

Abstract
The extracellular matrix in the lung must be destroyed for Mycobacterium tuberculosis— the agent that causes tuberculosis (TB)—to spread. The current paradigm proposes that this destruction occurs as a result of the action of pro-inflammatory cytokines, chemokines, immune cells, and lipids that mediate TB-associated necrosis in the lung. However, this view neglects the fact that lung matrix can only be degraded by proteases. We propose an original conceptual framework of TB immunopathology that may lead directly to treatments that involve inhibition of matrix metalloproteinase activity to hinder matrix destruction and reduce the morbidity and mortality associated with TB.
INTRODUCTION
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis (Mtb), affects about one third of the world's population (1). Although most infections are asymptomatic, active tuberculosis, which usually primarily involves the lungs, is often fatal. Despite the intensive biomedical research efforts of the last two decades, the standard TB treatment regime has remained unchanged for over 30 years. Although it is widely accepted that immunopathology—that is, an excessive host immune response to mycobacterial antigens—causes mortality in patients (2) and also drives the destruction of the extracellular matrix that is necessary for the spread of M. tuberculosis, understanding of this process remains surprisingly vague (3). A contributing factor is the imprecise terminology used to describe TB-related tissue damage, such as “immunopathology,” “delayed-type hypersensitivity” (a secondary, cell-mediated immune reaction that occurs several days after antigen exposure), and “caseous necrosis” (the accumulation of amorphous debris characteristic of TB infection). Patients die from this TB-associated immunopathology, which often worsens during initial treatment, and so mortality continues even after starting effective antituberculous agents. Fear of driving pathology also prevents the development of immunostimulatory strategies to reduce treatment duration from the current minimum of 6 months. Therefore, we need a better understanding of the mechanisms of TB-related pathology in order to reduce deaths from TB and to introduce short course treatment regimes.
HISTORIC TERMINOLOGY OF TB PATHOLOGY
TB is primarily a disease of the lung, which has an intricate structure of air-filled alveoli to permit gas exchange. Mtb is phagocytosed by macrophages, the central effector cells of the innate immune response. The microscopic pathology caused by Mtb in humans has been described as caseous necrosis for over 100 years because of the accumulation of material with a cheese-like appearance in the center of TB-associated granulomas (cell aggregates—in which lymphocytes surround infected macrophages—that are thought to form to isolate Mtb). Lung infection leads to cavitation, which is the development of large air-filled spaces within the lung where the intricate parenchymal structure has been completely destroyed (Fig. 1). Mtb then proliferates exponentially in the cavity essentially walled off from the host immune response (4), and each cavity may contain up to 109 mycobacteria (5). The current paradigm of TB pathology states that accumulating caseous material erodes into an airway, creating a cavity within which the Mtb proliferates freely (Fig. 2, blue arrows). This paradigm was developed from the rabbit model during seminal work in the 1960s by Lurie and Dannenberg, and they highlighted the likely involvement of proteases, lipases, and nucleases (6). However, in current TB reviews the involvement of proteases is completely neglected (2, 3, 7–10). The observation from a series of post-mortem studies that human cavities appear to begin in areas of lipoid pneumonia (regions of lung inflammation caused by the presence of lipid-rich material), not in well organized granulomas, has generally been overlooked (11). More recent conceptual models of TB imunopathology propose an imbalance between TH1 and TH2 responses (from two types of T helper cells) (12), excessive production of interleukin-17 (a cytokine that induces proinflammatory responses) (13), or a failure of regulatory T cells to limit immunopathology (14), but none address effector mechanisms of tissue damage.
Fig. 1. Matrix destruction.
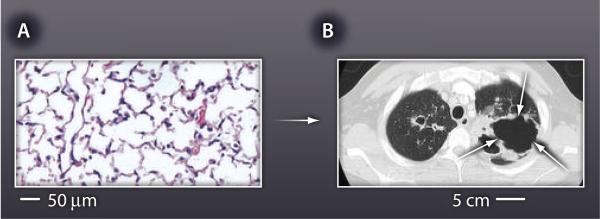
(A) Normal lung architecture is highly organized, supported by an intricate network of extracellular matrix. About 170 alveoli are present per cubic millimeter of healthy lung tissue in humans. (B) This matrix is destroyed during TB, resulting in formation of cavities that are often several centimeters across (white arrows on computerized tomography scan).
Fig. 2. Lung cavitation must result from extracellular matrix destruction.

The longstanding paradigm of TB immunopathology (blue arrows) states that cell death leads to the accumulation caseous necrosis in TB granulomas (black arrows), which then rupture into an airway, causing an air-filled cavity in the lung (white arrows on chest radiograph). This model does not explain how the extracellular matrix is degraded. We propose that two independent processes must be taking place: (A) cell death causing the accumulation of caseous material and (B) induction of protease activity to drive destruction of the extracellular matrix, which then results in cavity formation and transmission (red arrows).
All of these models overlook the fact that cavity formation must involve destruction of the lung extracellular matrix, which includes a complex, highly stable network of collagen fibrils (15) that supports the structure of the lung and that, when intact, regulates many aspects of inflammation (16). Matrix breakdown has historically been considered part of a single process of caseous necrosis (8), but the death of cells to cause caseation is likely to be a distinct process from extracellular matrix destruction (Fig. 2, red arrows). Indeed, the network of collagen fibrils can only be degraded by proteases. The cytokines, chemokines, T cells, and lipids that have been proposed as the mediators of caseous necrosis simply cannot degrade fibrillar collagens, the primary structural fibrils of the lung (15). The inescapable conclusion, we believe, is that matrix destruction that leads to lung cavitation in TB must result from protease activity.
LUNG MATRIX BIOCHEMISTRY PREDICTS A CENTRAL ROLE FOR METALLOPROTEASES
The human lung has evolved to be highly resistant to destruction of the extracellular matrix; for example, even after exposure to cigarette smoke for a lifetime, the majority of smokers have preserved lung function (17). Mtb must overcome this matrix protective environment to degrade the lung fibrils to permit transmission. Consequently, understanding the processes that drive destruction of the lung extracellular matrix is central to understanding TB immunopathology. The matrix metalloproteinases (MMPs), a family of proteases that are collectively able to degrade all components of the extracellular matrix, are the only enzymes known to cleave lung fibrillar collagens at neutral pH (18). MMPs are not stored in cells, except neutrophils, and their expression is tightly regulated at the level of gene transcription (19). Therefore, MMPs must be directly induced by Mtb as opposed to their protease activity being a by-product of cellular necrosis. Accumulating evidence implicates MMP activity in TB-related tissue destruction. For example, it was demonstrated in the 1970s that mycobacterial stimulation of guinea pig macrophages up-regulates collagenase expression (20). Virulent Mtb up-regulates MMP-1 (interstitial collagenase) more potently than does the weakened vaccine strain M. bovis BCG in primary human macrophages (21). In patients with TB, pulmonary epithelial cells and fibroblasts express MMP-1, regulated by a monocyte-dependent network (22, 23). In the zebrafish model of TB, M. marinum up-regulates epithelial cell MMP-9 secretion to generate a migration gradient for monocytes (24). Additionally, virulent M. marinum up-regulates collagenases more potently than an attenuated strain (25).
The mouse is the most widely used model of TB immunology, with key roles for CD4+ helper T cells, interferon-γ, and tumor necrosis factor–α first identified in mouse studies subsequently confirmed in man (2, 26). However, when infected with Mtb the mouse does not develop immunopathology similar to that in humans. Alveolar walls remain intact in areas of infection (26), and the mouse does not express an ortholog of MMP-1 in the lung (27, 28). Therefore, the mouse cannot be used to study MMP-1–driven matrix destruction in TB, and reliance on this model may have limited progress in understanding TB immunopathology. Despite the biochemical arguments for a central role for MMPs in TB pathology and the evidence that they play a role in destroying the lung matrix, translation to new therapies for TB has thus far been slow.
A NEW CONCEPTUAL FRAMEWORK OF TB IMMUNOPATHOLOGY
TB immunopathology is currently considered under the umbrella term of caseous necrosis, but it should be divided into fundamental processes. At least three mechanisms must be taking place to cause the immunopathology that results in caseous necrosis. First, Mtb infection is directly toxic to macrophages and causes cell necrosis (29). Second, several lines of evidence demonstrate that activated T-cells drive pathology (2). For example, in advanced HIV infection during which patients present with TB in the context of a low CD4+ cell count, caseating granulomas are not observed (2), but as the immune system reconstitutes during anti-retroviral therapy, pathology develops (30). Likewise, in the rabbit model presensitisation with serial injections of heat killed M. bovis to drive a delayed-type hypersensitivity response in the skin accelerates pathology and results in cavitation in the lungs (31). Third, MMP activity must be a final effector, causing destruction of the extracellular matrix. Considering these processes separately permits evaluation of where therapies can be most effectively targeted to limit immune-mediated tissue damage.
THE URGENT CLINICAL NEED TO LIMIT IMMUNOPATHOLOGY
TB continues to kill almost 2 million people per year (1), and these patients die from TB-related tissue destruction. Furthermore, the introduction of new anti-tuberculous drugs with rapid bactericidal activity, such as TMC207 (32), will increase the early release of Mtb antigen and consequently drive pathology. Ironically, very rapid killing of the pathogen may also kill the host if the mycobacterial load is high. Similarly, the early introduction of anti-retroviral therapy for patients with TB-HIV co-infection, which reduces mortality, will increase the incidence of TB-immune reconstitution inflammatory syndrome, which is characterized by tissue destruction (30). (This condition occurs when the immune system begins to recover and responds to Mtb infection, paradoxically causing worsening pathology.) New approaches to limit TB-related immunopathology, both in cavitatory and disseminated TB (extensive disease characterized by numerous small lesions in the lungs and other organs), are needed to reduce mortality and to permit immunostimulatory approaches to shorten treatment. If one breaks down TB pathology into its constitutive parts, MMPs emerge as the immunopathological mediators most readily targeted with orally available compounds.
MMP INHIBITION TO REDUCE IMMUNOPATHOLOGY IN TB
If MMPs are proven to be the final common effectors of matrix destruction in TB, MMP inhibition emerges as an attractive strategy to limit TB morbidity and mortality. Many MMP inhibitors were developed in the 1990s because they showed initial promise in the treatment of cancer (33). Although results of the first trials in that clinical context were disappointing, in part because of musculoskeletal side-effects, more selective inhibitors are now available. In addition, MMP inhibition in TB would be of relatively short duration. Tetracycline antibiotics are broad spectrum MMP inhibitors, acting both at a pretranscriptional level and also directly as enzyme inhibitors (34). Doxycycline (a type of tetracycline), prescribed at a sub-antimicrobial dose of 20 mg twice a day, is licensed by the U.S. Food and Drug Adminstration to limit MMP activity in periodontal disease (35). Doxycycline is cheap, safe, and widely available so could be readily deployed in resource-poor settings. A more potent, selective collagenase inhibitor, Ro32-3555, has passed phase III clinical trials for patients with arthritis (36) and so has the potential to prevent lung matrix destruction. Other MMP inhibitors have been developed that would merit study in appropriate animal models of TB (34). A central question is whether a specific MMP should be targeted or whether broad-spectrum MMP inhibition will be more efficacious.
In summary, extracellular matrix destruction is one of the critical pathological events in TB. Defining mechanisms of TB immunopathology identifies MMP activity as a potential therapeutic target to limit morbidity and mortality in TB.
Acknowledgments
P.E. and J.S.F. are grateful for support from the National Institute for Health Research Biomedical Research Centre funding scheme at Imperial College. We thank L. Ramakrishnan and J. Green for critically reviewing the manuscript and F. Mauri for providing the histological image. Funding: P.E. is supported by the UK National Institute for Medical Research. J.S.F.'s research on MMPs is principally supported by the Medical Research Council (UK) and The Wellcome Trust.
Footnotes
Competing interests: The authors declare that they have no competing financial interests.
Citation: P. Elkington, J. M. D'Armiento, J. S. Friedland, Tuberculosis immunopathology: The neglected role of extracellular matrix destruction. Sci. Transl. Med. 3, 71ps6 (2011).
REFERENCES AND NOTES
- 1.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 2.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell DG, Barry CE, 3rd, Flynn JL. Tuberculosis: what we don't know can, and does, hurt us. Science. 2010;328:852–856. doi: 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, Mathema B, Ramaswamy SV, Walther G, Steyn LM, Barry CE, 3rd, Bekker LG. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 2003;71:7099–7108. doi: 10.1128/IAI.71.12.7099-7108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helke KL, Mankowski JL, Manabe YC. Animal models of cavitation in pulmonary tuberculosis. Tuberculosis (Edinb.) 2006;86:337–348. doi: 10.1016/j.tube.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Dannenberg AM, Jr M. Sugimoto, Liquefaction of caseous foci in tuberculosis. Am. Rev. Respir. Dis. 1976;113:257–259. doi: 10.1164/arrd.1976.113.3.257. [DOI] [PubMed] [Google Scholar]
- 7.Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, Niemann S, Rohde KH. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe. 2010;8:68–76. doi: 10.1016/j.chom.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell DG. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 2007;5:39–47. doi: 10.1038/nrmicro1538. [DOI] [PubMed] [Google Scholar]
- 9.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009;10:943–948. doi: 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter RL, Jagannath C, Actor JK. Pathology of postprimary tuberculosis in humans and mice: contradiction of long-held beliefs. Tuberculosis (Edinb.) 2007;87:267–278. doi: 10.1016/j.tube.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Dheda K, Booth H, Huggett JF, Johnson MA, Zumla A, Rook GA. Lung remodeling in pulmonary tuberculosis. J. Infect. Dis. 2005;192:1201–1209. doi: 10.1086/444545. [DOI] [PubMed] [Google Scholar]
- 13.Desvignes L, Ernst JD. Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am. J. Respir. Crit. Care Med. 2006;173:803–810. doi: 10.1164/rccm.200508-1294OC. [DOI] [PubMed] [Google Scholar]
- 15.Davidson JM. Biochemistry and turnover of lung interstitium. Eur. Respir. J. 1990;3:1048–1063. [PubMed] [Google Scholar]
- 16.Sorokin L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010;10:712–723. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher C, Peto R. The natural history of chronic airflow obstruction. BMJ. 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parks WC, Wilson CL, López-Boado YS. Matrix metal-loproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 20.Wahl SM, Wahl LM, McCarthy JB, Chedid L, Mergenhagen SE. Macrophage activation by mycobacterial water soluble compounds and synthetic muramyl dipeptide. J. Immunol. 1979;122:2226–2231. [PubMed] [Google Scholar]
- 21.Elkington PT, Nuttall RK, Boyle JJ, O'Kane CM, Horncastle DE, Edwards DR, Friedland JS. Mycobacterium tuberculosis, but not vaccine BCG, specifically upregulates matrix metalloproteinase-1. Am. J. Respir. Crit. Care Med. 2005;172:1596–1604. doi: 10.1164/rccm.200505-753OC. [DOI] [PubMed] [Google Scholar]
- 22.Elkington PT, Emerson JE, Lopez-Pascua LD, O'Kane CM, Horncastle DE, Boyle JJ, Friedland JS. Mycobacterium tuberculosis up-regulates matrix metalloproteinase-1 secretion from human airway epithelial cells via a p38 MAPK switch. J. Immunol. 2005;175:5333–5340. doi: 10.4049/jimmunol.175.8.5333. [DOI] [PubMed] [Google Scholar]
- 23.O'Kane CM, Elkington PT, Friedland JS. Monocyte-dependent oncostatin M and TNF-α synergize to stimulate unopposed matrix metalloproteinase-1/3 secretion from human lung fibroblasts in tuberculosis. Eur. J. Immuno l. 2008;38:1321–1330. doi: 10.1002/eji.200737855. [DOI] [PubMed] [Google Scholar]
- 24.Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 2010;327:466–469. doi: 10.1126/science.1179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Sar AM, Spaink HP, Zakrzewska A, Bitter W, Meijer AH. Specificity of the zebrafish host transcriptome response to acute and chronic mycobacterial infection and the role of innate and adaptive immune components. Mol. Immunol. 2009;46:2317–2332. doi: 10.1016/j.molimm.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 26.North RJ, Jung YJ. Immunity to tuberculosis. Annu. Rev. Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 27.Balbín M, Fueyo A, Knäuper V, López JM, Alvarez J, Sánchez LM, Quesada V, Bordallo J, Murphy G, López-Otín C. Identification and enzymatic characterization of two diverging murine counterparts of human interstitial collagenase (MMP-1) expressed at sites of embryo implantation. J. Biol. Chem. 2001;276:10253–10262. doi: 10.1074/jbc.M009586200. [DOI] [PubMed] [Google Scholar]
- 28.Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Lett. 2004;563:129–134. doi: 10.1016/S0014-5793(04)00281-9. [DOI] [PubMed] [Google Scholar]
- 29.Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meintjes G, Lawn SD, Scano F, Maartens G, French MA, Worodria W, Elliott JH, Murdoch D, Wilkinson RJ, Seyler C, John L, van der Loeff MS, Reiss P, Lynen L, Janoff EN, Gilks C. R. ColebundersInternational Network for the Study of HIV-associated IRIS, Tuberculosis-associated immune reconstitution inflammatory syndrome: case definitions for use in resource-limited settings. Lancet Infect. Dis. 2008;8:516–523. doi: 10.1016/S1473-3099(08)70184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nedeltchev GG, Raghunand TR, Jassal MS, Lun S, Cheng QJ, Bishai WR. Extrapulmonary dissemination of Mycobacterium bovis but not Mycobacterium tuberculosis in a bronchoscopic rabbit model of cavitary tuberculosis. Infect. Immun. 2009;77:598–603. doi: 10.1128/IAI.01132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andries K, Verhasselt P, Guillemont J, Göhlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 33.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 34.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 35.Golub LM, McNamara TF, Ryan ME, Kohut B, Blieden T, Payonk G, Sipos T, Baron HJ. Adjunctive treatment with subantimicrobial doses of doxycycline: effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J. Clin. Periodontol. 2001;28:146–156. doi: 10.1034/j.1600-051x.2001.028002146.x. [DOI] [PubMed] [Google Scholar]
- 36.Hemmings FJ, Farhan M, Rowland J, Banken L, Jain R. Tolerability and pharmacokinetics of the collagenase-selective inhibitor Trocade in patients with rheumatoid arthritis. Rheumatology (Oxford) 2001;40:537–543. doi: 10.1093/rheumatology/40.5.537. [DOI] [PubMed] [Google Scholar]