

Abstract
In a recent experiment, the repair efficiency of DNA thymine cyclobutane dimers (T<>T) on UV excitation of 8-oxoG base paired either to C or A was reported. An electron transfer mechanism from an excited charge transfer state of 8-oxoG-C (or 8-oxoG-A) to T<>T was proposed and 8-oxoG-A was found to be 2 – 3 times more efficient than 8-oxoG-C in repair of T<>T. Intra base pair proton transfer (PT) in charge transfer (CT) excited states of the base pairs was proposed to quench the excited state and prevent T<>T repair. In this work, we investigate this process with TD-DFT calculations of the excited states of 8-oxoG-C and 8-oxoG-A base pairs in the Watson-Crick and Hoogsteen base pairs using long-range corrected density functional, ωB97XD/6–31G* method. Our gas phase calculations showed that CT excited state (1ππ*(CT)) of 8-oxoG-C appears at lower energy than the 8-oxoG-A. For 8-oxoG-C, TD-DFT calculations show the presence of a conical intersection (CI) between the lowest 1ππ*(PT-CT) excited state and the ground state which likely deactivates the CT excited state via a proton-coupled electron transfer (PCET) mechanism. The 1ππ*(PT-CT) excited state of 8-oxoG-A base pair lies at higher energy and its crossing with ground state is inhibited because of a high energy gap between 1ππ*(PT-CT) excited state and ground state. Thus the gas phase calculations suggest the 8-oxoG-A would have longer excited state lifetimes. When the effect of solvation is included using the PCM model, both 8-oxoG-A and 8-oxoG-C show large energy gaps between the ground state and both the excited CT and PT-CT states and suggest little difference would be found between the two base pairs in repair of the T<>T lesion. However, in the FC region the solvent effect is greatly diminished owing to the slow dielectric response time and smaller gaps would be expected.
Keywords: 8-oxo-7,8-dihydroguanine (8-oxoG); excited state; proton-coupled electron transfer; PCET; excited state electron-induced proton transfer; density functional; thymine dimer (T<>T)
1. Introduction
DNA, the carrier of genetic information for all cellular forms of life, is susceptible to specific types of DNA damage from ultraviolet (UV) light. The major photoproducts on UV radiation of cellular DNA are the cyclobutane pyrimidine dimers, such as, the thymine cyclobutane photodimer (T<>T) formed by the [2+2] cycloaddition of the stacked thymines.1–6 Using femtosecond time-resolved infrared spectroscopy Schreier et al.6 showed the formation of cyclobutane dimers in the oligodeoxynucleotide (dT)18 within ca. 1 picosecond after the UV excitation at 272 nm. In a very recent study, Pan et al.7 have determined the quantum yield for the formation of T<>T for a series of short DNA single-strand and base-paired hairpin structures possessing a single thymine-thymine step with flanking purines. These authors found that the quantum yield for the formation of T<>T photodimer was very strongly dependent upon the oxidation potential of the purines and decreasing in the order: hypoxanthine > adenine > guanine > deazaguanine.7 Mechanisms of repair of T<>T photodimers are of considerable interest3,8,9–18. Studies have shown that both: (i) direct photoreversal3,8 and (ii) indirect photoreversal9–14 mechanisms are possible. We note that nucleotide excision repair (NER) is also a versatile repair pathway which involves enzymatic removal of the UV-induced cyclobutane pyrimidine dimers.16–18
Very recently, Nguyen and Burrows studied the photoreversal of damaged T<>T present in DNA duplexes containing 8-oxo-7,8-dihydroguanine (8-oxoG). In their experiment, they exposed the damaged DNA to UV light that electronically excited an 8-oxoG which was placed at different positions with respect to a T<>T in DNA and monitored the repair rates of the T<>T.12–14 The results found suggest that the T<>T repair via photoreversal supported a catalytic role played by 8-oxoG. The mechanism involved the transfer of an excess electron from the excited 8-oxoG to the T<>T. This resulted in the cleavage of the T<>T cyclobutane ring which is followed by back electron transfer to 8-oxoG•+ restoring the original 8-oxoG. The repair rate was found to depend on both the location of the 8-oxoG and on its base-pairing partner in the damaged DNA. When 8-oxoG paired with cytosine (8-oxoG-C) in the Watson-crick conformation the repair was 2 – 3 times slower than the 8-oxoG paired with adenine (8-oxoGsyn-Aanti) in a Hoogsteen base pair.13,14 To account for the slow repair rate of T<>T by the excited 8-oxoG-C base pair it was speculated that a fast deactivation of the CT excited state in 8-oxoG-C base pair occurred via a proton-coupled electron transfer (PCET) mechanism. However, for 8-oxoGsyn-Aanti deactivation of the CT excited state through PCET was suggested to be less effective than found for 8-oxoG-C because the repair efficiency of nearby T<>T was higher for 8-oxoGsyn-Aanti than for 8-oxoG-C. Such fast deactivation mechanisms have been shown to occur within a picosecond and non-radiative decay of CT excited states in several DNA base pairs (G-C, A-T and a model base pair) through a PCET mechanism has already been proposed in the literature.19–25
8-oxoG is the most common oxidatively generated DNA damage caused by ionizing or UV radiation and usually results from •OH addition to guanine or nucleophilic addition of water to one-electron oxidized guanine.26–28 The mechanism of 8-oxoG formation in cellular DNA has been well documented recently by Cadet et al.29,30 In DNA, 8-oxoG base pairs with cytosine in the Watson-Crick hydrogen-bonded fashion and it can also base pair with adenine in the Hoogsteen base pair between 8-oxoG and adenine in which they adopt syn- and anti-conformations (8-oxoGsyn-Aanti) in duplex DNA.31,32
The standard reduction (SR) potential for the oxidation of guanine is the lowest (1.3 V) of all four main DNA bases. This has important implications in DNA since guanine acts as a “hole” sink and protects other bases from damage by one-electron oxidation. The SR potential for oxidation of 8-oxoG is (0.7 V)14 and it is significantly lower than that of guanine. Thus, 8-oxoG is a far better “hole” scavenger than guanine in DNA.33 While the excited states of the G-C base pair have been extensively studied by experiment and theory in order to better understand the deactivation mechanism of the charge transfer excited state19–25,34, theoretical studies of the excited states of 8-oxoG-C or 8-oxoG-A base pairs have not been performed. The study of the excited states of 8-oxoG-C or 8-oxoG-A base pairs are of interest to understand the effect of possible charge transfer excited state formation on the T<>T repair process. In this work, we have investigated the excited states of 8-oxoG-C and 8-oxoG-A (8-oxoGsyn-Aanti) base pairs using long-range corrected time-dependent density functional theory (TD-DFT).
The organization of the present study is as follows: In section 2, the details of the ground and excited states calculations using the ωB97XD density functional is given. In section 3.1, the accuracy of the chosen functional for excited state calculation is compared with the available CC2 excited state energies of the G-C and A-T base pairs. The geometries of the ground and excited states, vertical excitation energies of 8-oxoG-C and 8-oxoG-A base pairs and the potential energy profile of the decay path of the 8-oxoG moiety in the two base pairs in the gas phase and in solution are discussed in sections 3.2 to 4.0. The outcome of the results is discussed in section 4.1.
2. Method of Calculations
In this work, we employed ωB97XD density functional recently developed by Chai and Head-Gordon.35,36 ωB97XD is a long-range corrected hybrid density functional with damped atom–atom dispersion corrections and has been found more reliable for the calculation of the dispersion as well as charge transfer (CT) excited states than results found using earlier density functionals.35,36 The initial geometry of 8-oxoG-C was generated by placing 8-oxoG and C monomers in the hydrogen-bonded Watson-Crick conformation while the geometry of 8-oxoG-A base pair was generated by placing the 8-oxoG and A in a Hoogsteen base pair. We attached methyl group to N9 of guanine and adenine and N1 of cytosine in the base pairs to mimic the effect of sugar ring as these sites are attached to the sugar ring in the DNA. The initial geometries of 8-oxoG-C and 8-oxoG-A thus generated were used for the optimization of their ground and excited states using the ωB97XD/6–31G* method. The ground state optimized geometries were used for calculation of the vertical excited states using the TD-ωB97XD/6–31G* method. The potential energy surface (PES) for proton transfer in the lowest CT excited state in the vertical and adiabatic states were calculated by choosing N1-H and N7-H bond lengths of 8-oxoG as reaction coordinate in 8-oxoG-C and 8-oxoG-A base pairs, respectively. For adiabatic calculations, at each chosen N-H bond length on the PES, the CT excited state was fully optimized by only constraining the corresponding bond length. The effect of solvation on the excited states of these base pairs are discussed in section 4.0. All the calculations were carried out using Gaussian 09 suite of programs.37 JMOL38 and GaussView39 molecular modeling software were used to draw molecular structures and molecular orbitals. In the text, we label highest occupied molecular orbital (HOMO) as H and the lowest unoccupied molecular orbital (LUMO) as L.
3. Results and Discussion
3.1 Suitability of the ωB97XD method for excited state calculation
To test the suitability of the ωB97XD/6–31G* method for the excited state calculations involving charge transfer excited states, we considered guanine(G)-cytosine(C) and adenine(A)-thymine(T) base pairs as test cases and calculated their singlet vertical excitation energies. The calculated excitation energies of G-C and A-T base pairs by the TD-ωB97XD/6–31G* method were compared with those calculated in references 40 and 41 using the Coupled Cluster singles and doubles (CC2) method42, 43, see Tables 1 and 2.
Table 1.
Comparison of Methods: Vertical low-lying singlet excited state energies (ΔE) in eV and oscillator strength (f) of G-C base pair calculated with the CC2/TZVP and ωB97XD/6–31G* methods (ε = 1).
| State | G-C base pair | |||
|---|---|---|---|---|
| CC2/TZVPa | ωB97XD/6–31G*b | |||
| Transition | ΔE(f) | Transition | ΔE (f) | |
| S1 | G(π) → G(π*) | 4.88 (0.061) | G(π) → G(π*) | 5.22 (0.0616) |
| S2 | C(π) → C(π*) | 5.06 (0.063) | C(π) → C(π*) | 5.32 (0.1036) |
| S3 | G(π) → C(π*) (CT)c | 5.23 (0.028) | G(π) → C(π*) (CT)c | 5.45 (0.0114) |
| S4 | C(n) → C(π*) | 5.49 (0.001) | C(n) → C(π*) | 5.72 (0.0010) |
| S5 | C(π) → C(π*) | 5.50 (0.183) | C(π) → C(π*) | 5.92 (0.1238) S6)d |
| S6 | G(π) → G(π*) | 5.51 (0.425) | G(π) → G(π*) | 5.75 (0.4124) (S5)d |
| S7 | C(n) → C(π*) | 5.80 (0.000) | C(n) → C(π*) | 6.45 (0.0001) (S8)d |
| S8 | G(n) → G(π*) | 5.90 (0.000) | G(n) → G(π*) | 5.94 (0.0005) (S7)d |
Table 2.
Comparison of Methods: Vertical low-lying singlet excited state energies (ΔE) in eV and oscillator strength (f) of A-T base pair calculated with the CC2/TZVP and ωB97XD/6–31G* methods (ε = 1).
| State | A-T base pair | |||
|---|---|---|---|---|
| CC2/TZVPa | ωB97XD /6–31G*b | |||
| Transition | ΔE | Transition | ΔE (f) | |
| S1 | T(n) → T(π*) | 4.94 | T(n) → T(π*) | 5.33 (0.0001) |
| S2 | T(π) → T(π*) | 5.21 | T(π) → T(π*) | 5.41 (0.1777) |
| S3 | A(π) → A(π*) | 5.40 | A(π) → A(π*) | 5.51 (0.1132) |
| S4 | A(π) → A(π*) | 5.47 | A(π) → A(π*) | 5.57 (0.1947) |
| S5 | A(n) → A(π*) | 5.54 | A(n) → A(π*) | 5.63 (0.0003) |
| S6 | - | - | A(n) → A(π*) | 6.11 (0.0001) |
| S7 | A(π) → T(π*) | 6.04 | A(π) → T(π*) | 6.32 (0.0034) |
Ref. 36.
Present calculation.
For G-C base pair in Cs symmetry, the TD-ωB97XD/6–31G* method predicts the lowest two transitions (S1 and S2) as local excitations (LE) which are 1ππ* in nature. The corresponding transition energies are 5.22 eV and 5.32 eV, respectively, and the oscillator strengths of these states are 0.0616 and 0.1036, see Table 1. The third transition (S3) occurs at 5.45 eV with oscillator strength 0.0114 and it is also 1ππ*. It is due to the charge transfer from guanine to cytosine and designated as G(π)→C(π*) in Table 1. The S4 transition is a local excitation C(n)→C(π*) with excitation energy 5.72 eV. The S5, S6, S7 and S8 transitions are local excitations and they are 1ππ*, 1ππ*, 1nπ* and 1nπ* types and occur at excitation energies 5.75 eV, 5.92 eV, 5.94 eV, 6.44 eV and 6.45 eV, respectively. The excited states of G-C base pair in Cs symmetry was studied by Yamazaki and Taketsugu40 using the CC2/TZVP method. The CC2/TZVP method also predicts the lowest four transitions (S1 – S4) as 1ππ*(LE), 1ππ*(LE), 1ππ*(CT) and 1nπ*(LE) as calculated using the ωB97XD/6–31G* method. The CC2/TZVP calculated excitation energies are 4.88 eV, 5.06 eV, 5.23 eV and 5.49 eV, respectively, see Table 1. The CC2/TZVP method predicts S5 and S6 as local excitations C(π)→C(π*), and G(π)→G(π*) while ωB97XD/6–31G* method predicts these transitions as G(π)→G(π*) and C(π)→C(π*). The S7 and S8 transitions calculated by the CC2/TZVP method are C(n)→C(π*) and G(n)→G(π*). Thus, we see that the ωB97XD/6–31G* method predicts S1 – S4 transitions of G-C base pair in good agreement with those calculated by the CC2/TZVP method having a maximum difference of ca. 0.3 eV, see Table 1.
Using the CC2/TZVP method, Lange and Herbert41 calculated the six vertical singlet excitation energies of A-T base pair. The first transition (S1) of A-T base pair, predicted by the CC2/TZVP method is a local excitation T(n)→T(π*) and occurs at 4.94 eV, see Table 2. The S2 – S4 transitions are 1ππ* type local excitations having excitation energies 5.21 eV, 5.40 eV and 5.47 eV, respectively, while the S5 transition was predicted to be A(n)→A(π*) type. The 6th transition (labeled as S7 in Table 2) calculated by the CC2/TZVP method is a charge transfer excitation A(π)→T(π*) and has excitation energy 6.04 eV. The ωB97XD/6–31G* calculated excitation energies of A-T base pair matched very well with those calculated by the CC2/TZVP method and have a maximum difference of ca. 0.3 eV except the first transition (S1) for which the difference is ca. 0.4 eV. Thus, from these two test cases (G-C and A-T base pairs) we found that the ωB97XD /6–31G* method is suitable for the excited state calculations having the charge transfer excitation and the calculated excitation energies are in excellent agreement with those calculated by the CC2/TZVP method.40,41
3.2 Ground state geometry of 8-oxoG-C
The ground state (S0) optimized geometry of 8-oxoG-C base pair in the Watson-Crick hydrogen-bonded conformation, calculated by the ωB97XD/6–31G* method, is presented in Figure 1(a). The atom numbering of 8-oxoG-C is shown in Table 3. The optimized structure is perfectly planar except the methyl groups attached to N9(G) and N1(C) sites. The bond angles surrounding atoms N2(G) and N4(C) constituting the NH2 groups are 360 deg. which shows that NH2 groups are planar in the base pair. The ωB97XD/6–31G* calculated distances between O6(G)---N4(C), N1(G)---N3(C) and N2(G)---O2(C) in 8-oxoG-C involved in hydrogen bonding are 2.81 Å, 2.91 Å and 2.90 Å, respectively. These calculated distances are in excellent agreement with those determined experimentally by X-ray31 and the corresponding distances are 2.85 Å, 2.95 Å and 2.84 Å, respectively.
Figure 1.

The ωB97XD/6–31G* calculated optimized structures of 8-oxoG-C base pair in (a) ground state (S0), (b) 1ππ* LE, (c) 8-oxoG•+-C•−; 1ππ*(CT) (N1-H and N2-H in 8-oxoG were constrained during optimization) and (d) PT-CT 8-oxoG(-H)•-C(H)•; (1ππ*(CT)). The pink circle shows the location of proton in the base pairs.
Table 3.
Vertical lowest lying singlet excited state energies (ΔE) in eV and oscillator strengths (f) of 8-oxoG-C base pair calculated using the TD-ωB97XD/6–31G* method (ε = 1).
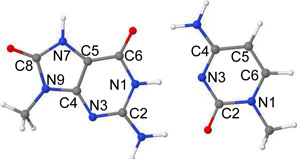 | |||||
| State | Transition | ΔE (eV) | f | Configuration | Naturea |
|---|---|---|---|---|---|
| S1 | 8-oxoG(π) → 8-oxoG(π*) | 4.79 | 0.1233 | ~35% | LE |
| S2 | 8-oxoG(π) → C(π*) | 5.17 | 0.0348 | ~40% | CT |
| S3 | C(π) → C(π*) | 5.26 | 0.1153 | ~38% | LE |
| S4 | 8-oxoG(π) → 8-oxoG(π*) | 5.78 | 0.3145 | ~46% | LE |
| S5 | C(n) → C(π*) | 5.81 | 0.0009 | ~21% | LE |
| S6 | C, 8-oxoG(π) → C(π*) | 5.91 | 0.1067 | ~40%; delocalized on C and 8OG | LE |
| S7 | 8-oxoG(n) → 8-oxoG(π*) | 6.19 | 0.0001 | ~19% | LE |
LE= local excitation; CT= charge transfer.
3.3 Vertical excited states of 8-oxoG-C
The lowest seven (S1 – S7) vertical singlet excited states of 8-oxoG-C base pair calculated using the TD-ωB97XD/6–31G* method are presented in Table 3 and the molecular orbitals involved in the excitation are presented in Figure 2. The optimized ground state (S0) geometry shown in Figure 1(a) is used for vertical excited state calculation. The TD-ωB97XD/6–31G* method predicts first transition (S1) as 1ππ*(LE) localized on 8-oxoG with an excitation energy 4.79 eV and oscillator strength 0.1233. This transition (S1) is dominated by H→L+2 configuration, see Figure 2. The second transition (S2) is a charge transfer excitation and 1ππ* in nature and occurs at 5.17 eV with oscillator strength 0.0348. The charge transfer excitation (S2) occurs between H→L and in this state 8-oxoG-C becomes 8-oxoG•+-C•−. The S3, S4 and S6 excitations are 1ππ*(LE) while S5 and S7 excitations are 1nπ*(LE), see Table 3. The S3, S4 and S6 excitations are dominated by H-2→L, H→L+3 and H-4→L and S5 and S7 excitations are dominated by H-10→L and H-5→L+2, see Figure 2. In comparison to charge transfer excitation (5.45 eV) in G-C base pair (Table 1), the charge transfer excitation in 8-oxoG-C occurs at lower energy (5.17 eV; Table 3) and it is the second transition while in G-C base pair it is the third transition. The oscillator strength in both the systems are relatively low compared to the oscillator strength of the local 1ππ* excitation. Thus, it is possible that these charge transfer states are populated by the nearby states having strong oscillator strengths and small energy difference, see Tables 1 and 2, respectively. Also, smaller charge transfer excitation of 8-oxoG-C than G-C is in accord with 8-oxoG being the site for primary oxidation instead of guanine in DNA. This is supported from the fact that the oxidation potential of 8-oxoG is ca. 0.6 V lower than G.12–14
Figure 2.

Vertical excitation energy in eV of 8-oxoG-C base pair obtained using the TD-ωB97XD/6–31G* method(ε = 1). The nature of the transition and molecular orbitals involved in the excitation (S1 – S7) are also given. H= HOMO (highest occupied molecular orbital); L= LUMO (lowest unoccupied molecular orbital). For details, see Table 3.
3.4 Excited state geometries of 8-oxoG-C
The ωB97XD/6–31G* optimized structure of locally excited 1ππ*(LE) state (S1) of 8-oxoG-C base pair is shown in Figure 1(b) and the optimized structure is planar. The hydrogen bonding distances between O6(G)---N4(C), N1(G)---N3(C) and N2(G)---O2(C) are 2.81 Å, 2.89 Å and 2.90 Å, respectively, which are similar to those calculated in the ground state, see Figure 1(a). Since the excitation is local to the 8-oxoG, there is no appreciable structural change in the cytosine ring as the bond lengths are almost identical to the ground state structure, see Figures 1(a) and 1(b). However, as expected the 8-oxoG ring shows some structural change in compare to the ground state structure. In the excited state, the C4–C5 bond length increases by ca. 0.07 Å, C8-N7 bond length increases by ca. 0.03 Å and C8-N9 bond length decreases by 0.03 Å, respectively.
Interestingly, the full optimization of the charge transfer excited state 1ππ*(CT) of 8-oxoG-C converges to the structure in which the N1 proton (H+) of 8-oxoG transferred to the N3 of cytosine. To scan the adiabatic surface from the initial state to the final proton transferred state (see Figure 1(d)), we constrained the bond lengths of N1-H and N2-H of 8-oxoG to their ground state equilibrium bond lengths (see Figure 1(a)) and optimized the structure in the charge transfer 1ππ*(CT) excited state using the TD-ωB97XD/6–31G* method. The resulting optimized structure is shown in Figure 1(c). Electron transfer from 8-oxoG to cytosine creates the charge transfer excited state. Thus, in the CT excited state 8-oxoG becomes a radical cation (8-oxoG•+) and cytosine becomes anion radical (C•−). On optimization of the excited 8-oxoG•+-C•− CT state, a large structural change occurs in the cytosine ring and creates a slightly non-planar overall structure. The NH2 group of the cytosine becomes pyramidal (non-planar) and the angle surrounding the N4(C) is 331.7 deg. The non-planarity in the NH2 group in the excited state has been found in earlier studies.40,44–46 The hydrogen bond distances between O6(G)---N4(C), N1(G)---N3(C) and N2(G)---O2(C) are 3.15 Å, 2.67 Å and 2.61 Å, respectively. In comparison to ground state, the N1(G)---N3(C) and N2(G)---O2(C) distances decrease by ca. 0.3 Å while the O6(G)---N4(C) distance substantially increases by ca. 0.3 Å. The C4–C5 bond of 8-oxoG lengthens by 0.04 Å, the N3-C4 and C4–C5 bonds of cytosine increase and decrease by 0.06 Å, respectively.
The optimized proton transferred charge transfer (PT-CT) excited state 1ππ* of 8-oxoG-C is shown in Figure 1(d). In this PT-CT excited state the base pair 8-oxoG-C becomes a diradical, i.e. 8-oxoG(-H)•-C(H)•. The structure is predicted to be planar by the ωB97XD/6–31G* method except for the NH2 group of cytosine which shows small non-planarity (sum of angles surrounding N4 is 342 deg). The hydrogen bond distances O6(G)---N4(C), N1(G)---N3(C) and N2(G)---O2(C) are 3.08 Å, 2.96 Å and 2.86 Å, respectively. In comparison to ground state, the N3-C4 bond of cytosine increases by 0.07 Å while C4–C5 bond of cytosine decreases by 0.07 Å.
3.5 Excited state PCET reaction in 8-oxoG-C
The TD-ωB97XD/6–31G* calculated potential energy profile for intra base pair proton transfer from N1 of 8-oxoG to N3 of cytosine (see Table 3 for atom numbering) in the ground (S0) and four lowest vertical excited states (1ππ*(LE), 1ππ*(CT), 1ππ*(LE) and1ππ*(LE)) of 8-oxoG-C is shown in Figure 3. Along with the vertical excited state potential energy profile, we also present the optimized lowest 1ππ*(LE)OPT and 1ππ*(CT)OPT excited states calculated along the minimum-energy path for the proton transfer from N1(8-oxoG) and N3(C) in 8-oxoG-C using the same level of theory.
Figure 3.

Potential energy surface (PES) profile of the ground (S0), the three lowest 1ππ*(LE) and the lowest 1ππ*(CT) excited states of 8-oxoG-C base pair calculated using the TD-ωB97XD/6–31G* method (ε = 1) considering N1-H of 8-oxoG as a reaction coordinate, see Figure 1(a) and Table 3. The optimized lowest 1ππ*(CT)OPT state is also shown. The ground state (S0’) surface is obtained from a single point energy calculation using the corresponding optimized geometries of 1ππ*(CT)OPT states. All the energies were calculated using the energy of the optimized ground state of 8-oxoG-C base pair, shown in Figure 1(a), as reference. All states are vertical except for those labeled OPT which are adiabatic.
The potential energy profile, calculated in the vertical excited states, shows that the energy of the ground state (S0) and three local excited states (1ππ*(LE)) increase with increasing N1-H distance, see Figure 3. However, the vertical charge transfer excited state (1ππ*(CT)) decreases with increasing N1-H distance. Therefore it is dissociative in nature and crosses the lowest local excited state (1ππ*(LE)) at N1-H distance around 1.25 Å, (see pink and green curves in Figure 3). Inspection of the vertical charge transfer excited state (1ππ*(CT); green curve) in Figure 3 shows that proton transfer from N1 of 8-oxoG to N3 of C is barrierless and exothermic in nature and predicts a shallow minimum around N1-H=1.8 Å. The energy gap between ground (S0) and proton transferred 1ππ*(CT) states at N1-H=1.8 Å is ca. 2.8 eV. This state (1ππ*(CT)) is biradical in nature, i.e. 8-oxoG(-H)•-C(H)•.
From Figure 3, it is evident that adiabatic lowest 1ππ*(LE)OPT (black curve) also shows bound nature as in the vertical state, i.e. the energy of this state (1ππ*(LE)OPT) rises with the increase of N1-H bond distance. The excited state geometry of 1ππ*(LE)OPT is given in Figure 1(b). The adiabatic charge transfer (1ππ*(CT)OPT; orange curve) is predicted to be the lowest excited state and in this state the structure is stabilized by a subsequent proton transfer from 8-oxoG to C without a substantial barrier. The TD-ωB97XD/6–31G* optimized geometry of the PT-CT excited state is shown in Figure 1(d). The optimization of this state shows that during proton transfer from 8-oxoG to C a small non-planarity in the structure takes place and after complete proton transfer the resulting structure (Figure 1(d)) becomes planar. The S0’ (ground state; lime color curve) is calculated using the corresponding 1ππ*(CT)OPT optimized excited state geometries. A crossing (conical intersection (CI)) is expected between the falling charge transfer 1ππ*(CT)OPT excited state and the continuous rising ground state S0’ curve. Such an intersection would efficiently deactivate the 1ππ*(CT)OPT back to the ground state.20,25 In the present calculation, we are not able to optimize the structure at the conical interaction because of the limitations of the TD-DFT method.47 However, several studies pointed out that TD-DFT can provide reliable estimates of the geometry and the energy of minimum energy CI for single excitations.4,40,48 Our calculations show the presence of the stabilized charge transfer excited state (1ππ*(CT)OPT) at 0.6 eV from the ground state, see points enclosed by the broken circle in Figure 3. This relatively small energy difference clearly suggests that an efficient relaxation of the charge transfer excited state to the ground state is possible in 8-oxoG-C Watson-Crick conformation that is driven by an excited state PCET mechanism. Similar mechanism has been proposed in the literature for the G-C base pair19–21,23–25, 40 and in another model base pair.22 It is interesting to note that for G-C base pair in the Watson-Crick conformation, Sobolewski et al.23 also calculated the energy gap (ca. 0.6 eV) between the proton transferred-charge transfer excited state and the ground state using the CC2 method as calculated for 8-oxoG-C in the present study.
3.6 Ground state geometry of 8-oxoG-A
The ground (S0) state geometry of 8-oxoG-A base pair in Hoogsteen base pair arrangement, optimized by the ωB97XD/6–31G* method, is given in Figure 4(a). The Hoogsteen 8-oxoG-A base pair, in its ground state, has two hydrogen bonds between O6(8-oxoG)---N6(A) and N7(8-oxoG)---N1(A). The corresponding crystallographic distances are 3.06 Å and 2.70 Å, respectively.32 These hydrogen bond distances calculated by the ωB97XD/6–31G* method are 2.92 Å and 2.82 Å, respectively. The calculated structure adopts perfectly planar structure except obviously for the hydrogens of the methyl groups at N9 sites of both the bases.
Figure 4.

ωB97XD/6–31G* calculated optimized structures of 8-oxoG-A base pair in (a) ground state (S0) and (b) proton transferred 8-oxoG(-H)•-A(H)•; (1ππ*(CT)). Pink circle in (a) and (b) shows the location of transferring proton.
3.7 Vertical excited states of 8-oxoG-A
The nine lowest (S1 – S9) vertical excitation energies (ΔE in eV), oscillator strength (f) and type of transition, calculated by the TD-ωB97XD/6–31G* level of theory for 8-oxoG-A, are presented in Table 4. Molecular orbitals involved in the excitations are shown in Figure 5. For 8-oxoG-A, the TD-ωB97XD/6–31G* method predicts eight transitions (S1 – S6, S8 and S9) as local excitations and S7 as charge transfer. The S1 and S2 excitations are 1ππ*(LE) with energies 4.61 eV and 5.44 eV and oscillator strengths 0.2128 and 0.0936, see Table 4. These transitions (S1 and S2) take place mainly between H→L and H→L+2 molecular orbitals localized on 8-oxoG, see Figure 5. The S3, S4 and S5 excitations are 1ππ*(LE), 1ππ*(LE) and 1nπ*(LE) and localized on adenine. The energies of these excitations (S3 – S5) are 5.47 eV, 5.57 eV and 5.66 eV, respectively. The S6 excitation is 1nπ* in nature and delocalized on A and 8-oxoG. The transition energy and oscillator strength for this excitation is 6.04 eV and 0.0003. The S7 transition is a charge transfer excitation 1ππ*(CT) with excitation energy 6.16 eV and oscillator strength 0.0016. This state (S7) is dominated by H→L+1 localized on 8-oxoG and adenine, see Figure 5. The 1ππ*(CT) excited state of 8-oxoG-A lies ca. 1 eV above the 1ππ*(CT) of 8-oxoG-C (Table 3). S8 and S9 transitions are 1nπ* and 1ππ* local excitations and occur at 6.19 eV and 6.56 eV with oscillator strength 0.0001 and 0.2895.
Table 4.
Vertical lowest lying singlet excited state energies (ΔE) in eV and oscillator strength (f) of 8-oxoG-A base pair calculated using the TD-ωB97XD/6–31G* method (ε = 1)
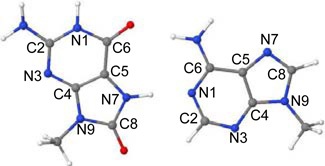 | |||||
| State | Transition | ΔE (eV) | f | Configuration | Naturea |
|---|---|---|---|---|---|
| S1 | 8-oxoG(π) → 8-oxoG(π*) | 4.61 | 0.2128 | ~48% | LE |
| S2 | 8-oxoG(π) → 8-oxoG(π*) | 5.44 | 0.0936 | ~38% | LE |
| S3 | A(π) → A(π*) | 5.47 | 0.2110 | ~41% | LE |
| S4 | A(π) → A(π*) | 5.57 | 0.2097 | ~33% | LE |
| S5 | A(n) → A(π*) | 5.66 | 0.0002 | ~40% | LE |
| S6 | A, 8-oxoG(n) → 8-oxoG(π*) | 6.04 | 0.0003 | ~21%; delocalized on 8-oxoG and A | LE |
| S7 | 8-oxoG(π) → A(π*) | 6.16 | 0.0016 | ~48% | CT |
| S8 | A(n) → A(π*) | 6.19 | 0.0001 | ~41% | LE |
| S9 | 8-oxoG(π) → 8-oxoG(π*) | 6.56 | 0.2895 | ~38% | LE |
LE= local excitation; CT= charge transfer.
Figure 5.

Vertical excitation energy in eV of 8-oxoG-A base pair obtained using the TD-ωB97XD/6–31G* method(ε = 1). The nature of the transition and molecular orbitals involved in the excitation (S1 – S9) are also given. For details, see Table 3.
3.8 Excited state geometries of 8-oxoG-A
The PT-CT excited state geometry of 8-oxoG-A base pair in Hoogsteen base pair arrangement, optimized by the ωB97XD/6–31G* method, are given in Figures 4(b). The PT-CT excited (1ππ*(CT)) state of 8-oxoG-A is biradical in nature (8-oxoG(-H)•-A(H)•). The geometry in this excited state was optimized by constraining the C2(A)-C8(oxoG)-C6(8-oxoG)-N6(A) dihedral angle to the ground state value (ca. 0 deg.) because during full optimization both the bases were tending to twist by more than 70 deg. and convergence could not be achieved. Further this motion would be hindered in a stacked DNA system. The TD-ωB97XD/6–31G* optimized geometry in this excited state shows large structural changes in the hydrogen bonding region. The O6(8-oxoG)---N6(A) and N7(8-oxoG)---N1(A) distances are appreciably increased by ca. 0.2 Å and ca. 0.3 Å in comparison to the corresponding ground state distances, see Figures 4(a) and (b). In the excited state, C4–C5, C5–C6 and C5-N7 bonds of 8-oxoG are slightly increased by 0.03 Å – 0.05 Å and N1-C2, C2-N3, N3-C4 and C4–C5 bonds in adenine are increased by 0.03 Å – 0.09 Å in comparison to the ground state distances.
3.9 Excited state PCET reaction in 8-oxoG-A
The potential energy profile of 8-oxoG-A base pair in the vertical excited state, considering the N7-H bond distance of 8-oxoG as the reaction coordinate, is shown in Figure 6. Figure 6 provides qualitative information about curve crossing as found for 8-oxoG-C in Figure 3.
Figure 6.

Vertical potential energy surface (PES) profile of the ground (S0), 1ππ*(LE), 1nπ*(LE) and the lowest 1ππ*(CT) excited states of 8-oxoG-A base pair calculated using the TD-ωB97XD/6–31G* method (ε = 1) considering N7-H of 8-oxoG as a reaction coordinate, see Figure 4(a) and Table 4. All the energies were calculated using the energy of the optimized ground state of 8-oxoG-A base pair, shown in Figure 4(a), as reference.
From Figure 6, it is evident that the energy of the ground (S0) state increases with the increase of N7-H bond distance of 8-oxoG in the base pair. The charge transfer excited state (1ππ*(CT)) in this case shows a barrierless proton transfer from N7 of 8-oxoG to N1 of adenine. The energy of this state decreases with increasing N7-H bond distance and beyond 1.4 Å, the PT-CT excited (1ππ*(CT)) state crosses the lowest 1ππ*(LE) (pink curve in Figure 6) and becomes the lowest excited state and stabilized at N7-H=1.8 Å, see Figure 6. The energy gap at this point (N7-H=1.8 Å) with respect to the ground state is ca. 3.3 eV which is about 0.5 eV higher than the energy gap calculated for the PT-CT excited state in 8-oxoG-C pair, see Figure 3.
In Figure 7, we present the energy level diagram of the optimized PT-CT excited state (1ππ*(CT)OPT) of 8-oxoG-A base pair. From Figure 7, we see that the optimized PT-CT excited state (1ππ*(CT)OPT) of 8-oxoG-A lies ca. 1.2 eV above the ground state. This energy gap (1.2 eV) is quite large in comparison to the 8-oxoG-C base pair (0.6 eV), Figure 3. The present calculations are in close agreement with those concluded by Sobolewski et al.23 Using the CC2 method Sobolewski et al.23 showed that for G-C base pair in specific hydrogen bonding conformations, the crossing between 1ππ*(CT) and ground state of G-C pair in the Watson-Crick conformation occurs at smaller energy (0.6 eV) than the other conformations.
Figure 7.

Energy level diagram of 1ππ*(CT) excited states of 8-oxoG-A base pair along with the optimized PT-CT excited state (1ππ*(CT)OPT) in the gas phase (ε = 1). For clarity other transitions are not shown. For details see Figures 5 and 6 and Table 4.
4.0 Effect of solvation on excited state PCET reactions in 8-oxoG-C and 8-oxoG-A
The effect of solvation on the excited state PCET reactions in 8-oxoG-C and 8-oxoG-A base pairs has been incorporated through the use of the polarizable continuum model (PCM) using the integral equation formalism (IEF). The ground state geometries of 8-oxoG-C and 8-oxoG-A base pairs in solution were optimized using the PCM-ωB97XD/6–31G* method and their vertical excited states were calculated. The calculated vertical excitation energies are presented in Table 5. From Table 5, it is evident that local excited states energies are not much affected by the solvation on comparison to their gas phase energies, see Tables 3 and 4. The CT excited state of 8-oxoG-C in solution is predicted to be 4th transition (S4) with energy 5.93 eV (Table 5) while in the gas phase it is the 2nd transition (S2) with energy 5.17 eV (Table 3). The CT excited states energies of oxoG-A in solution and in the gas phase are 6.03 eV and 6.16 eV, respectively, see Tables 4 and 5.
Table 5.
Vertical lowest lying singlet excited state energies (ΔE) in eV and oscillator strength (f) of 8-oxoG-C and 8-oxoG-C base pairs in solution (ε = 78.4) calculated using the PCM-TD-ωB97XD/6–31G* method.
| 8-oxoG-C | |||||
|---|---|---|---|---|---|
| State | Transition | ΔE (eV) | f | Configuration | Naturea |
| S1 | 8-oxoG(π) → 8-oxoG(π*) | 4.80 | 0.1570 | ~46% | LE |
| S2 | C(π) → C(π*) | 5.22 | 0.2096 | ~44% | LE |
| S3 | 8-oxoG(π) → 8-oxoG(π*) | 5.76 | 0.4277 | ~44% | LE |
| S4 | 8-oxoG(π) → C(π*) | 5.93 | 0.0012 | ~47% | CT |
| S5 | 8-oxoG(n), C(π) → C(π*) | 6.05 | 0.0396 | ~25%; delocalized on 8-oxoG and C | LE |
| 8-oxoG-A | |||||
| S1 | 8-oxoG(π) → 8-oxoG(π*) | 4.73 | 0.2531 | ~43% | LE |
| S2 | A(π) → A(π*) | 5.42 | 0.2498 | ~29% | LE |
| S3 | A(π) → A(π*) | 5.47 | 0.0688 | ~24% | LE |
| S4 | 8-oxoG(π) → 8-oxoG(π*) | 5.62 | 0.3621 | ~45% | LE |
| S5 | A(n) → A(π*) | 5.71 | 0.0004 | ~36% | LE |
| S6 | 8-oxoG(π) → A(π*) | 6.03 | 0.0011 | ~43% | CT |
| S7 | 8-oxoG(n) → 8-oxoG(π*) | 6.12 | 0.0001 | ~24% | LE |
LE= local excitation; CT= charge transfer.
The potential energy profile of solvated 8-oxoG-C and 8-oxoG-A base pairs in the vertical excited states are shown in Figures 8 and 9. For 8-oxoG-C base pair, the ground state (S0) energy in solution increases with the increase of the N1-H bond distance of 8-oxoG, see Figure 8. The energy of the 1ππ*(CT) excited state decreases with increasing N1-H bond distance and after (N1-H = 1.4 Å) it becomes the lowest excited state as found in the gas phase. The potential energy profile of the solvated 8-oxoG-A base pair is similar to the 8-oxoG-C base pair. The 1ππ*(CT) in 8-oxoG-A becomes the lowest after N7-H = 1.4 Å. The energy difference between ground state and the lowest PT-CT excited state at 1.8 Å in 8-oxoG-C and 8-oxoG-A base pairs are 4.56 eV and 4.28 eV, respectively, see Figures 8 and 9.
Figure 8.

Vertical potential energy surface (PES) profile of the ground (S0), 1ππ*(LE), 1nπ*(LE) and the lowest 1ππ*(CT) excited states of 8-oxoG-C base pair calculated using the PCM-TD-ωB97XD/6–31G* method in water solution (ε = 78.4) considering N1-H of 8-oxoG as a reaction coordinate. All the energies were calculated using the energy of the optimized ground state of 8-oxoG-C base pair as reference.
Figure 9.

Vertical potential energy surface (PES) profile of the ground (S0), 1ππ*(LE), 1nπ*(LE) and the lowest 1ππ*(CT) excited states of 8-oxoG-A base pair calculated using the PCM-TD-ωB97XD/6–31G* method in water solution (ε = 78.4) considering N7-H of 8-oxoG as a reaction coordinate. All the energies were calculated using the energy of the optimized ground state of 8-oxoG-A base pair as reference.
The energy level diagram of the optimized PT-CT excited state (1ππ*(CT)OPT) of 8-oxoG-C and 8-oxoG-A base pairs calculated in different dielectric constants (ε = 78.4 (water); ε = 4.0 and ε = 1.0 gas phase) in planar conformation are shown in Figures 10 and 11. The dielectric constant ε = 4.0 represents the dielectric within DNA.24 From these figures, it is evident that PT in the CT excited state is highly influenced by the dielectric constant of the surroundings. In water solution (ε = 78.4) the energy gap between the ground state and PT-CT excited state is large (ca. 3.3 eV) in both the cases and this gap decreases with decrease in the dielectrics of the medium. Recently, the excited state decay of C-C base pair in Watson-Crick conformation in chloroform (CHCl3) have been studied using the broad-band transient absorption spectroscopy and quantum mechanical calculations using PCM-TD-DFT by Biemann et al.49. This study also shows a large energy gap between the ground and PT-CT excited states and proposed the involvement of an intrastrand excimer for fast fluorescence decay. Similar conclusions were drawn for A-T base pair using the PCM-TD-DFT method by Improta and coworkers.50 Markovitsi51 and Kohler52 groups also support the excited states decay via intrastrand exciplexes in G-C DNA duplexes.
Figure 10.

Energy level diagram of 1ππ*(CT) excited states of 8-oxoG-C base pair along with the optimized PT-CT excited state (1ππ*(CT)OPT) in ε = 78.4 (water); 4.0 (DNA) and 1.0 (gas phase). For details see Figure 8 and Table 5. Calculations were done using the PCM-TD-ωB97XD/6–31G* method.
Figure 11.

Energy level diagram of 1ππ*(CT) excited states of 8-oxoG-A base pair along with the optimized PT-CT excited state (1ππ*(CT)OPT) in ε = 78.4 (water); 4.0 (DNA) and 1.0 (gas phase). For details see Figure 8 and Table 5. Calculations were done using the PCM-TD-ωB97XD/6–31G* method.
Schwalb and Temps24 reported the femtosecond time resolved experiment on G-C base pair in solution (CHCl3) and proposed the fast deactivation of G-C base pair through excited state PCET mechanism. The fluorescence lifetime of G-C pair in the Watson-Crick conformation reported to be 0.355 picoseconds by Schwalb and Temps24 in agreement with QM/MM molecular dynamics simulations (0.29 ± 0.05).25 Recently, Kohler and coworkers53 reported the fast deactivation in G-C oligonucleotide through an excited state PCET with the possibility of the formation of an intrastrand exciplex state. The recent electron spin resonance (ESR) experiment of one-electron oxidized G-C DNA oligomers confirms the interbase proton transfer from G•+ to C at 77 K.54 This was further supported by the theory by incorporating explicit water molecules constituting the first hydration layer around one-electron oxidized G-C base pair.55 Marwick and Doltsinis used nonadiabatic molecular dynamics simulations to calculate excited-state lifetimes of the G-C Watson-Crick pair in the gas phase and in aqueous solution and an excited state coupled proton-electron transfer from G to C along the central hydrogen bond is observed upon excitation of the ππ* state initially localized on G.25 Thus, it seems that excited state proton transfer in solution depends on several factors such as the dynamics of the solute-solvent interaction, including the hydrogen bonding between water and solute (first hydration layer) and location of the counter ions in DNA which are absent in the solvation model. The fast excited state deactivation in DNA is possibly mediated through exciplex states with significant CT character which enable proton transfer across base pairs as pointed out by Kohler and coworkers.53
4.1 Conclusions
This theoretical study was stimulated by the recent experiments of Nguyen and Burrows.12–14 These workers found that 8-oxoG-A mismatch base pair in DNA is 2 – 3 times more efficient than the 8-oxoG-C base pair in repair of the T<>T dimer. Our gas phase calculations suggest that the CT-excited state would rapidly decay via a barrierless transfer of the N1-H proton of 8-oxoG to N3 of cytosine, see Figure 3. The relatively small energy gap (ca. 0.6 eV) in the gas phase between ground and S1 state suggests a crossing between these two states (conical intersection) that rapidly deactivates the 1ππ*(CT) excited state as calculated for G-C base pair using multiconfigurational ab initio calculations and QM/MM molecular dynamics simulations.20,25 On the other hand, the 1ππ*(CT) excited state of 8-oxoG-A occurs at higher energy than the 8-oxoG-C pair. The larger energy gap (ca. 1.2 eV) between ground and excited state (Figure 7) would also inhibit the deactivation process through the PCET mechanism and enhance the life time of this state.
When the surrounding medium is included we find the situation changes. The excited PT-CT state in 8-oxoG-C and 8-oxoG-A base pairs lies at much higher energies from the ground state for water (3.3 eV for both at ε = 78.4) and for DNA (2.6 eV for 8-oxoG-C and 2.9 eV for 8-oxoG-A at ε = 4) than found for the gas phase calculations (0.6 eV for 8-oxoG-C and 1.2 eV for 8-oxoG-A at ε =1). These large energy gaps in solution lower the probability that fast deactivation through PCET as found in the gas phase for 8-oxoG-C occurs. Since both 8-oxoG-A and 8-oxoG-C show large energy gaps between the ground state and both the excited CT and PT-CT states we would expect longer lifetimes for both and little difference between the two base pairs in repair of the T<>T lesion. We note that the PCM-TDDFT highly destabilizes the CT excited state and would appear to prohibit the mechanism proposed by Burrows and coworkers.14 However, it is important to note that in the FC region, only the electronic cloud of the surrounding medium (solvent) responds so that the initial dielectric constant(ε) of the medium is about 2.15 Thus in the Franck-Condon (FC) region the solvent effect is minimized and solvent has a relatively small effect on the CT transitions and subsequent fast deactivation processes if they are short with respect to the dielectric response time (ca. 20 ps).
Deactivation in these solvated systems could result from one of the other mechanisms discussed in section 4.0. For example, formation of an intrastrand excimer could lead to either fast fluorescence decay or a deactivation process through exciplex states as suggested by several groups.49–52 The possibility of significant CT character in the exciplex that would enable proton transfer across base pairs has been suggested.53 Such a PCET mechanism in excited states has been suggested by several workers,24,25,53 however, a number of other recent works argue against this.49–52
Acknowledgments
This work was supported by the NIH NCI under Grant No. R01CA045424, and computational studies were supported by a computational facilities Grant, NSF CHE-0722689.
References
- 1.Beukers R, Berends W. Isolation and identification of the irradiation product of thymine. Biochim. Biophys. Acta. 1960;41:550–551. doi: 10.1016/0006-3002(60)90063-9. [DOI] [PubMed] [Google Scholar]
- 2.Taylor JS. Unraveling the molecular pathway from sunlight to skin cancer. Acc. Chem. Res. 1994;27:76–82. [Google Scholar]
- 3.Holman MR, Ito T, Rokita SE. Self-repair of thymine dimer in duplex DNA. J. Am. Chem. Soc. 2007;129:6–7. doi: 10.1021/ja0668365. [DOI] [PubMed] [Google Scholar]
- 4.Banyasz A, Douki T, Improta R, Gustavsson T, Onidas D, Vayá I, Perron M, Markovitsi D. Electronic excited states responsible for dimer formation upon UV absorption directly by thymine strands: Joint experimental and theoretical study. J. Am. Chem. Soc. 2012;134:14834–14845. doi: 10.1021/ja304069f. [DOI] [PubMed] [Google Scholar]
- 5.Schreier WJ, Kubon J, Regner N, Haiser K, Schrader TE, Zinth W, Clivio P, Gilch P. Thymine dimerization in DNA model systems: Cyclobutane photolesion is predominantly formed via the singlet channel. J. Am. Chem. Soc. 2009;131:5038–5039. doi: 10.1021/ja900436t. [DOI] [PubMed] [Google Scholar]
- 6.Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernández CE, Swaminathan VN, Carell T, Zinth W, Kohler B. Thymine dimerization in DNA is an ultrafast photoreaction. Science. 2007;315:625–629. doi: 10.1126/science.1135428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Z, Hariharan M, Arkin JD, Jalilov AS, McCullagh M, Schatz GC, Lewis FD. Electron donor acceptor interactions with flanking purines influence the efficiency of thymine photodimerization. J. Am. Chem. Soc. 2011;133:20793–20798. doi: 10.1021/ja205460f. [DOI] [PubMed] [Google Scholar]
- 8.Johns HE, Rapaport SA, Delbrück M. Photochemistry of thymine dimers. J. Mol. Biol. 1962;4:104–114. doi: 10.1016/s0022-2836(62)80042-4. [DOI] [PubMed] [Google Scholar]
- 9.Kao YT, Saxena C, Wang L, Sancar A, Zhong D. Direct observation of thymine dimer repair in DNA by photolyase. Proc. Natl. Acad. Sci U.S.A. 2005;102:16128–16132. doi: 10.1073/pnas.0506586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heil K, Pearson D, Carell T. Chemical investigation of light induced DNA bipyrimidine damage and repair. Chem. Soc. Rev. 2011;40:4271–4278. doi: 10.1039/c000407n. [DOI] [PubMed] [Google Scholar]
- 11.Pan Z, Chen J, Schreier WJ, Kohler B, Lewis FD. Thymine dimer photoreversal in purine-containing trinucleotides. J. Phys. Chem. B. 2012;116:698–704. doi: 10.1021/jp210575g. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen KV, Burrows CJ. Photorepair of cyclobutane pyrimidine dimers by 8-oxopurine nucleosides. J. Phys. Org. Chem. 2012;25:574–577. [Google Scholar]
- 13.Nguyen KV, Burrows CJ. A prebiotic role for 8-oxoguanosine as a flavin mimic in pyrimidine dimer photorepair. J. Am. Chem. Soc. 2011;133:14586–14589. doi: 10.1021/ja2072252. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen KV, Burrows CJ. Whence flavins? Redox-active ribonucleotides link metabolism and genome repair to the RNA world. Acc. Chem. Res. 2012;45:2151–2159. doi: 10.1021/ar300222j. [DOI] [PubMed] [Google Scholar]
- 15.Anusiewicz I, Świerszcz I, Skurski P, Simons J. Mechanism for repair of thymine dimers by photoexcitation of proximal 8-oxo-7,8-dihydroguanine. J. Phys. Chem. A. 2013;117:1240–1253. doi: 10.1021/jp305561u. [DOI] [PubMed] [Google Scholar]
- 16.van der Wees C, Jansen J, Vrieling H, van der Laarse A, Van Zeeland A, Mullenders L. Nucleotide excision repair in differentiated cells. Mutat. Res. 2007;614:16–23. doi: 10.1016/j.mrfmmm.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. Washington, DC, USA: ASM Press; 2006. [Google Scholar]
- 18.Cadet J, Mouret S, Ravanat J-L, Douki T. Photoinduced damage to cellular DNA: Direct and photosensitized reactions. Photochem. Photobiol. 2012;88:1048–1065. doi: 10.1111/j.1751-1097.2012.01200.x. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Sevilla MD. Proton-coupled electron transfer in DNA on formation of radiation-produced ion radicals. Chem. Rev. 2010;110:7002–7023. doi: 10.1021/cr100023g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groenhof G, Schäfer LV, Boggio-Pasqua M, Goette M, Grubmüller H, Robb MA. Ultrafast deactivation of an excited cytosine-guanine base pair in DNA. J. Am. Chem. Soc. 2007;129:6812–6819. doi: 10.1021/ja069176c. [DOI] [PubMed] [Google Scholar]
- 21.Perun S, Sobolewski AL, Domcke W. Role of electron-driven proton-transfer processes in the excited-state deactivation of the adenine-thymine base pair. J. Phys. Chem. A. 2006;110:9031–9038. doi: 10.1021/jp061945r. [DOI] [PubMed] [Google Scholar]
- 22.Schultz T, Samoylova E, Radloff W, Hertel IV, Sobolewski AL, Domcke W. Efficient deactivation of a model base pair via excited-state hydrogen transfer. Science. 2004;306:1765–1768. doi: 10.1126/science.1104038. [DOI] [PubMed] [Google Scholar]
- 23.Sobolewski AL, Domcke W, Hättig C. Tautomeric selectivity of the excited-state lifetime of guanine cytosine base pairs: The role of electron-driven proton-transfer processes. Proc. Natl. Acad. Sci U.S.A. 2005;102:17903–17906. doi: 10.1073/pnas.0504087102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwalb NK, Temps F. Ultrafast electronic relaxation in guanosine is promoted by hydrogen bonding with cytidine. J. Am. Chem. Soc. 2007;129:9272–9273. doi: 10.1021/ja073448+. [DOI] [PubMed] [Google Scholar]
- 25.Marwick PRL, Doltsinis NL. Ultrafast repair of irradiated DNA: Nonadiabatic ab initio simulations of the guanine-cytosine photocycle. J. Chem. Phys. 2007;126:175102–175106. doi: 10.1063/1.2728897. [DOI] [PubMed] [Google Scholar]
- 26.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Res. 2004;32:6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steenken S, Jovanovic SV, Bietti M, Bernhard K. The trap depth (in DNA) of 8-Oxo-7,8-dihydro-2’deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc. 2000;122:2373–2374. [Google Scholar]
- 28.Kasai H, Yamaizumi Z, Berger M, Cadet J. Photosensitized formation of 7,8-dihydro-8-oxo-2’-deoxyguanosine (8-Hydroxy-2’-deoxyguanosine) in DNA by riboflavin: A non singlet oxygen mediated reaction. J. Am. Chem. Soc. 1992;114:9692–9694. [Google Scholar]
- 29.Cadet J, Douki T, Ravanat J-L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008;41:1075–1083. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
- 30.Cadet J, Douki T, Ravanat J-L. Oxidatively generated base damage to cellular DNA. Free Radic. Biol. Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 31.Lipscomb LA, Peek ME, Morningstar ML, Verghis SM, Miller EM, Rich A, Essigmann JM, Williams LD. X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc. Natl. Acad. Sci USA. 1995;92:719–723. doi: 10.1073/pnas.92.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McAuley-Hecht KE, Leonard GA, Gibson NJ, Thomson JB, Watson WP, Hunter WN, Brown Tom. Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry. 1994;33:10266–10270. doi: 10.1021/bi00200a006. [DOI] [PubMed] [Google Scholar]
- 33.Cai Z, Sevilla MD. Electron and hole transfer from DNA base radicals to oxidized product of guanine in DNA. Radiat. Res. 2003;159:411–419. doi: 10.1667/0033-7587(2003)159[0411:eahtfd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 34.Crespo-Hernández CE, Cohen B, Hare PM, Kohler B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004;104:1977–2019. doi: 10.1021/cr0206770. [DOI] [PubMed] [Google Scholar]
- 35.Chai J-D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 36.Chai J-D, Head-Gordon M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008;128:084106. doi: 10.1063/1.2834918. [DOI] [PubMed] [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Wallingford CT: Gaussian, Inc.; 2010. [Google Scholar]
- 38.Jmol, An Open-Source Java Viewer for Chemical Structures in 3D. 2004 see http://jmol.sourceforge.net. [Google Scholar]
- 39.GaussView. Pittsburgh, PA: Gaussian, Inc.; 2003. [Google Scholar]
- 40.Yamazaki S, Taketsugu T. Photoreaction channels of the guanine–cytosine base pair explored by long-range corrected TDDFT calculations. Phys. Chem. Chem. Phys. 2012;14:8866–8877. doi: 10.1039/c2cp23867e. [DOI] [PubMed] [Google Scholar]
- 41.Lange AW, Herbert JM. Both intra- and interstrand charge-transfer excited states in aqueous B-DNA are present at energies comparable to, or just above, the 1ππ* excitonic bright states. J. Am. Chem. Soc. 2009;131:3913–3922. doi: 10.1021/ja808998q. [DOI] [PubMed] [Google Scholar]
- 42.Hättig C, Weigend F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000;113:5154–5161. [Google Scholar]
- 43.Christiansen O, Koch H, Jorgensen P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995;243:409–418. [Google Scholar]
- 44.Shukla MK, Mishra PC. A gas phase ab initio excited state geometry optimization study of thymine, cytosine and uracil. Chem. Phys. 1999;240:319–329. [Google Scholar]
- 45.Shukla MK, Mishra SK, Kumar A, Mishra PC. An ab initio study of excited states of guanine in the gas phase and aqueous media: Electronic transitions and mechanism of spectral oscillations. J. Comp. Chem. 2000;21:826–846. [Google Scholar]
- 46.Shukla MK, Leszczynski J. Electronic spectra, excited state structures and interactions of nucleic acid bases and base assemblies: A review. J. Biomol. Struct. Dyn. 2007;25:93–118. doi: 10.1080/07391102.2007.10507159. [DOI] [PubMed] [Google Scholar]
- 47.Levine BG, Ko C, Quenneville J, Martińez TJ. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 2006;104:1039–1051. [Google Scholar]
- 48.Tapavicza E, Tavernelli I, Rothlisberger U, Filippi C, Casida ME. Mixed time-dependent density-functional theory/classical trajectory surface hopping study of oxirane photochemistry. J. Chem. Phys. 2008;129:124108. doi: 10.1063/1.2978380. [DOI] [PubMed] [Google Scholar]
- 49.Biemann L, Kovalenko SA, Kleinermanns K, Mahrwald R, Markert M, Improta R. Excited state proton transfer is not involved in the ultrafast deactivation of guanine cytosine pair in solution. J. Am. Chem. Soc. 2011;133:19664–19667. doi: 10.1021/ja2089734. [DOI] [PubMed] [Google Scholar]
- 50.Dargiewicz M, Biczysko M, Improta R, Barone V. Solvent effects on electron-driven proton-transfer processes: adenine–thymine base pairs. Phys. Chem. Chem. Phys. 2012;14:8981–8989. doi: 10.1039/c2cp23890j. [DOI] [PubMed] [Google Scholar]
- 51.Miannay F-A, Bányász A, Gustavsson T, Markovitsi D. Ultrafast excited-state deactivation and energy transfer in guanine-cytosine DNA double helices. J. Am. Chem. Soc. 2007;129:14574–14575. doi: 10.1021/ja077100q. [DOI] [PubMed] [Google Scholar]
- 52.Crespo-Hernández CE, de La Harpe K, Kohler B. Ground-state recovery following UV excitation is much slower in G·C-DNA duplexes and hairpins than in mononucleotides. J. Am. Chem. Soc. 2008;130:10844–10845. doi: 10.1021/ja802183s. [DOI] [PubMed] [Google Scholar]
- 53.de La Harpe K, Crespo-Hernández CE, Kohler B. Deuterium isotope effect on excited-state dynamics in an alternating GC oligonucleotide. J. Am. Chem. Soc. 2009;131:17557–17559. doi: 10.1021/ja9076364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adhikary A, Khanduri D, Sevilla MD. Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J. Am. Chem. Soc. 2009;131:8614–8619. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar A, Sevilla MD. Influence of hydration on proton transfer in the guanine-cytosine radical cation (G•+–C) base pair: A density functional theory study. J. Phys. Chem. B. 2009;113:11359–11361. doi: 10.1021/jp903403d. [DOI] [PMC free article] [PubMed] [Google Scholar]