

Abstract
Voltage-gated K+ (KV) channels play critical roles in shaping neuronal signals. KV channels distributed in the perisomatic regions and thick dendrites of cortical pyramidal neurons have been extensively studied. However, the properties and regulation of KV channels distributed in the thin axons remain unknown. In this study, by performing somatic and axonal patch-clamp recordings from layer 5 pyramidal neurons of prefrontal cortical slices, we showed that the rapidly inactivating A-currents mediated the transient K+ currents evoked by action potential (AP) waveform command (KAP) at the soma, whereas the rapidly activating but slowly inactivating KV1-mediated D-currents dominated the KAP at the axon. In addition, activation of D1-like receptors for dopamine decreased the axonal K+ currents, as a result of an increase in the activity of cAMP–PKA pathway. In contrast, activation of D2-like receptors showed an opposite effect on the axonal K+ currents. Further experiments demonstrated that functional D1-like receptors were expressed at the main axon trunk and their activation could broaden the waveforms of axonal APs. Together, these results show that axonal KV channels were subjected to dopamine modulation, and this modulation could regulate the waveforms of propagating APs at the axon, suggesting an important role of dopaminergic modulation of axonal KV channels in regulating neuronal signalling.
Key points
Dopamine and its receptors in prefrontal cortex (PFC) play an important role in regulating synaptic transmission and PFC-mediated cognitive functions.
Considering that presynaptic action potential waveform can modulate postsynaptic responses, we investigated whether axonal K+ channels and action potential waveform are subjected to modulation by dopamine.
Patch-clamp recording from the axon of PFC pyramidal neurons showed that the activation of D1 and D2 dopamine receptors decreased and enhanced axonal K+ currents, respectively. Further experiments revealed that intracellular cAMP–PKA pathway was involved in this dopaminergic modulation of axonal K+ currents.
Recording from axons disconnected from the soma revealed that the dopaminergic modulation still occurred, indicating the presence of functional dopamine receptors along the axon.
We further demonstrate that axonal action potentials were substantially prolonged by D1 receptor activation. Taken together, our results reveal a new mechanism for dopaminergic modulation of neuronal signalling in PFC.
Introduction
Axon terminals containing dopamine (DA) are densely distributed in the prefrontal cortex (PFC), DA release from these terminals is involved in a variety of higher-order behavioural and cognitive processes, including attention and working memory (Goldman-Rakic, 1998; Lewis et al. 1998; Seamans & Yang, 2004). Alteration of dopaminergic modulation in PFC may lead to brain disorders such as schizophrenia and Parkinson's disease. Therefore, it is critical to reveal the underlying mechanisms of dopaminergic modulation in PFC neurons. After release, DA binds to its G-protein-coupled receptors, mainly D1-like and D2-like receptors (D1 and D2 for short), and regulates the activity of cAMP–PKA signalling pathway (Neve et al. 2004; Beaulieu & Gainetdinov, 2011). Activation of D1 receptors increases the production of cAMP and thus the activity of PKA, whereas activation of D2 receptors has an opposite effect; therefore, it is expected that activation of these receptors could produce distinct effects on ion channels, such as Na+ channels (Valentijn et al. 1991; Geijo-Barrientos & Pastore, 1995; Gorelova & Yang, 2000; Maurice et al. 2001) and K+ channels (Surmeier & Kitai, 1993; Harris-Warrick et al. 1995; Werner et al. 1996; Yang & Seamans, 1996; Dong & White, 2003; Dong et al. 2004, 2005; Witkowski et al. 2008), and consequently regulate the excitability of PFC neurons as well as their synaptic communication (Gao et al. 2001; Seamans et al. 2001; Neve et al. 2004; Rotaru et al. 2007; Di Pietro & Seamans, 2011).
As we know, KV channels play important roles in regulating AP waveforms and firing properties (including firing pattern and frequency). Changes in their biophysical properties may have a profound impact on the AP signalling in individual neurons and the operation of cortical circuits. AP waveform profiles, the repolarization phase in particular, in different compartments of a given neuron are determined by specific K+ channel subtypes. For example, in cortical pyramidal neurons, somatic APs are regulated by the rapidly activating and inactivating KV4-mediated A-type K+ currents (A-current; Kang et al. 2000; Kim et al. 2005; Yuan et al. 2005; Nerbonne et al. 2008) as well as the rapidly activating but slowly inactivating KV1-mediated K+ currents (known as D-current; Bekkers & Delaney, 2001; Mitterdorfer & Bean, 2002). In the axon initial segment and the main axonal trunk, the repolarization of APs is largely determined by KV1 channels that are selectively distributed in these compartments (Kole et al. 2007; Shu et al. 2007b; Goldberg et al. 2008). Inactivation of these axonal KV1 channels in response to somatic subthreshold depolarization can lead to broadening of axonal APs and an increase in neurotransmitter release, a new mode of neuronal signalling known as analog communication (Alle & Geiger, 2006; Shu et al. 2006). This mode of neuronal communication has been demonstrated to regulate the operation of microcircuits that mediate recurrent inhibition and thus contribute to the maintenance of a dynamic excitation–inhibition balance in the cerebral cortex (Zhu et al. 2011). Therefore, regulation of the axonal KV1 channels and AP waveforms may alter neuronal signalling and circuit functions.
Previous studies revealed that KV channels are subjected to DA modulation in the PFC. Application of DA could suppress K+ currents via the activation of D1 receptors and the downstream cAMP–PKA pathway in pyramidal neurons (Yang & Seamans, 1996; Yang et al. 1999; Dong & White, 2003) and fast-spiking parvalbumin-positive neurons in the PFC (Gorelova et al. 2002). Activation of D2 receptors and subsequent down-regulation of cAMP–PKA pathway may also contribute to DA modulation of K+ currents (Dong et al. 2004). Because of space-clamp problems with somatic whole-cell recording, macroscopic K+ currents recorded at the soma were most likely perisomatic. Therefore, it is still unknown whether K+ channels distributed at remote locations down the axon are subjected to dopaminergic modulation. Recent studies in cartwheel interneurons in the brain stem show that Ca2+ channels expressed at the axon initial segment can be modulated by DA release from nearby dopaminergic fibres, and this modulation can switch neuronal firing from bursts to tonic spiking (Bender et al. 2010, 2012); we thus speculate that DA may also modulate axonal KV channels and regulate AP conduction along cortical axons and subsequent neurotransmitter release.
In this study, we report that axonal K+ currents at the proximal unmyelinated segments of the main axon in PFC pyramidal neurons were modulated by DA receptors, with the activation of D1 and D2 receptors suppressing and enhancing K+ currents respectively, resulting from the differential regulation of the activity of intracellular cAMP–PKA signalling pathways. Further experiments revealed that dopaminergic modulation of axonal K+ currents led to changes in the waveforms of axonal APs, suggesting a new mechanism underlying dopaminergic modulation of neuronal signalling.
Methods
Ethical approval
The use and care of the experimental animals (Sprague-Dawley rats) in this project was approved by the Animal Advisory Committee at the Shanghai Institutes for Biological Sciences.
Slice preparation
Coronal slices from prefrontal cortex were prepared from rats (postnatal days 16–22) using protocols described previously (Shu et al. 2007b). In brief, the animals were initially anaesthetized with sodium pentobarbital (30 mg kg−1) and then decapitated. The intraperitoneal injectate volume of pentobarbital for each animal was approximately 0.3 ml, depending on the body weight. After decapitation, the brain was dissected out and immersed in an aerated (95% O2–5% CO2) and ice-cold (0°C) slicing solution that contained 213 mm sucrose as a substitute for 126 mm NaCl in normal ACSF. Slices were then incubated in the normal ACSF (in mm: 126 NaCl, 2.5 KCl, 2 MgSO4, 2 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, and 25 dextrose; 315 mosmol l−1; pH 7.4; 35°C). After 1 h incubation, slices were transferred to a recording chamber bathed with aerated ACSF at 36–36.5°C. Cortical neurons were visualized with an upright infrared differential interference contrast (IR-DIC) microscope (BX51WI; Olympus).
Electrophysiological recordings
Patch-clamp recordings were achieved from both the soma and the terminal bleb of the cut axon of layer 5 regular-spiking pyramidal neurons (Shu et al. 2006, 2007a,b; Hu et al. 2009). Patch pipettes had an impedance of 3–6 MΩ for somatic and 5–12 MΩ for axonal recordings when filled with the internal solution containing (in mm) 140 potassium gluconate, 3 KCl, 2 MgCl2, 2 Na2ATP, 0.3 Na3GTP, 10 Hepes, and 0.2 EGTA (288 mosmol l−1, pH 7.2 with KOH). In order to trace the axon and label the recorded cell, we added Alexa Fluor 488 (100 μm) and biocytin (0.2%) to the internal solution. The K+ currents and membrane potential were low-pass filtered at 10 kHz and sampled at a frequency of 50 kHz. The reversal potential (−109 mV) of K+ currents used for the measurement of the conductances was calculated from the Nernst equation.
After obtaining the somatic whole-cell recording, the fluorescent dye was able to diffuse into the recorded cell, allowing visualization of the complete neuronal structures including the dendrites and axons. These neuronal compartments were examined under the fluorescent microscope equipped with a 40× water-immersion objective and a magnifier of up to 2×. We frequently observed the blebs (4–5 μm in diameter) at the cut end of the axon. Patch-clamp recordings from these axon blebs were guided by switching back and forth between the fluorescent and differential interference contrast (DIC) images, tight-seal (GΩ seal) recordings, including intact whole-cell recording and outside-out patch recording, could be achieved easily from the blebs. For simultaneous recording from the soma and axon, patch pipettes for axonal recording were filled with a similar internal solution to those used for somatic recording except that no fluorescent dye was added. After recording, slices were transferred to 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB). Diaminobenzidine (DAB) staining was employed for visualization of the recorded neurons and measurement of the axon length. Signals were amplified and acquired by using the Multiclamp 700B amplifier and pClamp10 software (Molecular Devices, Sunnyvale, CA, USA). Access resistance was monitored frequently during the whole period of simultaneous somatic and axonal recordings. Recordings with series resistance (Rs) larger than 25 MΩ at the soma or 35 MΩ at the axonal bleb were discarded. Since the input resistance of the patches (including somatic nucleated patches, isolated and intact axonal blebs) was >20-fold larger than Rs, the Rs could only introduce a <5% error in the true voltage values. In addition, considering the high risk of damage of the axonal recording induced by the oscillation of Rs compensation, particularly during long recordings with pharmacological treatments, we chose not to apply the Rs compensation. During current-clamp recording, bridge balance and capacitance neutralization were carefully adjusted before and after every experimental protocol. The membrane potential values shown in the text and figures were corrected with a liquid junction potential of 15 mV.
To obtain the voltage waveforms of somatic and axonal APs, we placed a concentric electrode at layer 2/3 near the recorded cell and delivered single electrical shocks (0.1 ms in duration) to the slice. The dual somatic and axonal recording allowed simultaneous detection of somatic and axonal APs in response to the extracellular stimulation (Fig. 1D). To examine the types of K+ currents that mediated the somatic and axonal APs, we utilized the waveform of a somatic AP and its corresponding axonal AP (axon length: 137 μm) as voltage commands to evoke somatic and axonal KAP, respectively. Dual somatic and axonal recording was also employed to examine the effects of D1 receptor activation on the waveforms of somatic and axonal APs evoked by either somatic or axonal current injections (Fig. 8).
Figure 1. Distinct properties of somatic and axonal K+ currents evoked by step-voltage and AP-waveform commands.
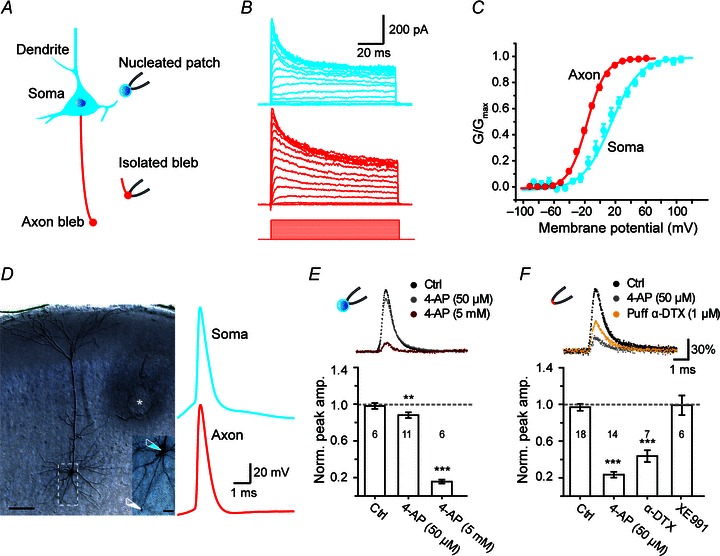
A, schematic diagram of somatic nucleated patch and isolated axon bleb recording from a pyramidal neuron. The somatodendritic and axonal compartments were shown in blue and red, respectively. B, somatic (blue) and axonal (red) K+ currents evoked by voltage steps (soma, from −90 to 110 mV; axon, from −90 to 60 mV; voltage steps for bleb recording were shown). C, activation curves of somatic and axonal K+ currents. D, left, DAB staining of a layer 5 pyramidal neuron after simultaneous recording from the soma and the axon bleb (scale bar, 100 μm). The area in the white box was enlarged for clarity (scale bar, 20 μm). * indicates the location where the stimulation electrode was placed. D, right, somatic and axonal APs evoked by extracellular stimulation (membrane potentials before the somatic and axonal action potentials were −77 mV and −73 mV, respectively). These APs were then used as voltage commands for somatic nucleated patch and axonal outside-out patch recordings. E, somatic nucleated patch recording. Top, the blockade of somatic KAP by 50 μm and 5 mm 4-AP. Bottom, bar graph of relative inhibition of the peak amplitude of somatic KAP by a low or high concentration of 4-AP. F, recording from outside-out patches excised from the axon blebs. Top, the blockade of axonal KAP by 50 μm 4-AP and 1 μmα-DTX. Bottom, bar graph of relative inhibition of the axonal KAP by α-DTX, 4-AP, and XE991. Number of patches indicated in the bar graphs. **P < 0.01, ***P < 0.001.
Figure 8. Modulation of AP waveforms at the soma and the axon by D1 receptor activation.
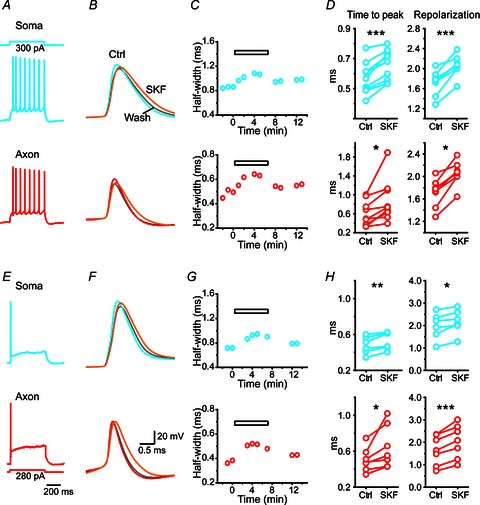
A, an example dual recording from the soma and the axon. Trains of APs were recorded at both recording sites in response to DC current injection (500 ms in duration) at the soma. B, expansion of the first APs during the AP train obtained in control (soma, blue; axon, red), SKF81297 (50 μm, orange) treatment and washout (grey). Note that bath application of SKF81297 broadened the waveforms of both somatic and axonal APs. C, time course of AP waveform changes shown in B. The horizontal bar indicates the treatment of SKF. D, group data showing significant increases in the duration of both rising (time to peak) and repolarizing phases of APs. Top, somatic APs; bottom, axonal APs. E, same recording as in A and B. Stimulating the axon with DC current injection only evoked single APs at both recording sites. F, expansion of APs showing the broadening induced by SKF81297. G, time course of AP waveform (half-width) changes shown in F. H, group data showing that both the rising and repolarizing phases were significantly prolonged by SKF81297. *P < 0.05; **P < 0.01, ***P < 0.001.
The outward K+ current was isolated by adding 200 μm CdCl2 and 1 μm TTX to the bath solution to block the voltage-gated Ca2+ and Na+ currents, respectively. For axonal recording, only TTX was used because no inward Ca2+ currents could be recorded at the axons (>100 μm away from the soma). To investigate the effect of Cd2+ on the voltage dependence of somatic K+ currents, we performed cell-attached recordings from the soma of pyramidal neurons in slices perfused with normal ACSF. Patch pipettes were filled with Ca2+-free ACSF containing TTX (1 μm) or TTX + CdCl2 (200 μm). Voltage-clamp protocols were applied on the assumption that the resting potential of the neuron remained constant at –70 mV. For all recordings, the leak currents were subtracted using an online P/5 procedure. We could obtain K+ currents from the regular outside-out patches excised from the axon blebs and also from giant outside-out patches: the somatic nucleated patches (Sather et al. 1992) and the isolated axon blebs. To obtain the isolated axon blebs, we made a small cut at the border between the layer 6 and white matter (Hu et al. 2009), blebs in the white-matter side were selected for recording. In these experiments, we used the internal solution that contained both the fluorescent dye (Alexa Fluor 488) and biocytin for the bleb recording to verify the success of axon bleb isolation from the soma. These isolated blebs most likely belonged to layer 5 pyramidal neurons because intact bleb recordings at the border between the layer 6 and white matter indicated that the soma was located at layer 5. Axon blebs of superficial pyramidal neurons formed at layer 4 and 5, possibly due to the cutting angle during slicing procedures.
SKF81297, SKF38393, SCH23390, SCH39166, quinpirole, eticlopride, PKI, H89, forskolin, XE991, ketanserin and biocytin were obtained from Tocris; DA, 4-AP, yohimbine, prazosin, propranolol and Way-100635 from Sigma. The KV1 channel blocker α-dendrotoxin (α-DTX) was obtained from Alomone.
Immunostaining
After 3 h fixation in 4% PFA and 4% sucrose (wt/vol) in 0.1 m PB (pH 7.4), brain slices were rinsed in 0.01 m PBS three times and then incubated in 0.5% Triton X-100 for 0.5 h and in blocking solution (10% BSA) for 1 h. Slices were then incubated with the primary antibody (rabbit, AB152, 1:1000, Millipore) against tyrosine hydroxylase (TH) overnight at 4°C. After a complete wash in PBS, the slices were incubated in the following secondary antibodies (1:1000; Invitrogen) for 2 h at room temperature, the Alexa Fluor 488 avidin conjugate (A-21370) and the Alexa Fluor 555 donkey anti-rabbit IgG (A-31570). For double staining of TH and dopamine β-hydroxylase (DBH), the rats (P15–18) were perfused with normal saline and followed by 4% PFA and 4% sucrose after deep anaesthesia with sodium pentobarbital. Cortical tissues containing the prefrontal cortex were dissected out and post-fixed in the same fixative for 2 h. Coronal sections (50 μm in thickness) were obtained by vibration section. After rinsing in 0.01 m PBS three times, these sections were incubated in 0.5% Triton X-100 for 0.5 h and in blocking solution (10% BSA) for 1 h, and then with the primary antibody against TH and DBH (mouse, MM-008-P, 1:25, Medimabs) overnight at 4°C. After a complete wash in PBS, the sections were incubated in the following secondary antibodies (1:1000; Invitrogen) for 2 h at room temperature, the Alexa Fluor 488 goat anti-rabbit IgG (A31627) and the Alexa Fluor 594 goat anti-mouse IgG (A31624). Finally the slices (or sections) were washed and then mounted on slides with fluoromount-G (Electron Microscopy Sciences, Hatfield, PA, USA).
Confocal images were taken with a laser scanning confocal microscope (Nikon A1R, Japan). With a 40× objective, we obtained z-stack images (interval of 1 μm) for the labelled cells. Single optical images were acquired with a 60× objective.
Statistical analysis
Data are presented as means ± SEM in the main text and figures. Student's t test was used to examine the significance of difference between data obtained before and after the drug treatment in each group. In addition, we also used one-way ANOVA and post hoc Tukey's test to examine the level of significance of difference among multiple groups. P values less than 0.05 were considered statistically significant.
The KAP was obtained by subtracting the baseline K+ current from the AP waveform-evoked K+ current. In order to examine the role of different K+ current types (A-, M- and D-currents) in mediating the KAP and the effect of DA receptor activation on KAP, we measured the peak amplitude and the integrated charge of KAP to compare the changes in these parameters before and after the application of channel blockers or DA receptor agonists. The peak amplitude of K+ currents evoked by AP waveforms or step voltage commands was measured as the difference between the peak and the baseline currents. The integrated charge of KAP was measured as the curve area of current traces above 2 × SD (standard deviation) of baseline noise unless otherwise stated. To examine whether DA receptor activation could have an effect on the AP waveform, we compared the AP peak amplitude, the half-width, the rise time (time to peak) and the repolarization time before and after the application of receptor agonists. The AP peak amplitude (or the rise time) was measured as the voltage (or time) difference between the AP threshold (dV dt−1= 20 V s−1) and the voltage deflection. The AP half-width was measured as the duration at the half-peak AP amplitude. The repolarization time was measured as the voltage drop from the peak to 10% of the peak amplitude.
Results
Somatic and axonal K+ currents
Layer 5 pyramidal neurons in the PFC were identified by their typical pyramidal-shaped somata and a long single apical dendrite extending to superficial layers. Somatic patch-clamp recordings were obtained from the soma or nucleated patches, whereas axonal patch-clamp recordings were obtained from the intact or isolated axon blebs (>100 μm away from the soma), which were the cut ends of axons formed during slicing procedures (Shu et al. 2006, 2007a,b; Hu et al. 2009).
We first characterized the electrophysiological properties of voltage-activated K+ currents at the soma and the main axon. To obtain pure somatic or axonal K+ currents, we performed somatic nucleated patch recording and isolated axon bleb recording (Fig. 1A; see Methods). In voltage-clamp mode, we applied a series of 100 ms depolarization pulses from −90 to 110 mV for nucleated patches or to 60 mV for the isolated axon blebs and calculated the peak conductance of KV channels (Fig. 1A–C). Comparison of the activation properties revealed significant differences between somatic and axonal K+ currents (Fig. 1C). Axonal K+ currents showed a more hyperpolarized half-activation voltage (V1/2) than somatic currents (−17.5 ± 1.2 vs. 9.1 ± 4.2 mV). The axonal K+ currents had a slope factor (k) of 13.0 ± 0.5 (n= 12 axon blebs), less than that of somatic currents (20.4 ± 1.6, n= 7 nucleated patches; Fig. 1C). Consistent with previous reports (Bekkers, 2000b; Kang et al. 2000; Kim et al. 2005; Yuan et al. 2005), these results suggested a differential distribution of KV channel subtypes at the somatic and axonal compartments.
To investigate the type of K+ currents and related channel subtypes that mediate the repolarization phases of APs, we performed voltage-clamp experiments using AP waveforms as voltage commands to evoke transient K+ currents (KAP; Fig. 1D–F). Using simultaneous whole-cell recording from the soma and the axon (137 μm from the soma), we obtained somatic and axonal APs evoked by electrical stimulation with a concentric electrode (Fig. 1D). These APs were then used as voltage commands in waveform-clamp recordings in nucleated patches and outside-out patches from axon blebs, respectively (Fig. 1D–F). For somatic KAP, bath application of 4-AP at a low concentration (50 μm) that selectively blocks KV1-mediated D-current caused a slight decrease in the peak amplitude (to 88.0 ± 3.1% of control, P < 0.01) and the integrated charge (to 94.4 ± 4.6% of control, P > 0.05, n= 11); however, with a high concentration of 4-AP (5 μm) that blocks A-type K+ currents, the peak amplitude and integrated charge were dramatically reduced to 15.7 ± 2.1% and 12.5 ± 4.0% (P < 0.001 for both parameters, n= 6; Fig. 1E), respectively. In contrast, for axonal KAP, 50 μm 4-AP reduced the peak amplitude to 23.4 ± 3.2% and the integrated charge of axonal KAP to 11.1 ± 2.7% of control (n= 14; Fig. 1F). Similarly, puff application of α-DTX (1 μm), a KV1 channel selective blocker, dramatically decreased the peak amplitude of axonal KAP to 41.3 ± 7.0% and integrated charge to 36.8 ± 7.6% (n= 7). Considering that KV7 channels distributed at the axon initial segment play an important role in determining AP threshold and neuronal firing properties (Pan et al. 2006; Shah et al. 2008), we further investigated their role in mediating the axonal KAP. Bath application of XE-991 (10 μm), a selective blocker for KV7-mediated M-current, showed no effect on both peak amplitude (99.0 ± 10.7% of control) and the integrated charge (106.0 ± 15.4% of control, n= 6) of the axonal KAP (Fig. 1F). Collectively, these results demonstrate that somatic KAP was dominated by the A-current, whereas axonal KAP was dominated by the KV1-mediated D-current.
Dopaminergic modulation of axonal K+ currents
In somatic whole-cell recording configuration, previous studies showed that the modulation of ion channels and neuronal activities in PFC by DA was normally mediated by D1 and D2 receptors (Gorelova & Yang, 2000; Maurice et al. 2001; Dong & White, 2003; Dong et al. 2004; Rotaru et al. 2007); in our experiments, we further examined the effect of DA, as well as selective D1 or D2 receptor agonists, on axonal K+ currents.
The K+ currents were elicited by stepping the voltage from –90 mV to a test potential of –10 mV, at which the evoked peak K+ conductance was approximately half of the maximum (Fig. 2A). We found a substantial suppression of the axonal K+ current (to 82.8 ± 1.5% of control, n= 7, P < 0.001) after the bath application of a specific D1 receptor agonist SKF81297 (50 μm; Fig. 2A, B and E). In contrast, application of a specific D2 receptor agonist quinpirole (1 μm) significantly enhanced axonal K+ current to 106.7 ± 2.1% (n= 8, P < 0.05; Fig. 2C and E). Interestingly, application of DA (50 μm) had no significant effect on the peak amplitude of K+ currents (to 96.4 ± 1.8%, n= 9, P= 0.09; Fig. 2D and E), probably resulting from the net effect of simultaneous activation of DA receptor subtypes including D1 and D2 receptors. These results indicate that the sign of dopaminergic modulation depends on DA receptor subtypes activated under certain conditions.
Figure 2. Modulation of axonal K+ current by the activation of D1 and D2 receptors.
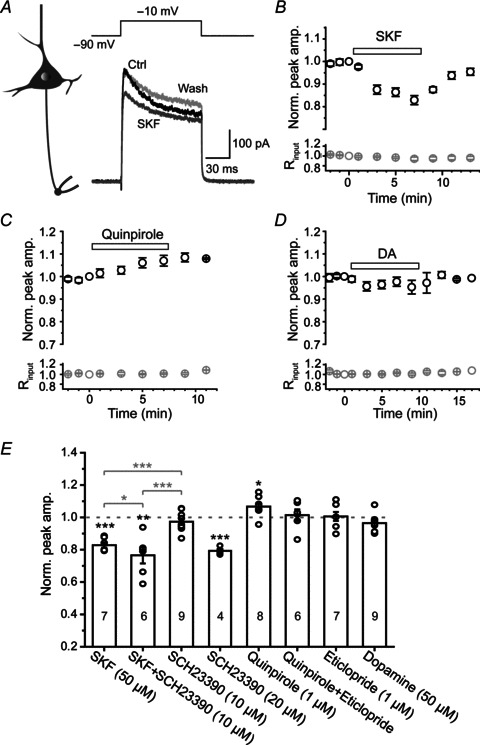
A, left, schematic diagram for axon bleb recording from a pyramidal neuron; right, example recording showing a decrease in peak amplitude of axonal K+ currents after bath application of D1-specific agonist SKF81297 (SKF, 50 μm, dark grey). B, time course of the inhibitory effect of SKF81297 (horizontal bar) on axonal K+ currents (n= 7). Bottom, changes in input resistance (Rinput) during the recording. C, time course of the facilitatory effect of D2-specific agonist quinpirole (1 μm) on axonal K+ currents (n= 8). D, no significant change in the peak amplitude of K+ currents after bath application of 50 μm DA (n= 9). E, bar graph of the effects of D1 and D2 agonists and antagonists on the peak amplitude of axonal K+ currents. Paired t test (black asterisks) was used for the comparison of data before (control) and during drug application in individual groups. One-way ANOVA and post hoc Tukey's test (grey asterisks) were used to examine the significance level of difference among groups (SKF, SFK+SCH23390 and SCH23390; quinpirole, quinpirole+eticlopride and eticlopride). No asterisk will be shown if P > 0.05. *P < 0.05; **P < 0.01, ***P < 0.001.
We next examined whether the specific modulatory effects of D1 and D2 receptor activation on K+ currents could be blocked by their corresponding antagonists (Fig. 2E). Interestingly, bath application of the D1 receptor antagonist SCH23390 at a concentration of 10 μm, which showed no detectable effect on the baseline K+ currents, could block the inhibitory effect of SKF81297 on somatic (Supplemental Fig. 1) but not axonal K+ currents (76.5 ± 5.0%, one-way ANOVA, P < 0.001; Fig. 2E). Increasing the concentration of SCH23390 to 20 μm substantially decreased the axonal K+ currents (Fig. 2E), indicating that SCH23390 may have non-specific effects on other receptors. We also used another D1 antagonist SCH39166 in subsequent experiments (Fig. 5). In contrast, we found that the facilitation of axonal K+ currents induced by D2 receptor agonist quinpirole was blocked by the pretreatment of D2 receptor antagonist eticlopride at a concentration of 1 μm (paired t test: before and after quinpirole application, P > 0.05; post hoc Tukey's test: comparing quinpirole and quinpirole+eticlopride, P > 0.05; Fig. 2E).
Figure 5. Opposite effects of D1 and D2 receptor activation on the axonal KAP.
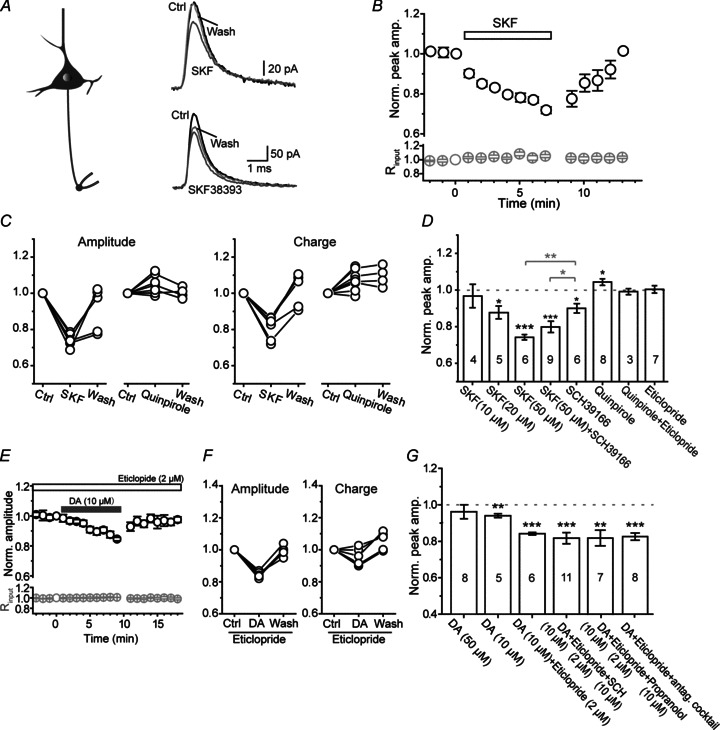
A, left, schematic diagram of intact bleb recording from a pyramidal neuron; right, example recording showing an inhibitory effect of D1 agonist SKF81297 (50 μm) and SKF38393 (10 μm) on axonal KAP (evoked by the same axonal AP waveform shown in Fig. 1D). B, group data showing the time course of the inhibitory effect of SKF81297 on axonal KAP (n= 6). C, group data showing the reversible effect of SKF81297 (50 μm) and quinpirole (D2 agonist, 1 μm) on the peak amplitude and the integrated charge of KAP. D, group data showing that D1 and D2 agonists significantly decreased and increased the peak amplitude of KAP, respectively. D1 antagonist SCH39166 (20 μm) could not block the inhibitory effect of SKF81297; D2 antagonist eticlopride (1 μm) blocked the facilitatory effect of quinpirole. E, group data showing the time course of the depressant effect of DA (10 μm) on axonal KAP in the presence of D2 receptor antagonist eticlopride (2 μm). F, group data showing the reversible effect of DA (10 μm) in the presence of eticlopride. G, group data showing the effects of DA in the presence of antagonists of DA, adrenergic or serotonergic receptors. Note that the depressant effect of DA on KAP was enhanced by eticlopride, but SCH23390 failed to block this effect. *P < 0.05, **P < 0.01, ***P < 0.001.
Taken together, these results showed that axonal KV channels were subjected to modulation by DA receptors, and that activation of D1 and D2 receptors inhibited and facilitated axonal K+ current, respectively.
Role of cAMP–PKA signalling pathway
Next, we examined the downstream intracellular pathways responsible for the modulatory effects of DA receptors on axonal K+ currents. Previous studies revealed that, at the soma, dopaminergic modulation of K+ currents was attributed to the activation of the cAMP–PKA signalling pathway (Valentijn et al. 1991; Greif et al. 1995; Dong & White, 2003; Dong et al. 2004). We therefore investigated whether this pathway was involved in dopaminergic modulation of the axonal K+ currents.
Bath application of forskolin (50 μm), an adenylate cyclase agonist, significantly suppressed axonal K+ currents (Fig. 3A). The peak amplitude was reversibly decreased to 77.4 ± 1.6% (n= 6, P < 0.001) in the presence of forskolin, and this depression could be abolished when the patch pipette was loaded with the PKA inhibitor PKI (100 μm; n= 5; Fig. 3C), suggesting that PKA activation is necessary in modulating axonal K+ currents. Bath application of 10 μm H-89, a selective PKA inhibitor, elicited no changes in the peak amplitude of axonal K+ currents (99.8 ± 2.6%, n= 8; Fig. 3B–C); but pretreatment with H-89 abolished the inhibitory effect of D1 receptor activation by its agonist SKF81297 (50 μm) on K+ currents (n= 4; Fig. 3B–C). Together, these results corroborate the idea that the activation of D1 receptors may activate cAMP–PKA signalling pathway and the elevated activity of PKA may subsequently inhibit axonal K+ currents.
Figure 3. The modulation of axonal K+ currents by D1 activation was mediated by cAMP–PKA pathway.
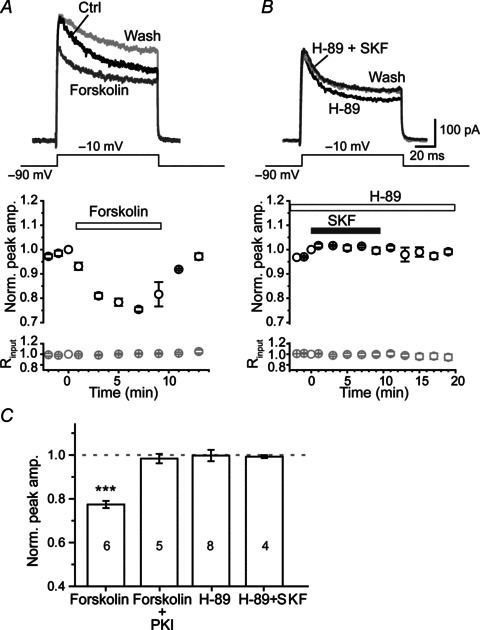
A, top, in intact axon bleb recording, K+ currents were reversibly reduced by the bath application of 50 μm forskolin (dark grey). A, bottom, group data showing the time course of peak K+ currents in the presence of forskolin (n= 6). B, bath application of the selective PKA inhibitor H-89 (10 μm) blocked the inhibitory effect of SKF81297 (50 μm) on axonal K+ currents. C, group data showing that the inhibitory effect of forskolin was abolished by adding 100 μm PKI to the pipette solution, and that the effect of SKF81297 on axonal K+ currents was blocked by pretreatment with 10 μm H-89. ***P < 0.001.
Dopaminergic modulation of KV channels could result in changes in channel properties; we therefore investigated the changes in the voltage-dependent properties of axonal K+ current upon PKA activation. In this experiment, we obtained K+ currents from isolated axon blebs to avoid contamination with K+ currents from somatodendritic compartments. K+ currents were elicited by 100 ms voltage steps (from −90 to 60 mV, 10 mV per step). We found that bath application of forskolin (50 μm) showed no changes in the maximum conductance of KV channels (Fig. 4A). However, the activation curve of axonal K+ currents showed a rightward (depolarizing) shift after forskolin application and, consistently, a leftward (hyperpolarizing) shift when using pipette solution containing 100 μm PKI (Fig. 4B and C), indicating constitutive phosphorylation of the channels. The V1/2 values for forskolin and PKI groups were −11.2 ± 0.8 (n= 10) and −23.2 ± 1.5 mV (n= 8), respectively, significantly different from that under control conditions (−17.5 ± 1.2 mV, n= 12; P < 0.001 and P < 0.01 respectively). Application of forskolin and PKI showed no significant change in the slope of the activation curve (13.0 ± 0.5 for control, 13.7 ± 0.7 for forskolin and 13.9 ± 0.8 for PKI; Fig. 4D). Unlike the opposite shift of activation curves, both forskolin and PKI shifted the inactivation curve toward hyperpolarizing membrane potential levels. The V1/2 values for channel inactivation were −77.5 ± 1.5 (n= 6) for forskolin and −80.6 ± 1.5 mV (n= 6) for PKI, significantly different from that in control conditions (−72.7 ± 1.0 mV, n= 11; Fig. 4B and C). Again, the slope factor showed no significant change (6.5 ± 0.3 for control, 5.7 ± 0.4 for forskolin and 5.6 ± 0.2 for PKI). Together these results demonstrate that the cAMP–PKA system was intact in the axon even isolated from the somatodendritic compartments, changes in its activity could regulate the properties of axonal K+ channels, with an increase (or decrease) in cAMP–PKA pathway depolarizing (or hyperpolarizing) the V1/2 for channel activation.
Figure 4. Voltage dependence of axonal K+ currents (at isolated blebs) was subject to modulation by intracellular cAMP–PKA pathway.
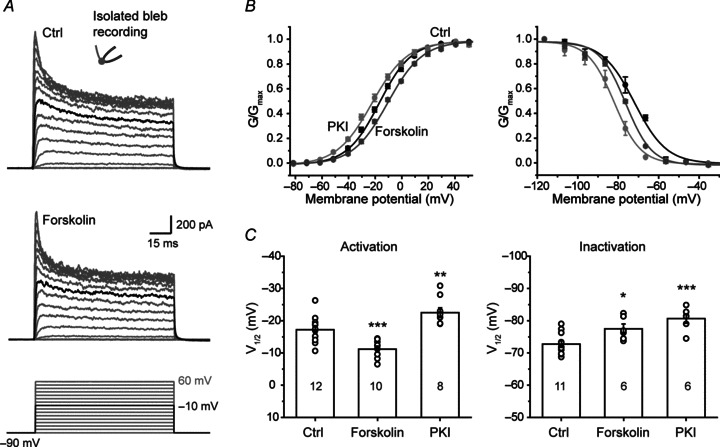
A, top and middle, example traces of axonal K+ currents evoked by voltage steps from −90 to 60 mV before and after the application of forskolin. Black traces indicate the currents evoked by stepping the voltage from −90 to −10 mV. Bottom, voltage commands. B, activation and inactivation curves under different conditions. The steady-state inactivation of K+ currents was examined by applying a series of 5-s-long hyperpolarizing steps (from a holding potential of −25 mV to −115 mV). Forskolin was applied through bath perfusion. PKI (100 μm) was applied through dialysis using PKI-containing pipette solution. Note the right and left shift of activation curves by forskolin and PKI, respectively. C, V1/2 values of activation and inactivation curves under different conditions. *P < 0.05; **P < 0.01, ***P < 0.001.
Dopaminergic modulation of AP waveform-evoked axonal K+ current
Next, we investigated whether K+ currents activated during APs were subjected to modulation by DA receptors. In this experiment, we obtained whole-cell recordings from intact axon blebs and used the AP waveform as voltage commands to evoke KAP. We found that the bath application of the D1 receptor agonist SKF81297 (50 μm) reduced the KAP (Fig. 5A and B) to a similar degree to its effect on K+ currents evoked by step-voltage command (Fig. 2). The peak amplitude and integrated charge of KAP were decreased by 25.8 ± 1.5% (P < 0.001) and 19.0 ± 2.7% (P < 0.001), respectively (n= 6; Fig. 5C and D). The inhibitory effect of SKF81297 on KAP was dose dependent. At 20 μm, SKF81297 still significantly decreased the peak amplitude of KAP (to 87.2% of control, n= 5, P < 0.05), but at 10 μm there was no effect on KAP (96.7% of control, n= 4, P > 0.05; Fig. 5D). In addition, another D1 receptor agonist SKF38393 (10 μm) also significantly suppressed the peak amplitude of axonal KAP to 83.2 ± 4.5% (n= 7, P < 0.01; Fig. 5A) and slightly decreased the integrated charge to 90.3 ± 4.6% of control (P > 0.05). To examine whether this dopaminergic modulation occurs only in early developmental stages, we performed similar experiments in young adult animals (P28–32). We found that the peak amplitude and the integrated charge of axonal KAP were decreased to 67.5 ± 5.7% and 71.8 ± 5.1% of control (n= 7, P < 0.01 for both, data not shown) in the presence of SKF81297 at 50 μm, indicating the dopaminergic modulation of axonal K+ channels also occurs in adult animals.
In contrast, the activation of D2 receptors by quinpirole (1 μm) increased the peak amplitude of KAP and the integrated charge by 4.3 ± 1.8% (P= 0.047, n= 8) and 7.3 ± 2.0% (P < 0.01), respectively (Fig. 5C and D). The facilitatory effect of quinpirole was blocked by D2 receptor antagonist eticlopride (1 μm; Fig. 5D). Again, the D1 receptor antagonist SCH23390 (10 μm) failed to block the inhibitory effect of SKF81297 on KAP (also see Fig. 2). Considering that SCH23390 may have non-specific effects on other receptors, we applied another D1 receptor antagonist, SCH39166, and found that bath application of SCH39166 at a concentration of 20 μm also failed to block the effect of SKF81297 (from 74.2 ± 1.5% to 80.4 ± 2.8%, n= 9, post hoc Tukey's test, P= 0.22; Fig. 5D).
In order to further verify the depressant effect of D1 receptor activation on axonal KAP, we examined the effect of DA on KAP in the presence of the D2 receptor blocker eticlopride (2 μm). We found that the suppression of the peak amplitude of KAP by DA was increased from 5.9 ± 1.1% (n= 5) to 15.8 ± 0.7% (n= 6, P < 0.001; Fig. 5E–G), indirectly indicating the existence of D1 receptor-mediated modulation. Again, the inhibitory effect of DA could not be blocked by SCH23390. In the presence of eticlopride and SCH23390, the peak amplitude of KAP showed a decrease of 18.3 ± 3.1% (n= 11, P < 0.001; Fig. 5G. See Discussion). Previous studies suggested that DA and its receptor agonists could also activate adrenergic and serotonergic receptors (Briggs et al. 1991; Cornil et al. 2002). To rule out the possibility that DA-induced reduction of axonal K+ currents resulted from the interaction of DA and adrenergic or serotonergic receptors, we examined whether the antagonists of these receptors could block the effect of DA. Considering the involvement of the cAMP–PKA pathway in the DA-induced reduction in K+ currents (Fig. 3), we first examined the effect of propranolol, a β-adrenergic receptor antagonist. In the presence of propranolol (10 μm) and eticlopride (2 μm), DA application still caused a significant reduction in the peak amplitude of KAP (to 81.8 ± 4.3%, n= 7, P < 0.01; Fig. 5G). Next, we used a cocktail of antagonists for adrenergic (1 μmα2 receptor antagonist yohimbine and 1 μmα1 antagonist prazosin) and serotonergic receptors (1 μm 5-HT1A antagonist Way-100635 and 10 μm 5-HT2 antagonist Ketanserin). Again, DA (also in the presence of eticlopride) suppressed axonal KAP to a similar degree (82.6 ± 2.0%, n= 8, P < 0.001; Fig. 5G). These results suggest that DA-induced modulation of axonal K+ currents was not attributable to crosstalk between DA and adrenergic or serotonergic receptors.
Since the α-DTX-sensitive K+ currents dominated KAP, KV1 channels could be the main target of dopaminergic modulation. However, our experiments further demonstrated that, in the presence of α-DTX (100 nm), the remaining K+ currents were also inhibited by D1 receptor activation (by 27.8 ± 3.8% of peak amplitude, n= 5, P < 0.01; Supplemental Fig. 2). Together, these results show that K+ currents activated during APs were subjected to the dopaminergic modulation; the ‘sign’ of the modulation (depressing or facilitating) was dependent on the subtypes of DA receptors activated.
Presence of functional D1 receptors at the main axonal trunk
To investigate whether DA receptors expressed at the main axonal trunk, we performed pharmacological experiments in axon blebs that were disconnected from the soma but with a segment of axon attached (see Methods; Fig. 6A). We found that D1 receptor activation reversibly decreased KAP (Fig. 6B–D), the peak amplitude and integrated charge of KAP decreased to 70.0 ± 1.9% (P < 0.001) and 86.4 ± 1.4% of control (P < 0.001, n= 8; Fig. 6C–E), respectively. In contrast, D2 receptor activation showed little effect on axonal KAP in this preparation (n= 6; Fig. 6D and E). We next investigated whether D1 receptor activation could have an effect on the voltage dependence of axonal K+ channels. In the presence of 50 μm SKF81297, the activation and inactivation curves showed depolarizing and hyperpolarizing shifts, respectively (Fig. 6F and G), consistent with the effect of forskolin (Fig. 4B). The activation V1/2 (−12.6 ± 1.8 mV) and the slope factor (16.6 ± 1.1, n= 8) were significantly different from those of controls (−17.5 ± 1.2 mV and 13.0 ± 0.5, n= 12; P < 0.05 and P < 0.01; Fig. 6F). The inactivation V1/2 was significantly hyperpolarized in the presence of SKF81297 (−72.7 ± 1.0 mV in control vs.−79.5 ± 1.2 mV in SKF, P < 0.001), while the slope factors showed no significant difference (6.5 ± 0.3 vs. 7.0 ± 0.2, P > 0.05; Fig. 6G), respectively. Taken together, these results indicate the presence of functional D1 receptors at the main axonal trunk, activation of these receptors was sufficient to modulate axonal K+ channels.
Figure 6. Dopaminergic modulation of KAP was intact in isolated axons.
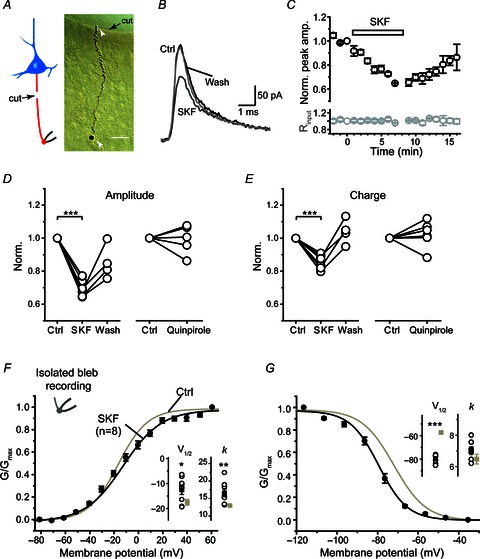
A, left, schematic diagram of the isolated axon bleb recording; right, DAB staining of an isolated axon bleb connected with a segment of axon (180 μm in length). The axon bleb (white arrow) was mechanically isolated from the main axon trunk before recording (see Methods). The white arrowhead indicates the proximal end of the isolated axon. The black arrow indicates the edge of the cut. Scale bar, 20 μm. B, example recording from the isolated axon bleb shown in A. Note the inhibitory effect of SKF81297 (50 μm) on KAP. C, group data showing the time course of the inhibitory effect of SKF81297 (n= 8). D and E, group data showing the effect of D1 and D2 receptor agonists on the peak amplitude and the integrated charge of KAP. F and G, SKF81297 shifted the activation and inactivation curves of K+ currents obtained in isolated axon blebs. Inset, comparison of V1/2 and slope factors (k) in control with those in SKF81297 (n= 8). *P < 0.05; **P < 0.01, ***P < 0.001.
In addition, we found that the TH-containing fibres and terminals were closely adjacent to the layer 5 pyramidal neuron axons (n= 6; Fig. 7A), suggesting a dopaminergic innervation of these axons. Although our immunostaining experiments also revealed that a large proportion of the TH-containing fibres (64.5 ± 4.0% of total fibre length, n= 7 from 3 rats) in deep cortical layers were also DBH-positive (noradrenergic axons; Fig. 7B), co-release of DA and noradrenaline can occur at these double-stained axons (Devoto et al. 2001, 2004). Whether the release of noradrenaline can regulate the properties of axonal channels remains to be examined.
Figure 7. Dopaminergic axons were closely adjacent to pyramidal cell axons.

A, projection of confocal z-stack images showing a biocytin-labelled pyramidal cell (green) and TH-containing axons (red, antibody staining). Note the axon bleb of the labelled cell. Scale bar, 25 μm. Section thickness, 12 μm. Inset, a single optical section from the boxed region. The white arrow indicates a crossing point of the pyramidal neuron axon and a TH-positive fibre. Scale bar, 5 μm. B, co-localization of TH (red) and DBH (green) in deep cortical layers. Scale bar, 40 μm, Section thickness, 50 μm.
Dopaminergic modulation of AP waveforms
In cortical pyramidal neurons, KV1 channels are selectively distributed in the axon and mediate the D-current; inactivation of this current after modest depolarization may result in broadening of axonal APs and thus enhancement of synaptic transmission, a potential mechanism for presynaptic membrane potential-dependent analog communication between cortical neurons (Kole et al. 2007; Shu et al. 2007b). Therefore, we further examined whether dopaminergic modulation of axonal K+ current could shape the waveform of axonal AP.
We performed dual whole-cell recording at the soma and the axon bleb in PFC pyramidal neurons. First, we injected depolarizing step currents through the somatic whole-cell recording electrode to evoke trains of APs. Dual recording allowed simultaneous monitoring of these APs at the soma and the axon. We found that waveforms of both somatic and axonal APs were broadened after the application of the D1 agonist SKF81297 (50 μm; Fig. 8A–D). Close examination of the AP waveforms during the AP train revealed that SKF81297 showed a stronger effect on the first axonal AP compared with its effect on the first corresponding somatic AP (Supplemental Fig. 3). This difference might be attributable to differential distribution of channel subtypes at the soma and the axon. The half-width of axonal APs increased from 1.30 ± 0.20 to 1.76 ± 0.24 ms (P < 0.01, n= 9). Although the peak amplitude of APs decreased from 52.9 ± 8.8 to 42.0 ± 6.0 mV (P < 0.05), the integrated area increased from 51.0 ± 8.0 to 57.0 ± 8.5 mV ms (P < 0.01; Fig. 8A–D). The increase in AP duration mainly resulted from the broadening of the repolarization phase because the rising phase (time to peak) was much shorter than the falling phase (repolarization time) and only increased from 0.60 ± 0.09 to 0.87 ± 0.15 ms (P < 0.05), while the falling phase increased from 1.94 ± 0.35 to 2.51 ± 0.34 ms (P < 0.05; Fig. 8A–D). The changes in the rising phase and the peak amplitude may mainly result from the dopaminergic modulation of Na+ channels (Surmeier & Kitai, 1993; Yang & Seamans, 1996; Maurice et al. 2001; Rotaru et al. 2007). Similar results were observed in somatic APs, as shown by an increase in the half-width (from 1.29 ± 0.07 to 1.49 ± 0.08 ms, P < 0.001) and a decrease in AP peak amplitude (from 73.9 ± 2.3 to 71.7 ± 2.5 mV, P > 0.05). The integrated area of somatic APs increased from 77.5 ± 3.3 to 87.7 ± 4.6 mV ms (P < 0.001, n= 9; Fig. 8A–D). The waveform changes of somatic APs were consistent with the dopaminergic modulation of K+ currents at the soma (Supplemental Fig. 1; Surmeier & Kitai, 1993; Dong & White, 2003).
Since APs recorded at the axon blebs (>100 μm away from the soma) were propagating APs that arrived at the bleb, their waveforms could be influenced by somatodendritic APs. To further determine the direct effect of D1 receptor activation on axonal APs, we injected step current locally at the axon bleb to evoke axonal APs as well as backpropagating APs at the soma (Fig. 8E). Consistent with previous findings (Shu et al. 2007b), only one AP per step current stimulation was observed. The waveforms of axonal APs were also broadened in the presence of SKF81297 (50 μm), the AP half-width increased from 1.13 ± 0.19 ms to 1.45 ± 0.23 ms (P < 0.001, n= 7) and the integrated voltage area increased from 51.8 ± 7.5 to 58.5 ± 8.0 mV ms (P < 0.05; Fig. 8E–H). Similar results were obtained for backpropagating APs that arrived at the soma: the half-width increased from 1.30 ± 0.15 to 1.49 ± 0.16 ms (P < 0.01) and the integrated voltage area increased from 84.4 ± 8.4 to 93.1.7 ± 8.6 mV ms (P < 0.05). In contrast, the D2 receptor agonist quinpirole had no significant effect on the waveforms of APs evoked at the soma or the axon bleb (Supplemental Fig. 4), reflecting its relatively weak effect on KAP in both intact axon and isolated axon bleb.
Together, these results show that local axonal APs were subjected to modulation by DA receptors, a mechanism that may contribute to the dopaminergic modulation of neuronal signalling at the PFC and participate in the regulation of cognitive functions.
Discussion
In this study, we found that A- and D-currents dominated the K+ currents activated during somatic and axonal APs, respectively, and axonal K+ channels were subjected to modulation by D1 and D2 dopamine receptors via cAMP–PKA pathways. In addition, we showed that this dopaminergic modulation could shape AP waveforms, particularly their repolarizing phases. Since changes in AP duration are tightly coupled with synaptic strength, these results point to a new mechanism for dopaminergic modulation of synaptic transmission.
Previous studies mainly focused on whole-cell K+ currents using somatic recordings (Yang & Seamans, 1996; Yang et al. 1999; Gorelova et al. 2002; Dong & White, 2003; Dong et al. 2004). In this study, by performing patch-clamp recording from isolated axon blebs combined with somatic nucleated patch recording, we revealed distinct biophysical properties of somatic and axonal K+ channels, with axonal K+ channels activating at much lower activation threshold (Fig. 1C). Using AP waveforms as voltage commands, we further demonstrated that the rapidly activating and inactivating A-currents dominated the somatic KAP, whereas the slowly inactivating D-current dominated the axonal KAP. The complete blockade of somatic KAP by millimolar 4-AP indicated a dominant role of A-currents in mediating the repolarization of somatic APs. In contrast, micromolar 4-AP and α-DTX dramatically reduced the axonal KAP, indicating that KV1-mediated D-currents played a dominant role in controlling the repolarization of axonal APs. These results were consistent with previous reports showing that α-DTX prolongs axonal but not somatic APs (Kole et al. 2007; Shu et al. 2007b). The inactivation of these axonal α-DTX-sensitive currents upon subthreshold depolarization could cause broadening of axonal APs and thus mediated membrane potential-dependent analog signalling between cortical neurons (Shu et al. 2006, 2007b; Kole et al. 2007). Although KV7-mediated M-currents exist at the axon initial segment and play important roles in determining the AP threshold and firing properties (Pan et al. 2006; Shah et al. 2008), our results revealed no contribution from KV7 channels to axonal KAP (Fig. 1F). In this study, we did not investigate the K+ channel subtypes that mediate the dendritic KAP. Since the properties of single K+ channels are similar along the somatodendritic axis (Bekkers, 2000a), the A-currents may also dominate the dendritic KAP. Previous studies showed that the presence of Cd2+ may shift the activation curve of K+ currents (Song et al. 1998), in order to exclude the possibility that Cd2+ may have an effect on the voltage dependence of somatic K+ currents, we therefore performed cell-attached recording from the soma with patch pipettes filled with or without Cd2+. However, we found no shift in activation curves (Supplemental Fig. 5), suggesting that the differential voltage dependence of somatic and axonal channels is attributable to selectively distributed channel subtypes.
Dopamine levels at the PFC play important roles in several high-order cognitive functions such as attention and working memory (Goldman-Rakic, 1998; Lewis et al. 1998; Seamans & Yang, 2004); disruption of dopaminergic system may cause brain disorders including depression and schizophrenia. Dopaminergic modulation of ion channels through dopamine receptors and related intracellular signalling pathways is important for neuronal signalling and circuit operation. The channel subtypes KV4 and KV1 could be regulated by channel phosphorylation via protein kinases (Jonas & Kaczmarek, 1996; Johnson et al. 2009; Lin et al. 2010). For example, K+ currents mediated by KV1.2 were increased by PKA activation in oocytes (Drain et al. 1994), HEK293 cells (Connors et al. 2008) and Chinese hamster ovary (Bosma et al. 1993). In cortical pyramidal neurons, macroscopic K+ currents obtained with whole-cell recording at the soma should contain currents activated at different cellular compartments, including the soma, dendrite and the axon. Due to space-clamp problems, the whole-cell macroscopic K+ currents mainly reflect the activation of perisomatic channels. We demonstrate in this study that not only perisomatic K+ channels but also those located at remote axonal segments were subjected to dopaminergic modulation. At the intact axon blebs, activation of D1 and D2 receptors inhibited and enhanced axonal K+ currents, respectively. Reducing PKA activity could block D1-induced inhibition of K+ currents. A complete cAMP–PKA signalling pathway may exist in the axon because forskolin and PKI could efficiently change the voltage-dependent properties of K+ currents in the mechanically isolated axon blebs. Interestingly, although forskolin and PKI showed opposite effects on the activation curve, they both shifted the inactivation curve toward hyperpolarizing membrane potential levels (Fig. 4). Similar to the effect of forskolin, activation of D1 receptors shifted the activation and inactivation curves towards depolarizing and hyperpolarizing potentials, respectively (Fig. 6). These results suggest that the activation and inactivation gates of the K+ channels could be differentially regulated by the cAMP–PKA system.
Previous studies showed that D1 and D2 receptors are expressed in the somatodendritic compartments (Vincent et al. 1993; Smiley et al. 1994; Santana et al. 2009). In layer 5 of PFC, D1 and D2 receptors tend to express in distinct subpopulation of pyramidal neurons. Neurons with compact apical dendrites express D1 receptors, whereas those with thick-tufted apical dendrites express D2 receptors (Gee et al. 2012; Seong & Carter, 2012). In this study, we obtained recordings from large layer 5 pyramidal neurons regardless of their dendritic morphology and revealed the expression of D1-like receptors at their axons. Due to the lack of selective D1 and D5 receptor antagonists, we were not able to identify the receptor subtype that mediated the SKF-induced suppression of axonal K+ channels in this study. The expression of D1-like receptors in the axon was supported by several lines of evidence. Firstly, the inhibitory effect of DA could be enhanced by the presence of D2 antagonist, indicating that D2 antagonist unmasked the effect of DA-activated D1-like receptors. Secondly, D1 agonists (including both SKF81297 and SKF38393) inhibited the axonal K+ current to a similar extent to DA. Thirdly, similar to the effect of forskolin, SKF changed the voltage-dependent properties of axonal K+ currents, excluding the possibility of its non-specific effect on channels. Finally, in agreement with the activation of D1 receptors, the inhibitory effect of SKF81297 could be abolished by the PKA inhibitor H-89, indicating the involvement of the PKA pathway in mediating the effect of SKF. Previous findings suggested that DA or its receptor agonist could also activate certain types of adrenergic and serotonergic receptors (Briggs et al. 1991; Cornil et al. 2002); however, our results demonstrate that DA-induced modulation of axonal K+ currents still occurred in the presence of antagonists for these monoamine receptors (Fig. 5G), suggesting that the effect of DA could be mediated by its own receptors.
The failure of D1 antagonists in blocking SKF- and DA-induced current inhibition could be attributed to their non-specific effects on other receptors, such as serotonin receptors (Millan et al. 2001). In our experiments, at a relatively higher concentration, D1 antagonists also significantly decreased the axonal K+ currents (Figs 2 and 5), suggesting a non-specific effect. Surprisingly, when we used a low concentration of SCH23390 that showed no obvious effect on baseline K+ currents, SKF-induced reduction of K+ currents in the soma was diminished (Supplementary Fig. 1), but not that at the axon (Fig. 2E). One explanation for the inconsistency of these results is the existence of a new D1-like receptor at the axon that is insensitive to the antagonist SCH23390. Unlike D2-like receptors, unique splice variants of D1-like receptors cannot exist because genes encoding D1 and D5 receptors are intronless; however, a new D1-like receptor (known as DAMB) with low sequence identity with other D1 receptors has been identified in Drosophila mushroom body (Han et al. 1996). Although it remains unknown whether DAMB orthologues or other D1-like receptors exist in mammalian brain, it is possible to isolate new receptors based on sequence homology with invertebrate receptors (for example, the isolation of human 5-HT7 receptor). Another explanation is the existence of DA receptor heteromers (e.g. D1–D2, D1–D3, D2–D5; Lee et al. 2004; Fiorentini et al. 2010; Hasbi et al. 2010) in the axon. Heteromers may have distinct pharmacological properties from D1 receptors. Future studies are needed to investigate the identity of axonal DA receptors and the difference between somatic and axonal receptors. Previous findings have revealed that the axon can express subunits or subtypes of neurotransmitter receptors distinct from those distributed in the somatodendritic compartments. For example, α2 subunit-containing GABAA receptors predominantly distributed in the axon (Nusser et al. 1996), and 5-HT1A and D3 receptors also target to the axon initial segment (Czyrak et al. 2003; Bender et al. 2010). Similarly, it is possible that axonal DA receptors may differ from those distributed in the somatodendritic compartments. Whether axonal K+ channels are subjected to regulation by other neuromodulators (such as serotonin and noradrenaline) remains to be further examined. Interestingly, our immunostaining results revealed that some of the TH-containing fibres also expressed DBH; co-release of dopamine and noradrenaline may occur in these fibres (Devoto et al. 2001, 2004) and subsequently modulate axonal channels.
The modulation of K+ currents by dopamine receptors was consistent with changes in AP waveforms at both the soma and the axon. Considering that somatic and axonal KAP were dominated by A- and D-currents, respectively, broadening of somatic and axonal APs upon D1 receptor activation reflected the modulation of corresponding channel subtypes. Since AP backpropagation plays an important role in mediating spike-timing-dependent synaptic plasticity, changes in AP waveforms at the somatodendritic compartments may influence the time window for this type of plasticity. At the axon, AP waveform changes may cause alteration of synaptic strength. In mossy fibre boutons, progressively inactivating A-current during high-frequency firing could cause an increase in AP duration and result in an enhancement of neurotransmitter release (Geiger & Jonas, 2000); in the pyramidal neuron axons, propagation of subthreshold depolarization from the soma could cause inactivation of KV1 channels and broadening of axonal APs, which may subsequently enhance synaptic transmission (Kole et al. 2007; Shu et al. 2007b) and regulate the dynamic balance between cortical excitation and inhibition (Zhu et al. 2011). Therefore, dopaminergic modulation of axonal K+ currents revealed in this study represents a new mechanism for dopaminergic regulation of neuronal signalling and supports the ‘waveform coding’ hypothesis (de Polavieja et al. 2005; Shu et al. 2006; Juusola et al. 2007; Shu, 2011). The opposite effects of D1 and D2 receptors and the absence of an effect of DA (50 μm) on axonal K+ current suggest that a balance between activities of these receptors may finely tune the neuronal and circuit activities at the PFC and thus regulate higher-order behavioural and cognitive functions; disruption of this balance may result in brain disorders such as schizophrenia and Parkinson's disease.
Acknowledgments
This work was supported by the 973 Program (2011CBA00400), the National Natural Science Foundation of China Project (31025012), the SA-SIBS Scholarship Program, the Hundreds of Talents Program, the Strategic Priority Research Program (XDA01020304) and the Knowledge Innovation Project (KSCX2-YW-R-102) from Chinese Academy of Sciences.
Glossary
- AP
action potential
- DA
dopamine
- KAP
K+ current evoked by action potential waveform
- KV (channels)
voltage-gated K+ (channels)
- PFC
prefrontal cortex
- TH
tyrosine hydroxylase
Additional information
Author contributions
J.Y. performed the electrophysiological recordings and data analysis; M.Y. helped in axon bleb recording and data analysis; C.T. and Y.W. performed immunostaining; Y.S. conceived and designed the research. Y.S. and J.Y. wrote the paper. All authors read and approved the final version of the paper.
Disclosures
None.
Supplementary material
Supplemental Figures
References
- Alle H, Geiger JR. Combined analog and action potential coding in hippocampal mossy fibers. Science. 2006;311:1290–1293. doi: 10.1126/science.1119055. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Bekkers JM. Distribution and activation of voltage-gated potassium channels in cell-attached and outside-out patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol. 2000a;525:611–620. doi: 10.1111/j.1469-7793.2000.t01-2-00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM. Properties of voltage-gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol. 2000b;525:593–609. doi: 10.1111/j.1469-7793.2000.t01-1-00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Delaney AJ. Modulation of excitability by α-dendrotoxin-sensitive potassium channels in neocortical pyramidal neurons. J Neurosci. 2001;21:6553–6560. doi: 10.1523/JNEUROSCI.21-17-06553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Ford CP, Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Uebele VN, Renger JJ, Trussell LO. Control of firing patterns through modulation of axon initial segment T-type calcium channels. J Physiol. 2012;590:109–118. doi: 10.1113/jphysiol.2011.218768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma MM, Allen ML, Martin TM, Tempel BL. PKA-dependent regulation of mKv1.1, a mouse Shaker-like potassium channel gene, when stably expressed in CHO cells. J Neurosci. 1993;13:5242–5250. doi: 10.1523/JNEUROSCI.13-12-05242.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, Pollock NJ, Frail DE, Paxson CL, Rakowski RF, Kang CH, Kebabian JW. Activation of the 5-HT1C receptor expressed in Xenopus oocytes by the benzazepines SCH 23390 and SKF 38393. Br J Pharmacol. 1991;104:1038–1044. doi: 10.1111/j.1476-5381.1991.tb12546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors EC, Ballif BA, Morielli AD. Homeostatic regulation of Kv1.2 potassium channel trafficking by cyclic AMP. J Biol Chem. 2008;283:3445–3453. doi: 10.1074/jbc.M708875200. [DOI] [PubMed] [Google Scholar]
- Cornil CA, Balthazart J, Motte P, Massotte L, Seutin V. Dopamine activates noradrenergic receptors in the preoptic area. J Neurosci. 2002;22:9320–9330. doi: 10.1523/JNEUROSCI.22-21-09320.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyrak A, Czepiel K, Mackowiak M, Chocyk A, Wedzony K. Serotonin 5-HT1A receptors might control the output of cortical glutamatergic neurons in rat cingulate cortex. Brain Res. 2003;989:42–51. doi: 10.1016/s0006-8993(03)03352-3. [DOI] [PubMed] [Google Scholar]
- de Polavieja GG, Harsch A, Kleppe I, Robinson HP, Juusola M. Stimulus history reliably shapes action potential waveforms of cortical neurons. J Neurosci. 2005;25:5657–5665. doi: 10.1523/JNEUROSCI.0242-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devoto P, Flore G, Pani L, Gessa G. Evidence for co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex. Mol Psychiatr. 2001;6:657–664. doi: 10.1038/sj.mp.4000904. [DOI] [PubMed] [Google Scholar]
- Devoto P, Flore G, Pira L, Longu G, Gessa GL. Alpha2-adrenoceptor mediated co-release of dopamine and noradrenaline from noradrenergic neurons’ in the cerebral cortex. J Neurochem. 2004;88:1003–1009. doi: 10.1046/j.1471-4159.2003.02239.x. [DOI] [PubMed] [Google Scholar]
- Di Pietro NC, Seamans JK. Dopamine and serotonin interactively modulate prefrontal cortex neurons in vitro. Biol Psychiatry. 2011;69:1204–1211. doi: 10.1016/j.biopsych.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Dong Y, Cooper D, Nasif F, Hu XT, White FJ. Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci. 2004;24:3077–3085. doi: 10.1523/JNEUROSCI.4715-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci. 2003;23:2686–2695. doi: 10.1523/JNEUROSCI.23-07-02686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+ channel inactivation gating by the cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Busi C, Spano P, Missale C. Dimerization of dopamine D1 and D3 receptors in the regulation of striatal function. Curr Opin Pharmacol. 2010;10:87–92. doi: 10.1016/j.coph.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS. Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci. 2012;32:4959–4971. doi: 10.1523/JNEUROSCI.5835-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Geijo-Barrientos E, Pastore C. The effects of dopamine on the subthreshold electrophysiological responses of rat prefrontal cortex neurons in vitro. Eur J Neurosci. 1995;7:358–366. doi: 10.1111/j.1460-9568.1995.tb00331.x. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The cortical dopamine system: role in memory and cognition. Adv Pharmacol. 1998;42:707–711. doi: 10.1016/s1054-3589(08)60846-7. [DOI] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88:3150–3166. doi: 10.1152/jn.00335.2002. [DOI] [PubMed] [Google Scholar]
- Gorelova NA, Yang CR. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol. 2000;84:75–87. doi: 10.1152/jn.2000.84.1.75. [DOI] [PubMed] [Google Scholar]
- Greif GJ, Lin YJ, Freedman JE. Role of cyclic AMP in dopamine modulation of potassium channels on rat striatal neurons: regulation of a subconductance state. Synapse. 1995;21:275–277. doi: 10.1002/syn.890210303. [DOI] [PubMed] [Google Scholar]
- Han KA, Millar NS, Grotewiel MS, Davis RL. DAMB, a novel dopamine receptor expressed specifically in Drosophila mushroom bodies. Neuron. 1996;16:1127–1135. doi: 10.1016/s0896-6273(00)80139-7. [DOI] [PubMed] [Google Scholar]
- Harris-Warrick RM, Coniglio LM, Barazangi N, Guckenheimer J, Gueron S. Dopamine modulation of transient potassium current evokes phase shifts in a central pattern generator network. J Neurosci. 1995;15:342–358. doi: 10.1523/JNEUROSCI.15-01-00342.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A, O’Dowd BF, George SR. Heteromerization of dopamine D2 receptors with dopamine D1 or D5 receptors generates intracellular calcium signaling by different mechanisms. Curr Opin Pharmacol. 2010;10:93–99. doi: 10.1016/j.coph.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Hughes MF, Schriemer DC, Walsh EJ, Walsh MP, Cole WC. Identification and functional characterization of protein kinase A-catalyzed phosphorylation of potassium channel Kv1.2 at serine 449. J Biol Chem. 2009;284:16562–16574. doi: 10.1074/jbc.M109.010918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarek LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Juusola M, Robinson HP, de Polavieja GG. Coding with spike shapes and graded potentials in cortical networks. Bioessays. 2007;29:178–187. doi: 10.1002/bies.20532. [DOI] [PubMed] [Google Scholar]
- Kang J, Huguenard JR, Prince DA. Voltage-gated potassium channels activated during action potentials in layer V neocortical pyramidal neurons. J Neurophysiol. 2000;83:70–80. doi: 10.1152/jn.2000.83.1.70. [DOI] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Sesack SR, Levey AI, Rosenberg DR. Dopamine axons in primate prefrontal cortex: specificity of distribution, synaptic targets, and development. Adv Pharmacol. 1998;42:703–706. doi: 10.1016/s1054-3589(08)60845-5. [DOI] [PubMed] [Google Scholar]
- Lin L, Sun W, Wikenheiser AM, Kung F, Hoffman DA. KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol Cell Neurosci. 2010;43:315–325. doi: 10.1016/j.mcn.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D. The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human serotonin2C receptors. Psychopharmacology (Berl) 2001;156:58–62. doi: 10.1007/s002130100742. [DOI] [PubMed] [Google Scholar]
- Mitterdorfer J, Bean BP. Potassium currents during the action potential of hippocampal CA3 neurons. J Neurosci. 2002;22:10106–10115. doi: 10.1523/JNEUROSCI.22-23-10106.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Benke D, Fritschy JM, Somogyi P. Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1996;93:11939–11944. doi: 10.1073/pnas.93.21.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru DC, Lewis DA, Gonzalez-Burgos G. Dopamine D1 receptor activation regulates sodium channel-dependent EPSP amplification in rat prefrontal cortex pyramidal neurons. J Physiol. 2007;581:981–1000. doi: 10.1113/jphysiol.2007.130864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana N, Mengod G, Artigas F. Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex. 2009;19:849–860. doi: 10.1093/cercor/bhn134. [DOI] [PubMed] [Google Scholar]
- Sather W, Dieudonne S, MacDonald JF, Ascher P. Activation and desensitization of N-methyl-D-aspartate receptors in nucleated outside-out patches from mouse neurones. J Physiol. 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Seong HJ, Carter AG. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci. 2012;32:10516–10521. doi: 10.1523/JNEUROSCI.1367-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y. Neuronal signaling in central nervous system. Sheng Li Xue Bao. 2011;63:1–8. [PubMed] [Google Scholar]
- Shu Y, Duque A, Yu Y, Haider B, McCormick DA. Properties of action-potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J Neurophysiol. 2007a;97:746–760. doi: 10.1152/jn.00922.2006. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Shu Y, Yu Y, Yang J, McCormick DA. Selective control of cortical axonal spikes by a slowly inactivating K+ current. Proc Natl Acad Sci U S A. 2007b;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci U S A. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ. Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci. 1998;18:3124–3137. doi: 10.1523/JNEUROSCI.18-09-03124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. Prog Brain Res. 1993;99:309–324. doi: 10.1016/s0079-6123(08)61354-0. [DOI] [PubMed] [Google Scholar]
- Valentijn JA, Louiset E, Vaudry H, Cazin L. Dopamine-induced inhibition of action potentials in cultured frog pituitary melanotrophs is mediated through activation of potassium channels and inhibition of calcium and sodium channels. Neuroscience. 1991;42:29–39. doi: 10.1016/0306-4522(91)90147-g. [DOI] [PubMed] [Google Scholar]
- Vincent SL, Khan Y, Benes FM. Cellular distribution of dopamine D1 and D2 receptors in rat medial prefrontal cortex. J Neurosci. 1993;13:2551–2564. doi: 10.1523/JNEUROSCI.13-06-02551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner P, Hussy N, Buell G, Jones KA, North RA. D2, D3, and D4 dopamine receptors couple to G protein- regulated potassium channels in Xenopus oocytes. Mol Pharmacol. 1996;49:656–661. [PubMed] [Google Scholar]
- Witkowski G, Szulczyk B, Rola R, Szulczyk P. D1 dopaminergic control of G protein-dependent inward rectifier K+ (GIRK)-like channel current in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2008;155:53–63. doi: 10.1016/j.neuroscience.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK, Gorelova N. Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology. 1999;21:161–194. doi: 10.1016/S0893-133X(98)00112-2. [DOI] [PubMed] [Google Scholar]
- Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci. 2005;25:9185–9194. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Jiang M, Yang M, Hou H, Shu Y. Membrane potential-dependent modulation of recurrent inhibition in rat neocortex. PLoS Biol. 2011;9:e1001032. doi: 10.1371/journal.pbio.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.