13 | 25.30 ± 1.23 |
| Failures (%) | 30.15 ± 4.43 | 37.29 ± 3.15 |
| Variance/mean (pA) | 6.27 ± 1.7 | 6.47 ± 1.49 |
| Probability of connection (%) | 44.44 ± 2.77 | 41.66 ± 2.77 |
Data were collected from double patch clamp recordings. Perforated patch recordings were established onto FSIs and whole-cell patch clamp recordings to MSNs. All parameters were measured in voltage clamp. Data are presented as mean ± SEM. No significant differences were observed between the two genotypes by Student's t tests.
Next, we examined PPRs at FSI–MSN synapses, as PV has been shown previously to affect short-term modulation of synaptic plasticity (Caillard et al. 2000; Vreugdehil et al. 2003; Müller et al. 2007, Eggermann & Jonas, 2011). PPRs of IPSCs were determined at different ISIs from 5 to 200 ms, a time range over which presynaptic Ca2+ buffers are expected to modulate facilitation or depression depending on their Ca2+-binding kinetics. At an ISI of 50 ms, PPR was 0.69 ± 0.04 (n= 5; from four mice) in WT MSNs, whereas a clear facilitation (1.24 ± 0.07, n= 6; from four mice; P < 0.001) was observed in PV−/− neurons (Fig. 5E and F). Analysis of PPR at six different ISIs (5, 10, 20, 50, 100 and 200 ms) demonstrated that PV reduces the amplitude of the second IPSC in a narrow temporal window between 20 and 50 ms (Fig. 5F). Analysis of the PPR-failure ratio revealed a mirror-like image of the PPR plot (Fig. 5I, compare with Fig. 5F) with a significantly decreased number of failures of the second IPSC in PV−/− FSI–MSN pairs at 50 ms ISI (Fig. 5I). This further indicated a presynaptic origin of the lower PPR values at WT FSI–MSN synapses and demonstrated that PPR facilitation observed in the PV−/− group was dependent on a reduction in the failure rate due to the absence of PV. Next, we indirectly tested the effect of PV on presynaptic Ca2+ by analysis of IPSCs during trains (eight APs) of presynaptic stimuli evoked at 50 Hz and recording from MSNs (Fig. 5G). In WT pairs the normalized IPSC amplitudes were all decreased: to 75.9 ± 5.84 and 61.69 ± 7.06% for the 2nd and 3rd IPSC (n= 5; from four mice), respectively, and reached values of approximately 30% at the end of the eight AP train (Figs 5G and H). Recordings from PV−/− pairs (n= 5; from four mice) revealed a slight facilitation of the 2nd and 3rd IPSC to 109.18 ± 2.61 and 107.88 ± 4.84%, respectively (Fig. 5G, arrows) and differences between WT and PV−/− pairs were significant (P < 0.05). From the 4th IPSC on, depression also prevailed in PV−/− pairs and the differences between genotypes disappeared. This demonstrated that PV is responsible for the short-term depression or, more precisely, for preventing facilitation during the early period (20–40 ms) of 50 Hz AP trains, while from 60 ms on, the depression is probably independent of PV (Fig. 5H).
Finally, to confirm the presynaptic nature of PPR depression being mediated by the presence of PV in FSI terminals, we analysed the square inverse of the CV (CV−2). According to Clements (1990), if a large difference is observed between CV−2 calculated from data recorded under control (1st IPSC) and test conditions (2nd IPSC), a presynaptic site of modulation is presumed to be strongly implicated. Thus, we calculated the ratio between CV−2 values for the 1st (CV−21-IPSC) and 2nd (CV−22-IPSC) IPSC at 50 ms ISI, where differences between genotypes were most pronounced (Fig. 5F). Thus, a reduction in release probability during the 2nd IPSC would induce a CV−2 ratio less than unity, although an increase in release probability would result in a CV−2 ratio larger than unity. When comparing those CV−2 values for each pair, we observed that in the WT group the ratio was 0.53 ± 0.15 (n= 5), confirming a reduction in probability of release for the 2nd IPSC. The same calculation for the PV−/− group resulted in a value of 1.49 ± 0.33 (n= 5; P < 0.05 vs. WT). This indicates that PV diminished CV−2 for the 2nd IPSC and that its absence increased it. In conclusion, PV decreases the probability of transmitter release preferentially at an ISI around 50 ms.
Discussion
Here we provide evidence that the absence of PV in PV−/− mice modifies two aspects of FSI function in the striatum. First, PV decreases the rhythmicity of spontaneous FSI spiking as well as the output frequency during depolarizations through the coupling of Ca2+ dynamics and SK channel activation. Second, at presynaptic terminals of FSI axons, PV prevents fast short-term facilitation in a narrow time window of 20–50 ms at FSI to MSN synapses.
Modulatory effect of PV on FSI excitability
We observed that FSIs generated continuous spiking at low frequencies around 10 Hz. Absence of PV modified the regularity and spike-timing of FSIs during both spontaneous and evoked spiking modes. Alteration of the regularity and spike timing of the spontaneous activity pattern in PV−/− demonstrated that endogenous Ca2+ buffering-dependent processes tightly control basal firing properties. This is further confirmed by the significant increase in electroresponsiveness of PV−/− FSIs during depolarizations. Indeed, a reduction in Ca2+ buffering causes a faster deactivation of Ca2+-dependent potassium channels (Gall et al. 2003). Similar changes in FSI excitability occur either when blocking SK channels or when PV is deleted in PV−/− FSIs, and suggest a tight correlation between Ca2+ buffering and spiking control. SK channels exert a dynamic control of spontaneous neuronal firing and are essential for maintaining the high precision of intrinsic pacemaker activity (Wolfart et al. 2001, Deister et al. 2009). We also present a mathematical model for FSIs (Bischop et al. 2012) that includes SK channel currents and Ca2+ dynamics. Our model demonstrates that the observed alterations of FSI electroresponsiveness are directly linked to the decreased Ca2+-buffering capacity caused by the absence of PV. When challenged with a decreased cytosolic Ca2+-buffering capacity, the model correctly predicts an increase in output frequency and an increase in rhythmicity, both of which are observed experimentally. Thus, PV appears to affect SK channel activation by acting as a passive Ca2+ buffer. Indeed, through the direct control of the SK channel current by the Ca2+ transient, spike repolarization speeds up when the Ca2+-buffering capacity is decreased.
FSIs present voltage-dependent subthreshold oscillations in the gamma frequency range occurring in a narrow voltage range just negative to threshold (Jonas et al. 2004). These oscillations can trigger spikes at the oscillatory frequency, if the membrane is sufficiently depolarized (Llinás et al. 1991). In rat striatal FSIs, these oscillations occur during sustained depolarizations (Tepper et al. 2010). These oscillations are mainly dependent on voltage-gated K+ and Na+ conductances and underlie a stuttering firing pattern (Bracci et al. 2003; Golomb et al. 2007; Sciamanna & Wilson, 2011). In mouse FSIs, which show regular spiking during depolarizations (Freiman et al. 2006), oscillations appear at more hyperpolarized potentials, as demonstrated in our FSI recordings of spontaneous activity during perforated patch recordings in slices. We hypothesize that in mouse FSIs, during somatic Ca2+ transients associated with APs, PV is implicated in providing arrhythmicity by modulating those intrinsic oscillations via SK activation. Thus PV, as a Ca2+ source at this time point, would delay the next AP and thereby break the rhythm, which is in good agreement with the larger CVISI value in PV-expressing FSIs. This hypothesis is supported by our theoretical model showing that PV, acting through the modulation of SK channel activation, modifies the rhythmicity of FSI firing by delaying the onset of sustained regular firing. Note that the stuttering pattern observed in our deterministic model is a direct consequence of the intrinsic dynamical properties of the system (Ermentrout & Wechselberger, 2009).
Presynaptic short-term plasticity at the FSI–MSN synapse
FSI connections onto MSNs mediate inhibitory responses that show depression at short ISI with high reliability in both slices (Koós et al. 2004) and organotypic cultures (Gustafson et al. 2006). Ca2+ buffers presumably reduce residual Ca2+ at the time of the second stimulus and the extent of the effect is determined by their Ca2+ binding kinetics (Neher, 1998). Two lines of evidence are presented to show how PV is able to regulate the extent of such depression: (1) the switch from facilitation to depression in the presence of PV only appeared at a restricted temporal window of 20–50 ms that is strongly correlated with changes in the failure rate ratio and (2) single IPSC kinetics and failures were independent of the presence or absence of PV. These results are consistent with the idea that in FSI–MSN presynaptic terminals, PV acts as a slow-onset Ca2+ buffer, unable to affect rapid Ca2+ transients associated with single IPSCs, but, in contrast, is involved in a delayed buffering of residual Ca2+ as observed in cerebellar interneurons (Caillard et al. 2000; Colin et al. 2005). Thus, our results are consistent with changes induced by PV in other preparations, namely the acceleration of the first phase of Ca2+ decay, but not the amplitude of the spike-triggered presynaptic Ca2+ transients (Collin et al. 2005; Müller et al. 2007).
What might be the functional impact of presynaptic PV in FSI-to-MSN synapses? Our results suggest that PV tunes the inhibitory impact of FSI terminals onto MSNs at specific frequencies and consequently influences the regularity at which MSNs finally fire. FSI recordings in vivo have shown that FSIs are the unique element capable of following high-frequency inputs. This allows the MSN output to function at intermediate frequencies around 20 Hz, but not at higher frequencies (Schulz et al. 2011). This could be linked to the fact that PV causes IPSC depression at 20–50 Hz and not at higher frequencies, thus acting at presynaptic FSI terminals as a filter of MSN final inhibition.
Implications of PV as a modulator of FSI function and its consequences on striatal physiology
This study highlights two functional facets of a CBP that is selectively expressed within a circuit of a defined neuronal subpopulation (FSI). Indeed, at the core of the striatal network, special conditions for handling intracellular Ca2+ are imposed to FSIs at two connected phases of physiological activity: during (1) the generation of stereotypic spike output patterns and (2) when transmitter release is controlled at specific frequencies.
Our results on striatal FSIs suggest a tight association between SK channel and PV function, resulting in a faithful translation of intracellular calcium dynamics in voltage fluctuations. Interestingly, in other neuronal types, SK channels have been linked to voltage-dependent Ca2+ channels and SERCA pumps, constituting a triad permitting neurons to generate oscillatory activities (Cueni et al. 2008). Thus, in striatal FSIs, PV emerges as a ‘fourth element’ of this Ca2+ signalling dependent mechanism by increasing irregularity to the final oscillatory spiking pattern.
In vivo and modelling studies have shown that FSIs present irregular activity patterns that are (1) usually not synchronized between neighbouring cells (Berke et al. 2004); (2) complex, idiosyncratic and uncoordinated by cortical inputs (Berke, 2008); and (3) not synchronized by gap junctions (Hjorth et al. 2009). Altogether, this suggests that FSI spiking is mainly (or mostly) determined by intrinsic mechanisms rather than dictated by chemical and electrical synapses. Thus, the irregular pattern of striatal FSI spiking emerges as an intrinsic property of the FSI unit, observed here for the first time in vitro, and modulated by a PV/SK channel-dependent mechanism.
What could be the physiological relevance of this particular behaviour? First, at the cellular level, those in vivo FSI irregular patterns seem to cause subtle shifts in MSN spike timing rather than to induce profound changes in MSN firing rates (Berke, 2011). The observed subthreshold high-frequency component of MSN membrane voltage (Mahon et al. 2006) is assumed to be essential to tune the MSN spike-timing, and could probably be explained by the fact that one MSN is contacted by many (4–27) FSIs (Koós & Tepper, 1999). Secondly, this behaviour might be relevant at the network level because specific spiking patterns of FSIs might control information flow from different afferent inputs (Akam & Kullmann, 2010) and the formation of assemblies of synchronized MSNs (Humphries et al. 2009). In striatum, those oscillatory activities remain a potential mechanism that is still to be elucidated, either by targeting in vivo FSI neuromodulation by SK blockers or by studying striatal FSIs in PV−/− mice during task-related recordings.
By contrast, PV might act as a regulator of the extent of inhibition by which it avoids uncontrolled feed-forward inhibition, thereby resulting in the dying out of MSN activity, which would then cause a rupture of the signal expected to reach the output nuclei. Hence, PV can be seen as a homeostatic regulator of output MSN gain. It is noticeable that FSI inhibition in dopamine-depleted models seems to be reorganized by the generation of more synapses onto D2-MSNs (Gittis et al. 2011). This switch in inhibition leading to more synchrony is probably behind the enhanced low-frequency rhythmic bursting observed in downstream nuclei of the indirect pathway that characterizes Parkinsonian motor symptoms (Bevan et al. 2002; Burkhardt et al. 2007). This pathological rhythm covers a range from 15 to 30 Hz, a value strikingly near to the frequency window at which FSIs allow MSN spiking, in accordance with our results. Indeed, our study on PV−/− FSIs suggests that more inhibition of FSIs on D2-MSNs by PV's absence can effectively lead to the establishment of such oscillatory frequencies related to the induction of bradykinesia. In addition, because FSI density and/or FSI firing has not only been described to be altered in Parkinson's disease models, but also in models of Huntington's disease, dystonia, Tourette syndrome and drug-induced motor impairment (see Gittis & Kreitzer, 2012 for a review), a better understanding of this PV SK-mediated FSI fine tuning also under pathological conditions might help to unravel disease-linked mechanisms in further studies. One could envisage expressing PV under the control of an FSI-specific promoter using either lentivirus or adeno-associated virus-mediated transfection for both in vitro and in vivo studies. Alternatively, a transgenic mouse model could be developed, where re-expression of PV in a PV-deficient background could be regulated in a cell-specific and inducible way, as demonstrated before for the proto-oncogene protein N-myc (Sun et al. 2012). The same approach could be used for SK channel modulation to investigate its putative contribution to FSI disturbances. These models are expected to reveal whether such manipulations may help to restore normal striatal micro-circuit functioning in the various disease models.
Translational perspective
Fast spiking interneurons (FSIs) modulate output of the striatum and are implicated in severe motor disorders such as Parkinsonism, dystonia, Tourette syndrome and drug-induced motor impairment. Selective expression of the calcium-binding protein parvalbumin (PV) in FSIs raises questions about how PV controls FSI Ca2+ dynamics. Here we report a novel mechanism in FSIs that links PV-Ca2+ buffering and spiking as a result of the activation of small conductance (SK) Ca2+-dependent K+ channels. We also show that at the presynaptic terminals, PV prevents synaptic facilitation at narrow frequencies at FSI to striatal output neuron synapses. Our data show that PV is a key element in providing rhythm generation in FSIs as well as filtering striatal output. Thus, FSI neuromodulation, via PV and/or SK channels, are an interesting target to control the establishment of oscillatory frequencies related to the induction or worsening of pathology-related motor rhythms.
Acknowledgments
D.O. was supported by a postdoctoral fellowship from Fonds de la Recherche Scientifique, FRS-FNRS, Belgium. We are grateful to Fonds Defay and Jerome Lejeune Foundation for help to improve our scientific equipment platform. This study was also supported by Fondation Médicale Reine Elisabeth (FMRE-Belgium), FRS–FNRS (Belgium), research funds from ULB, the Interuniversity Attraction Poles Program (IUAP – P7/10) from the Belgian State, Federal Office for Scientific, Technical and Cultural Affairs, and Action de Recherche Concertée from the CFWB. We thank Dr Hannah Monyer, Heidelberg, for providing PV-EGFP mice, Laetitia Cuvelier, Brussels, for help with immunocytochemical labellings, Simone Eichenberger, Fribourg, for handling of the PV−/− mice colony, the Light Microscopy Facility (LiMiF), for help with confocal images and Dr Jacsue Kehoe, Paris, for careful reading of the manuscript.
Glossary
- AHP
after-hyperpolarization
- AP
action potential
- BK
large conductance Ca2+-dependent K+ channels
- CBP
calcium-binding protein
- CV
coefficient of variation
- FSIs
fast spiking interneurons
- D1, D2
type 1 and type 2 dopamine receptors
- EGFP
enhanced green fluorescent protein
- FWHM
full-width at half maximum
- IPSCs
inhibitory postsynaptic currents
- ir
immunoreactive
- ISI
inter-spike interval
- MSNs
medium-sized spiny neurons
- PPR
paired-pulse ratio
- PV
parvalbumin
- PV−/−
PV knockout mice
- SK
small conductance Ca2+-dependent K+ channels
- WT
wild type mice
Author contributions
D.O., S.N.S. and D.G conceived and designed the experiments. D.O. collected and analysed the data. D.O., D.B.P., B.S., S.N.S. and D.G. interpreted the data. D.O. drafted the manuscript and D.O., D.B.P., B.S., S.N.S. and D.G. critically revised the manuscript. All authors approved the final version of the manuscript. All experiments were performed in the Laboratory of Neurophysiology, Université Libre de Bruxelles.
References
- Akam T, Kullmann DM. Oscillations and filtering networks support flexible routing of information. Neuron. 2010;67:308–320. doi: 10.1016/j.neuron.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allene C, Picardo MA, Becq H, Miyoshi G, Fishell G, Cossart R. Dynamic changes in interneuron morphophysiological properties mark the maturation of hippocampal network activity. J Neurosci. 2012;32:6688–6698. doi: 10.1523/JNEUROSCI.0081-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- Berke JD. Uncoordinated firing rate changes of striatal fast-spiking interneurons during behavioral task performance. J Neurosci. 2008;28:10075–10080. doi: 10.1523/JNEUROSCI.2192-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD. Functional properties of striatal fast-spiking interneurons. Front Syst Neurosci. 2011;5:45. doi: 10.3389/fnsys.2011.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD, Okatan M, Skurski J, Eichenbaum HB. Oscillatory entrainment of striatal neurons in freely moving rats. Neuron. 2004;43:883–896. doi: 10.1016/j.neuron.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Booth PA, Eaton SA, Bolam JP. Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci. 1998;18:9438–9452. doi: 10.1523/JNEUROSCI.18-22-09438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus–external globus pallidus network. Trends Neurosci. 2002;25:525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Bischop DP, Orduz D, Lambot L, Schiffmann SN, Gall D. Control of neuronal excitability by calcium binding proteins: a new mathematical model for striatal fast-spiking interneurons. Front Mol Neurosci. 2012;5:78. doi: 10.3389/fnmol.2012.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, Calabresi P. Dopamine excites fast-spiking interneurons in the striatum. J Neurophysiol. 2002;87:2190–2194. doi: 10.1152/jn.00754.2001. [DOI] [PubMed] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, Calabresi P. Voltage-dependent membrane potential oscillations of rat striatal fast-spiking interneurons. J Physiol. 2003;549:121–130. doi: 10.1113/jphysiol.2003.040857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JM, Constantinidis C, Anstrom KK, Roberts DCS, Woodward DJ. Synchronous oscillations and phase reorganization in the basal ganglia during akinesia induced by high-dose haloperidol. Eur J Neurosci. 2007;26:1912–1924. doi: 10.1111/j.1460-9568.2007.05813.x. [DOI] [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A. Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci U S A. 2000;97:13372–13377. doi: 10.1073/pnas.230362997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD. A statistical test for demonstrating a presynaptic site of action for a modulator of synaptic amplitude. J Neurosci Methods. 1990;31:75–88. doi: 10.1016/0165-0270(90)90012-5. [DOI] [PubMed] [Google Scholar]
- Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, Marty A, Llano I. Developmental changes in parvalbumin regulate presynaptic Ca2+ signalling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- D’Angelo E, Filippi GD, Rossi P, Taglietti V. Synaptic excitation of individual rat cerebellar granule cells in situ: evidence for the role of NMDA receptors. J Physiol. 1995;484:397–413. doi: 10.1113/jphysiol.1995.sp020673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deister CA, Chan CS, Surmeier DJ, Wilson CJ. Calcium-activated SK channels influence voltage-gated ion channels to determine the precision of firing in globus pallidus neurons. J Neurosci. 2009;29:8452–8461. doi: 10.1523/JNEUROSCI.0576-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Jonas P. How the ‘slow’ Ca2+ buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat Neurosci. 2011;15:20–22. doi: 10.1038/nn.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol. 1999;82:2476–2489. doi: 10.1152/jn.1999.82.5.2476. [DOI] [PubMed] [Google Scholar]
- Ermentrout B, Wechselberger M. Canards, clusters, and synchronization in a weakly coupled interneuron model. SIAM J Appl Dyn Syst. 2009;8:253–278. [Google Scholar]
- Freiman I, Anton A, Monyer H, Urbanski MJ, Szabo B. Analysis of the effects of cannabinoids on identified synaptic connections in the caudate-putamen by paired recordings in transgenic mice. J Physiol. 2006;575:789–806. doi: 10.1113/jphysiol.2006.114272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall D, Roussel C, Susa I, D’Angelo E, Rossi P, Bearzatto B, Galas MC, Blum D, Schurmans S, Schiffmann SN. Altered neuronal excitability in cerebellar granule cells of mice lacking calretinin. J Neurosci. 2003;23:9320–9327. doi: 10.1523/JNEUROSCI.23-28-09320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernert M, Hamann M, Bennay M, Löscher W, Richter A. Deficit of striatal parvalbumin-reactive GABAergic interneurons and decreased basal ganglia output in a genetic rodent model of idiopathic paroxysmal dystonia. J Neurosci. 2000;20:7052–7058. doi: 10.1523/JNEUROSCI.20-18-07052.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Finkbeiner S, Kreitzer AC. Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron. 2011;71:858–868. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Kreitzer AC. Striatal microcircuits and movement disorders. Trends Neurosci. 2012;35:557–564. doi: 10.1016/j.tins.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb D, Donner K, Shacham L, Shlosberg D, Amitai Y, Hansel D. Mechanisms of firing patterns in fast-spiking cortical interneurons. PLoS Comput Biol. 2007;3:e156. doi: 10.1371/journal.pcbi.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson N, Gireesh-Dharmaraj E, Czubayko U, Blackwell KT, Plenz D. A comparative voltage and current-clamp analysis of feedback and feedforward synaptic transmission in the striatal microcircuit in vitro. J Neurophysiol. 2006;95:737–752. doi: 10.1152/jn.00802.2005. [DOI] [PubMed] [Google Scholar]
- Hjorth J, Blackwell KT, Hellgren Kotaleski J. Gap junctions between striatal fast-spiking interneurons regulate spiking activity and synchronization as a function of cortical activity. J Neurosci. 2009;29:5276–5286. doi: 10.1523/JNEUROSCI.6031-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MD, Wood R, Gurney K. Dopamine-modulated dynamic cell assemblies generated by the GABAergic striatal microcircuit. Neural Netw. 2009;22:1174–1188. doi: 10.1016/j.neunet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, Miles R. Interneuron fiversity series: Fast in, fast out—Temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 2004;27:30–40. doi: 10.1016/j.tins.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kalanithi PSA, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, Schwartz ML, Leckman JF, Vaccarino FM. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102:13307–13312. doi: 10.1073/pnas.0502624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kosaka T, Heizmann CW. Parvalbumin-immunoreactive neurons in the rat neostriatum: a light and electron microscopic study. Brain Res. 1990;536:1–15. doi: 10.1016/0006-8993(90)90002-s. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM, Wilson CJ. Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci. 2004;24:7916–7922. doi: 10.1523/JNEUROSCI.2163-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Schwaller B, Neher E. Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol. 2000;525:419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR, Grace AA, Yarom Y. In vitro neurons in mammalian cortical layer 4 exhibit intrinsic oscillatory activity in the 10- to 50-Hz frequency range. Proc Natl Acad Sci U S A. 1991;88:897–901. doi: 10.1073/pnas.88.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Sadikot AF. GABA promotes survival but not proliferation of parvalbumin-immunoreactive interneurons in rodent neostriatum: an in vivo study with stereology. Neuroscience. 2001;104:93–103. doi: 10.1016/s0306-4522(01)00038-0. [DOI] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G, Charpier S. Distinct patterns of striatal medium spiny neuron activity during the natural sleep-wake cycle. J Neurosci. 2006;26:12587–12595. doi: 10.1523/JNEUROSCI.3987-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Moine CL, Charpier S, Gonon F. Feedforwa-rd inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci. 2005;25:3857–3869. doi: 10.1523/JNEUROSCI.5027-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AH, Katona I, Blatow M, Rozov A, Monyer H. In vivo labelling of parvalbumin-positive interneurons and analysis of electrical coupling in identified neurons. J Neurosci. 2002;22:7055–7064. doi: 10.1523/JNEUROSCI.22-16-07055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Orduz D, Llano I. Recurrent axon collaterals underlie facilitating synapses between cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 2007;104:17831–17836. doi: 10.1073/pnas.0707489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux M, Mahon S, Deniau JM, Charpier S. Integration and propagation of somatosensory responses in the corticostriatal pathway: an intracellular study in vivo. J Physiol. 2011;589:263–281. doi: 10.1113/jphysiol.2010.199646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planert H, Szydlowski SN, Hjorth JJJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast-spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499–3507. doi: 10.1523/JNEUROSCI.5139-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenz D, Kitai ST. Up and down states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex–striatum–substantia nigra organotypic cultures. J Neurosci. 1998;18:266–283. doi: 10.1523/JNEUROSCI.18-01-00266.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes in C. Cambridge: Cambridge University Press; 1992. [Google Scholar]
- Ramanathan S, Hanley JJ, Deniau JM, Bolam JP. Synaptic convergence of motor and somatosensory cortical afferents onto GABAergic interneurons in the rat striatum. J Neurosci. 2002;22:8158–8169. doi: 10.1523/JNEUROSCI.22-18-08158.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JM, Pitcher TL, Savanthrapadian S, Wickens JR, Oswald MJ, Reynolds JNJ. Enhanced high-frequency membrane potential fluctuations control spike output in striatal fast-spiking interneurones in vivo. J Physiol. 2011;589:4365–4381. doi: 10.1113/jphysiol.2011.212944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. The continuing disappearance of “pure” Ca2+ buffers. Cell Mol Life Sci. 2009;66:275–300. doi: 10.1007/s00018-008-8564-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol. 2010;2:a004051. doi: 10.1101/cshperspect.a004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Dick J, Dhoot G, Carroll S, Vrbova G, Nicotera P, Pette D, Wyss A, Bluethmann H, Hunziker W, Celio MR. Prolonged contraction–relaxation cycle of fast-twitch muscles in parvalbumin knockout mice. Am J Physiol. 1999;276:C395–C403. doi: 10.1152/ajpcell.1999.276.2.C395. [DOI] [PubMed] [Google Scholar]
- Schwaller B, Meyer M, Schiffmann S. ‘New’ functions for ‘old’ proteins: the role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 2002;1:241–258. doi: 10.1080/147342202320883551. [DOI] [PubMed] [Google Scholar]
- Sciamanna G, Wilson CJ. The ionic mechanism of gamma resonance in rat striatal fast-spiking neurons. J Neurophysiol. 2011;106:2936–2949. doi: 10.1152/jn.00280.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaght SJ, Paz T, Chavez M, Deniau JM, Mahon S, Charpier S. On the activity of the corticostriatal networks during spike-and-wave discharges in a genetic model of absence epilepsy. J Neurosci. 2004;24:6816–6825. doi: 10.1523/JNEUROSCI.1449-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Hirzel K, D’hoedt D, Pedarzani P. Matching molecules to function: neuronal Ca2+-activated K+ channels and afterhyperpolarizations. Toxicon. 2004;43:933–949. doi: 10.1016/j.toxicon.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Sun R, Zhao K, Shen R, Cai L, Yang X, Kuang Y, Mao J, Huang F, Wang Z, Fei J. Inducible and reversible regulation of endogenous gene in mouse. Nucleic Acids Res. 2012;40:e166. doi: 10.1093/nar/gks738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YP, Llano I. Modulation by K+ channels of action potential-evoked intracellular Ca2+ concentration rises in rat cerebellar basket cell axons. J Physiol. 1999;520:65–78. doi: 10.1111/j.1469-7793.1999.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koós T, Ibáñez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. doi: 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreugdenhil M, Jefferys JGR, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol. 2003;89:1414–1422. doi: 10.1152/jn.00576.2002. [DOI] [PubMed] [Google Scholar]
- Wiltschko AB, Pettibone JR, Berke JD. Opposite effects of stimulant and antipsychotic drugs on striatal fast-spiking interneurons. Neuropsychopharmacology. 2010;35:1261–1270. doi: 10.1038/npp.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]