

Abstract
Protein glycosylation is involved in a broad range of biological processes that regulate protein function and control cell fate. As aberrant glycosylation has been found to be implicated in numerous diseases, the study and large-scale characterization of protein glycosylation is of great interest not only to the biological and biomedical research community, but also to the pharmaceutical and biotechnology industry. Due to the complex chemical structure and differing chemical properties of the protein/peptide and glycan moieties, the analysis and structural characterization of glycoproteins has been proven to be a difficult task. Large-scale endeavors have been further limited by the dynamic outcome of the glycosylation process itself, and, occasionally, by the low abundance of glycoproteins in biological samples. Recent advances in mass spectrometry (MS) instrumentation, and progress in miniaturized technologies for sample handling, enrichment and separation, have resulted in robust and compelling analysis strategies that effectively address the challenges of the glycoproteome. This review summarizes the key steps that are involved in the development of efficient glycoproteomic analysis methods, and the latest innovations that led to successful strategies for the characterization of glycoproteins and their corresponding glycans. As a follow-up to this work, we review innovative capillary and microfluidic-MS workflows for the identification, sequencing, and characterization of glycoconjugates.
1. Introduction
Glycosylation is one of the most important post-translational modifications (PTM) of proteins, playing crucial roles in biological processes such as cell recognition, cell–cell interaction, signaling, embryonic development and recognition of hormones and toxins. It is estimated that ∼50 % of all proteins in mammalian cells are glycosylated at any given time [1]. As abnormal protein glycosylation has been correlated with several diseases such as cancer, inflammatory diseases, and congenital disorders, etc. [2-5], glycosylated proteins have found utility as valuable targets for developing novel vaccines, and as useful biomarkers for early detection and diagnosis of a disease. As a result, the study of the glycoproteome has received particular attention in the recent past.
Most PTMs are introduced by enzymes that target specific amino acids. In the case of glycosylation, the Asn, Ser and Thr residues are mostly affected. Glycosylation can impact the charge, conformation and stability of proteins, introducing heterogeneity into a protein as a direct consequence of various possible glycoforms. Compared to other PTMs, the analysis of protein glycosylation is a particularly challenging enterprise, for several reasons. To create the complete picture of a glycoprotein, information on the peptide sequence, glycosylation site and glycan structure must be generated. However, glycosylation does not conform to a single structure and does not rely on an underlying template. The glycans attached to proteins in humans are composed of seven different monosaccharides: mannose (Man), glucose (Glc), galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), fucose (Fuc) and sialic acids (SA) or neuraminic acids (NeuNAc) [3]. They can be linked in linear or branching chains of various sequences and lengths. While glycans such as Glc, Gal and Man have identical mass and charge, they represent different stereoisomers of the same underlying chemical structure, and their permutation in a glycan moiety results in a broad range of possible different glycoforms.
Because of the complexity and heterogeneity of glycans, large-scale analysis of glycosylated proteins has been restrained by the lack of a suitable technology. The use of any single analytical method has not been found to be sufficient to provide full compositional and structural details. Thus, a range of multiple analytical methods and strategies have been proposed [3]. Due to demonstrated sensitivity, selectivity, ability to map the sites of glycosylation and throughput, the combination of high-resolution separations (both in capillary and microfluidic format) with MS detection has evolved into the tool of choice for the analysis of glycans and glycopeptides. Recent advances in analytical methodologies and instrumentation have enabled improved processing of glycoproteomic samples (enrichment, proteolytic digestion, glycan release and capillary separations) and advanced mass spectrometric characterization (elucidation of both amino acid and glycan sequences and identification of glycosylation site). The development of soft ionization techniques such as electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI), as well as of sophisticated ion activation/fragmentation techniques such as electron transfer dissociation (ETD) or electron capture dissociation (ECD) that successfully complement conventional collision induced dissociation (CID), has resulted in a significant expansion of glycosylation analysis by MS. This review aims to highlight the critical issues involved in the analysis of glycoproteins and to provide an update on the current status and recent developments in capillary and microfluidic technologies with MS detection as applied to glycoproteomics. The first part is dedicated to reviewing the essential steps that are involved in processing glycoproteomic samples for successful MS characterization, while the second part to highlighting the strategies that have been incorporated into all-inclusive workflows that enable a comprehensive exploration of the complex glycoproteome.
2. Types of glycoproteins
The variable composition, linkage, branching points, and configuration of monosaccharides that result in the structural diversity of glycans, and the presence of various degrees of glycosylation at different glycosylation sites on glycoproteins, are the main reasons for the complexity of analytical approaches that have been developed for the study of this PTM [6]. Four main types of protein-linked glycans are known, including: (i) N-linked glycans, with the glycan attached to the amino group of Asn residues, via a GlcNAc, in a consensus sequence of Asn-Xxx-Ser/Thr (where Xxx can be any amino acid except Pro); (ii) O-linked glycans, with the glycan attached to Ser, Thr and rarely to hydroxylysine and hydroxyproline; (iii) C-glycans, with the glycan (Man) attached to Trp residues by a C-C bond in a consensus sequence of Trp-Xxx-Xxx-Trp or Trp-Ser/Thr-Xxx-Cys; and (iv) glycosylphosphatidylinositol (GPI) anchors, with the glycan attached to the carboxyl terminus of certain membrane-associated proteins by a phosphoethanolamine bridge with mannose [7]. The two most common forms of protein glycosylation include N- and O-glycosylation of Asn and Ser/Thr residues, respectively, but other amino acid residues, such as Cys or Lys, may also be glycosylated [8]. Both N- and O- linked glycoforms are characterized by complex branched 3D structures that are greatly diverse in form and size. Common core structures are provided in Figure 1. N-linked glycans contain a common trimannosyl-chitobiose core (Man3GlcNAc2) with one or more antennae attached to each of the two outer or inner mannose units (Figure 1A) [8]. Based on the location and nature of the monosaccharides added to the core, N-linked glycans can be further classified into: (i) the “high mannose” or “oligomannose” type (Man5–9GlcNAc2) N-glycans that have only mannose residues added to the core; (ii) the “complex type” N-glycans that contain N-acetyllactosamine (Galb1–3/4GlcNAc) in their antennal region; and (iii) the “hybrid type” N-glycans that contain both mannose residues and N-acetyllactosamine attached to the trimannosyl chitobiose core residues. Some relevant structures are provided in Figure 2. O-linked glycans, on the other hand, are characterized by the stepwise addition of sugar residues directly to a protein. In mammals, normally, the initiating step is the addition of GalNAc to Ser/Thr residues, although other monosaccharide units, such as GlcNAc, or mannose-linked oligosaccharides, have been reported to be involved in O-glycosidic linkages to hydroxyl amino acids [8]. Subsequent addition of Gal and/or GlcNAc leads to the formation of the common O-glycan core structures (Figure 1B). Biosynthesis of complex N- and O-glycans is completed by a variety of capping reactions, the most important in mammals being sialylation and fucosylation [9]. Sialic acid residues, primarily N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc), confer a net negative charge on otherwise neutral glycans. Glycans can be further modified by methylation, phosphorylation and sulfation, which can occur at internal or terminal positions in the glycan structure [3].
Figure 1.
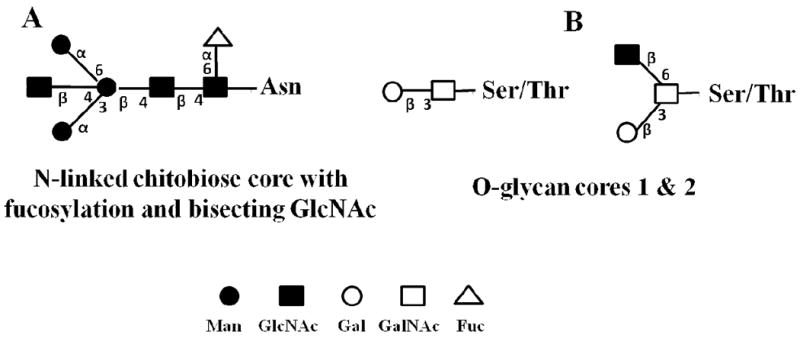
Common glycoprotein core structures. (A) N-linked glycoproteins; (B) O-linked glycoproteins. Annotation: numbers along the chemical bonds indicate the linkage in the glycan chain, i.e., 1→3, 1→4 and 1→6; α and β indicate the two stereoisomers of the anomeric carbon (reprinted with permission from reference [66]).
Figure 2.
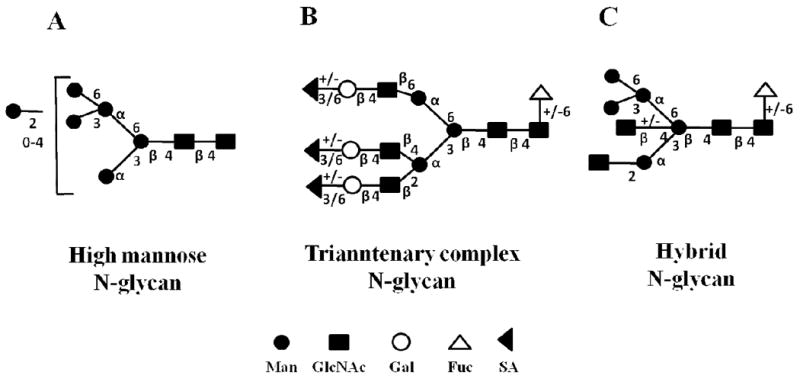
N-glycan structures. (A) High-mannose; (B) Complex; and (C) Hybrid. Annotation: same as in Figure 1; possible extensions of the glycan chain are provided (adapted from reference [8]).
3. Sample enrichment techniques for the analysis of glycoproteins
In the complex cellular milieu, proteins exist in a broad range of isoforms and carry a variety of posttranslational modifications. As a result, enrichment either at the glycoprotein or glycopeptide level, or both, is essential for a successful analysis. One of the most common methods used to isolate glycosylated proteins takes advantage of the capability of lectins to selectively bind complex carbohydrate structures and discriminate between subtly different glycan forms. The use of lectins for glycoprotein enrichment and detection has been extensively reviewed [10, 11]. Some of the most common lectins in use are: (a) ConA (Concavalin A), LCA (Lens culinaris agglutinin) and AAL (Aleuria aurantia lectin) for N-glycan enrichment; (b) HPA (Helix pomatia agarose), AIL or Jacalin (Artocarpus integrifolia lectin) and PNA (Peanut agglutinin) for O-glycan enrichment; and (c) WGA (Wheat germ agglutinin), SNA (Sambucus nigra agglutinin) and MAL (Maackia amurensis lectin) for the enrichment of sialylated glycoconjugates. Lectin enrichment may target a broad class of glycans (e.g., Con A is frequently used for the enrichment of high-mannose N-linked glycans [10]), or only glycans with specific structures (e.g., L-PHA, L-phytohemagglutinin, has been used for the targeted enrichment of β(1,6)-branched N-linked glycans [12]). To extend the range of glycoproteins that can be targeted within one analysis, serial lectin enrichment techniques have been developed [13-15]. For example, Hancock, Hincapie and co-workers have described a multi-lectin affinity chromatography (M-LAC) approach incorporating commercial ConA, Jacalin and WGA for the analysis of N-linked, O-linked and sialylated glycoconjugates, and have demonstrated this technology for cancer biomarker discovery in serum or plasma at the 10-100 ng/mL level [13, 16-18].
A frequently used alternative to lectin enrichment involves the use of hydrazide chemistry to perform solid phase extraction of glycopeptides (SPEG). N- or O-linked glycoproteins or glycopeptides are oxidized (with periodate) at the carbohydrate cis-diol groups to aldehydes that are further reacted with bead-immobilized hydrazine groups to form covalent hydrazone bonds. The immobilized glycoproteins can be digested directly on the hydrazide column to remove the non-glycosylated peptides. Specific N- or O-linked glycopeptides can be further released with the aid of various glycosidases. This method also enables intermediate stable isotope labeling of the α-amino groups of the immobilized peptides to facilitate accurate quantitative analysis and comparisons [19-23]. A comparison of glyocopeptide vs. glycoprotein enrichment by hydrazide chemistry on magnetic beads has revealed differences between the two methods in specificity, reproducibility and number of detectable glycopeptides from human plasma [24]. While hydrazide chemistry has the disadvantage of destroying the glycan moiety for enabling the attachment of peptides or proteins, it allows easier recovery of peptides for MS analysis, and it has been shown to be complementary to lectin enrichment [25].
To improve the effectiveness of the enrichment process, especially for the analysis of minute amounts of sample, a broad variety of alternative approaches have been explored. Lectin immobilization can occur through physical adsorption, chemical binding or affinity interactions. Traditionally, agarose-bound lectins have been used for glycoprotein enrichment. Novotny and co-workers have developed silica-bound lectin microcolumns that were used for the identification of 271 glyco-proteins from 20-30 μL of serum via nano-LC-MS/MS [14, 26]. Alternatively, Feng et al. have developed nanoscale chelating ConA monolithic capillary columns that enabled the reproducible detection of twice as many N-linked glycoproteins in human urine samples as conventional ConA columns [27]. Due to easy removal from the system, magnetic bead/nanoparticle-based enrichment technologies have been also pursued. Boronic-acid functionalized magnetic nanoparticles [28], or biotinylated lectins captured on paramagnetic streptavidin beads [12, 29], have shown promising capabilities for replacing traditional agarose lectin columns. To take advantage of chromatographic separation power, several techniques have been advanced as versatile methodologies for the enrichment and analysis of glycoproteins, including graphitic column chromatography [30], size exclusion chromatography (SEC) [31], hollow fiber flow field-flow fractionation [32] and hydrophilic interaction chromatography (HILIC) [33-35]. The sequential use of reversed phase POROS R2 and graphitic microcolumns (0.5 mm long, nL bed volumes) enabled the separation of glyco- from non-glycopeptides from gel-separated glycoproteins [30]. SEC-based enrichment takes advantage of the substantial difference in size between non-glycosylated and glycosylated peptides. A three-fold increase in the number of identified glycopeptides was experienced when using SEC enrichment prior to nano-LC-MS/MS analysis of N-linked glycopeptides [31]. HILIC chromatography presents particular interest, as it can be used for the separation of both glycopeptides and glycans, and can be coupled on-line to ESI-MS or off-line to MALDI-MS. Glycans are retained on HILIC particles by a combination of interactions involving hydrogen bonding, ionic interactions and dipole-dipole interactions. HILIC-based methods can be used for sample preparation, i.e., for glycopeptide enrichment prior to MALDI-MS analysis [33], or, for performing chromatographic separations of complex peptide/glycopeptide mixtures prior to on-line ESI-MS analysis. Oligosaccharide detection limits of 1 fmol have been demonstrated with nanoscale HILIC techniques [35].
4. Glycan release from glycoproteins
The presence of the glycan moiety on a protein can impede proteolysis with site-specific endopeptidases and reduce protein coverage by peptide mapping, ultimately, decreasing the number of identified proteins during proteomic studies. To facilitate the separate analysis of proteins and their attached glycan moieties, the release of glycans from proteins by chemical or enzymatic methods is frequently sought. Enzymatic or chemical methods have been developed with the ultimate goal of achieving reproducible and quantitative release, without introducing procedural artifacts [36]. For N-linked glycoproteins, this task is typically accomplished by peptide N-glycanase (PNGase) enzymes (or N-glycosidases) such as PNGase F [Peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase], PNGase A or endoglycosidases H or D (Endo H, Endo D) [37, 38]. One of the most frequently used enzymes is PNGase F, which is a broad spectrum endoglycosidase that cleaves the sugar moiety between the innermost GlcNAc of high mannose, hybrid and complex oligosaccharides, and the Asn residues from N-linked glycopeptides and proteins. Microwave assisted enzymatic digestion [39] and pressure cycling technology (PCT) have been shown to reduce deglycosylation times from hours to minutes [40]. Unlike PNGase F, PNGase A is able to cleave N-linked oligosaccharides that also contain a core α1,3 fucose at the innermost GlcNAc, but works on glycopeptides only and not on glycoproteins. Endo H is an endoglycosidase that is specific for the cleavage of N-linked mannose-rich or hybrid, but not complex, oligosaccharides from glycoproteins, while Endo D displays complementary specificity for the removal of complex sugar chains. In the case of O-linked glycans, due to a lack of broad specificity enzymes such as the N-glycanases [36], enzymatic release is not a straightforward process and can be accomplished only with a combination of endoglycosidases. Therefore, the release of O-glycans has been rather pursued by chemical means. Traditional approaches involve β-elimination (in the presence of NaBH4) and hydrazinolysis (with anhydrous hydrazine). However, these methods are not specific to O-linked glycans as they work on N-linked glycans, as well. Moreover, β-elimination (with possible incorporation of tags) involves harsh alkaline conditions and the need for excessive cleanup steps that result in sample losses. Hydrazynolysis results in simultaneous degradation of the protein backbone [41]. Milder procedures, ammonia or alkylamine-based, have been developed as alternative methods [42, 43]. Side reactions often accompany, though, such chemical release methods. Recently, an improved proteolytic enzymatic/β-elimination release method of O-glycans, at the microscale level, has been developed by Goetz et al. [36]. The method involves proteolytic digestion with multiple enzymes (trypsin and pronase) to help expose the O-glycosylation sites that are buried within the 3D structure of proteins, O-glycan release with an ammonia-borane complex, and immediate solid-phase permethylation with iodomethane on a spin-column packed with sodium hydroxide mesh beads. Alternatively, Hanisch et al. have developed a cyclic method for the sequential release of glycan moieties from the non-reducing end of the glycan chain [41]. Desialylated O-linked glycoproteins were immobilized on POROS beads, oxidized with NaIO4 to aldehydic moieties at vicinally hydroxylated sugar carbons, and treated with NH3 or NaOH for the cleavage of the oxidized sugar. Up to four oxidation/elimination cycles seemed to be enough for processing the majority of glycan structures. The method has had only a minimal impact on the structural integrity of the peptide core.
5. Proteolytic digestion
Glycoproteins are enzymatically digested to generate peptides with m/z values that fall in the analysis range of most available mass analyzers (<4000 m/z). Typically, trypsin is used to generate Lys/Arg terminating peptides that favor the formation of multiply charged peptides from which informative tandem mass spectra can be acquired. However, due to a rather substantial increase in the mass of a peptide upon glycosylation, such peptides may fall outside of the preferred range of mass analysis. In addition, the large glycan groups may prohibit trypsin access to the Lys and Arg residues that are found close to the glycosylation site, further hampering the digestion process. As a result, other broad substrate specificity enzymes such as pepsin (cleaves preferentially at the carboxylic groups of aromatic amino acids such as Phe, Trp and Tyr), thermolysin (cleaves preferentially peptide bonds containing hydrophobic amino acids), proteinase K (cleaves preferentially at the carboxylic group of aliphatic and aromatic residues), or a combination of enzymes, have been used to complement or corroborate data generated by tryptic digestion. Chen et al. have shown that the combined use of pepsin and thermolysin enabled the identification of 317 glyco-sites additional to the 622 identified by tryptic digestion alone [44]. Alternatively, to rather facilitate the generation of large mass peptides that can accommodate multiple charges for favoring precursor peptide ion activation/fragmentation via ETD, endoproteinase Lys-C (cleaves at the carboxyl end of Lys) was used for exploring glycosylation in model proteins [45]. An interesting glycoproteomic analysis strategy, based on the digestion of glycoproteins with pronase that produces glycopeptides with mono to hepto amino acid residues, was developed (pronase is a mixture of exo- and endoproteases capable of hydrolyzing essentially all peptide bonds). Progression of the digestion process results in the generation of progressively shorter lengths of amino acid sequences attached to the sugar moiety [46-49]. The method was found to be particularly useful for the analysis of N-linked glycans from bacterial glycoproteins that are not easily released by other available enzymes [47]. Lebrilla and co-workers have advanced the method of pronase digestion by immobilizing pronase to sepharose beads. Time-course analysis (90 min-24 h) of the digestion products of N- and O-linked glycoproteins enabled tracking the successive removal of amino acid residues from the peptide attached to the glycan moiety, and an in depth mass spectrometric characterization of the glycopeptides by infrared multiphoton dissociation (IRMPD)/Fourier-transform ion cyclotron resonance (FTICR)-MS [48, 49].
6. Capillary separation systems for the analysis of glycoproteins
After preliminary sample processing, the last stage of sample analysis prior to MS detection involves in most cases a nano-LC separation of glycopeptides. Typically, ∼10-15 cm long capillaries, ∼75-100 μm i.d., packed with reversed phase packing materials, are used. Performing this stage of analysis in a capillary format is particularly important to improving detection limits, especially in cases where the sample is available only in limited amounts (e.g., 10 μL of serum). Alternative strategies to conventional reversed phase nano-LC, such as capillary electrophoresis (CE), capillary electrochromatography (CEC), monolithic LC/CEC and capillary HILIC have been, however, explored. Due to the high resolving power and capability to separate +/- charged analytes, a variety of CE methodologies (capillary coatings, separation conditions, and CE-MS interfaces) have been developed for the analysis of intact, purified and underivatized glycoproteins. In particular, the characterization of glycoprotein heterogeneity, and the analysis of released carbohydrates from glycoproteins, was pursued [50-55]. However, the integration of CE in workflows that involve the analysis of complex samples has not yet been accomplished. For example, Thakur et al. have developed a 20 min long CE-MS method, on a high resolution/high mass accuracy linear trap quadrupole (LTQ)-FT-MS instrument, that enabled the identification of 60 different glycoforms containing up to nine sialic acids from recombinant human chorionic gonadrophin (r-αhCG). The separate analysis of released glycans and glycopeptides, by LC-MS, confirmed the location of specific glycan compositions associated with the two glycopeptides of r-αhCG [54]. To further explore the capabilities of capillary separations in glycoprotein analysis, Zhong and El Rassi have developed monolithic capillary columns to perform lectin affinity and normal-phase nano-LC and CEC of glycoconjugates [56, 57]. Several neutral diol methacrylate-based monoliths, as well as silica monoliths with polar functionality (2CN-OH), were prepared and demonstrated for the LC and CEC analysis of high-mannose and hybrid glycans released from standard glycoproteins. LC silica monoliths with immobilized ConA and WGA proved to be useful for the isolation and fractionation of glycoproteins, glycopeptides and glycans. HILIC-PLOT (porous layer open tubular) columns of only 10 μm i.d. have been developed for ultra-trace analysis [34]. The high sensitivity of such columns was demonstrated by the capability to perform an MS6 characterization of glycan structures from only 10 fmol of neutral and sialylated glycans (detection limit was 0.3 fmol), and the capability to confidently identify 28 N-linked glycans from only 3 ng of PNGase F digest of ovalbumin via LC-MS/MS.
7. MS techniques for the analysis of glycoproteins
The MS analysis of glycoproteins revolves around developing strategies for identifying the glycosylated protein substrates, identifying the glycosylation site, and characterizing the glycan structures (i.e., composition, sequence and linkage). Recent work makes use of the entire plethora of mass spectrometry instrumentation equipped preponderantly with ESI and MALDI sources (ion trap, quadrupole, time-of-flight (TOF), FTICR, ion mobility or hybrid instruments).
7.1. Glycopeptide ionization
ESI and MALDI are commonly used for the MS analysis of carbohydrates and glycoconjugates, and extensive reviews on these topics have been published [58-63]. The MS analysis of glycocopeptides is complicated by their heterogeneous structure that dictates the use of different experimental conditions for achieving optimized ionization. The hydrophilic nature of the native glycans limits their surface activity and reduces ionization efficiency. Neutral and basic glycoconjugates (amino sugar containing) may be analyzed as protonated, positively charged ions, while acidic glyconjugates (sialic acid and sulfate containing) as negatively charged, deprotonated ions, respectively. Alkaline/alkaline earth adduct ions can be analyzed in (+) ion mode. Derivatization by permethylation [64, 65] is commonly used to increase the hydrophobicity, surface activity and volatility of carbohydrates and glycopeptides, and to improve ionization efficiency. In addition, permethylation facilitates MS analysis in (+) ion mode as either protonated or sodiated ions, and increases chemical stability for the generation of more informative tandem mass spectra. Other common methodologies that improve the ionization/MS analysis of glycans involve: (a) reductive amination of glycans containing a reducing end and of N-linked glycans released enzymatically from proteins; and (b) methyl esterification or enzymatic removal of sialic acids [63].
7.2. Tandem MS
To identify the sequence of amino acids in peptides, the sequence of sugars in the glycan moiety, and the glycosylation site, tandem MS must be performed. The tandem MS analysis of glycopeptides is, however, complicated by the dissimilar chemical properties of the peptide and glycan moieties. In addition, glycoconjugates encompass a distribution of various glycoforms that build on a N-/O-linked core structure, a particular mass being common not to one, but to several positional isomers. The outcome of performing ESI tandem MS on glycopeptides via conventional CID is largely dependent on the energies involved in the ion fragmentation process, on the glycopeptide structure and on its charge state [62, 66, 67]. At relatively low CID energies, the tandem mass spectrum of glycopeptides is dominated by ions generated from the sequential loss of sugar residues, and, eventually, the intact deglycosylated peptide. Ions generated from the fragmentation of the peptide backbone are absent or observed only in low abundance, and the balance between glycosidic vs. peptide backbone fragmentation products dependents on the glycopeptide structure. The abundance of peptide fragments may be increased, however, at somewhat higher CID energies, at the expense of losing carbohydrate sequence information due to complete fragmentation [62, 66]. Information related to the stereochemistry (e.g., the presence of specific Glu, Gal or Man residues), linkage (e.g., 1→4, 1→3 or 1→6), and branching patterns (e.g., linear vs. branched) of sugar residues cannot be, however, obtained from such mass spectra. Such information can be generated only by the cross-ring fragmentation of the carbohydrate moiety that is generally achieved only at high collision energies (order of keV). This is typically accomplished with MALDI-TOF/TOF-MS instrumentation [62, 66]. The nomenclature for the glycoconjugate product ions generated by tandem MS is provided in Figure 3 [66]. The B, C, Y and Z ions are produced by the fragmentation of the glycosidic bonds, while the A and X ions are produced by the fragmentation of the glycosidic rings. Ions X, Y and Z contain the reducing ends of the sugar, while ions A, B, and C the non-reducing ends, respectively. The A and X ions are particularly useful for determining the glycan linkages. The nomenclature for the peptide product ions generated by tandem MS is provided in Figure 4. The b and y ions and produced by the fragmentation of the relatively week, protonated amide linkages, typically, by low energy CID (order of ∼100 eV). The a, c, x and z ions are produced by the fragmentation of the peptide backbone at high collision energies. Ions a, b and c contain the N-terminus, while ions x, y and z contain the C-terminus of a peptide, respectively [68].
Figure 3.
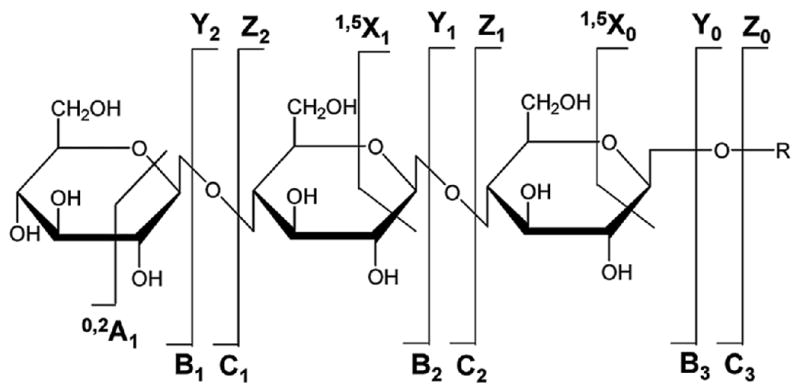
Nomenclature for glycopeptide fragmentation. Legend: B, C, Y and Z ions are produced by the fragmentation of the glycosidic bond; A and X ions are produced by the fragmentation of the glycosidic ring (reprinted with permission from reference [66]).
Figure 4.

Nomenclature for peptide fragmentation. Legend: a, b and y ions are produced by CID; c and z ions are produced by ECD or ETD (reprinted with permission from reference [83]).
A relevant tandem mass spectrum of a doubly charged glycopeptide analyzed on a Q-TOF mass spectrometer, with low energy settings in the ion source and the collision cell, is provided in Figure 5 (unpublished work, Lazar, A.C.). The peptide backbone is maintained intact (see the glycopeptide parent at m/z 2796.0 and the Y1 fragment at m/z 1392.6), while the glycosidic bonds are fragmented and provide information related to the sequence of sugar residues. The tandem mass spectrum of the same glycopeptide generated at higher fragmentation energies is provided in Figure 6. By using energetic conditions in the MS ion source, the glycan residue is mostly removed to generate a glycopeptide fragment that contains only one GlcNAc attached to the peptide (the Y1 ion). In fact, all glycoforms attached to the peptide collapse into the Y1 form, which is the most stable fragment ion generated in the ion source. The singly charged Y1 ion can be further isolated in the quadrupole section of the Q-TOF mass spectrometer and fragmented in the collision cell. The tandem mass spectrum displays the presence of the precursor Y1 ion (m/z 1392.8), the completely deglycosylated peptide ion (m/z 1189.6), the marker ion for HexNAc carbohydrates (m/z 204.1), and the relevant a, b, and y peptide fragment ions.
Figure 5.
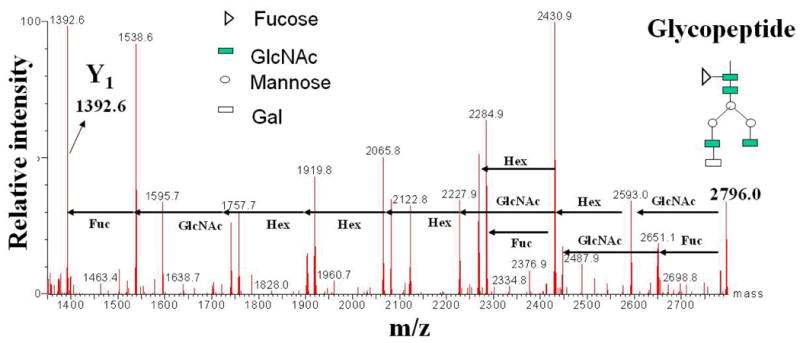
ESI tandem mass spectrum of an N-linked glycopeptide at low energy CID (glycan sequencing). The peptide backbone remains intact, while the glycosidic bonds are fragmented. The mass of the glycan can be determined from the difference between the m/z of the precursor ion (m/z 2796.0) and the m/z of the Y1 ion (m/z 1392.6). The mass of the glycan and the fragmentation pattern allows for the confirmation of the glycoform structure.
Figure 6.
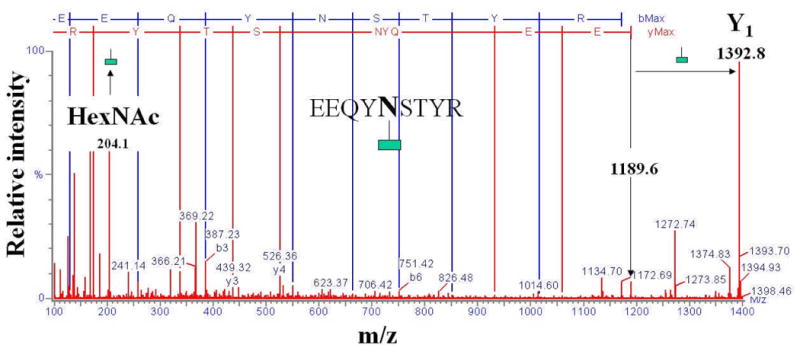
ESI tandem mass spectrum of an N-linked glycopeptide at higher energy CID (peptide sequencing). The glycan moiety is removed in the MS ion source. The tandem mass spectrum is generated by the sequencing of the Y1 ion precursor of the tryptic glycopeptide (m/z 1392.8). The peptide sequence is EEQYNSTYR (m/z 1189.6). A GlcNAc is preserved on the Asn residue. The difference between the precursor ion mass and the mass of the peptide corresponds to the mass of the GlcNAc residue (204.1 Da). The spectrum contains b, y, and a ions indicative of the peptide sequence, several internal fragments, and deamminated and/or dehydrated b, y, and a ions.
Typically, large scale LC-MS analysis of complex glycoprotein cellular extracts is performed either on the deglycosylated peptides or on the released glycans, to identify the presence of specific glycoproteins or to elucidate the structure of the present glycans, respectively. A specific glycan modification cannot be, however, always assigned to a specific glycosylation site, especially when using CID-MS alone. Alternative ion fragmentation techniques such as ETD or ECD may provide, however, a successful alternative to this problem (see following section of the manuscript). Nevertheless, selective identification of glycopeptides with CID-MS can be performed with specialized MS scanning functions, i.e, neutral loss and precursor ion scanning. Neutral loss scans enable the detection of glycopeptide ions carrying characteristic labile moieties, of a specific mass, that are easily lost as neutral fragments upon CID (e.g., HexmHexNAcn). Combinations of neutral loss and MSn scans provide peptide structural information and may enable the identification of the glycan attachment site. Alternatively, precursor ion scans enable the detection of glycopeptide ions that generate a specific oxonium product ion of the type Hex+ (C6H11O5+, m/z 163.06), HexNAc+ (C8H14NO5+, m/z 204.09), HexHexNAc+ (C14H24NO10+, m/z 366.14), SA+ (protonated neuraminic acid, m/z 292.1), or SA+-H2O (protonated dehydrated neuraminic acid, m/z 274.1) [69].
To capitalize on the developments of MS instrumentation, a variety of other MS ionization and fragmentation strategies have been explored for the analysis of carbohydrates and glycopeptides, i.e., IRMPD, photodissociation, ECD and ETD. In IRMPD, the absorption of infrared light causes vibrational excitation of ions and bond fragmentation. Similar to CID, glycopeptides will generate abundant glycosidic bond cleavage products, but low abundance peptide backbone cleavage products [70]. The analysis of O-linked glycopeptides by ESI-IRMPD has revealed that the glycan and peptide backbone fragmentation process is controlled by the composition and the charge (H+ or Na+) of the parent glycopeptide [71]. The photodissociation (157 nm) of N-linked glycopeptides has also shown that both MALDI-TOF/TOF-MS and ESI-MS, on a linear ion trap, will generate fragmentation of both peptide and glycan moieties, but the nature of the fragments will dependent on the charge state (1+, 2+) of the parent glycopeptide [72]. Techniques such as ECD and ETD rely on alternative ion activation/fragmentation strategies. Irradiation with a low energy (<0.1 eV) beam of electrons in the case of ECD [73], and with radical anions in the case of ETD [74, 75], favor a rather abundant and homogeneous peptide backbone fragmentation process at the N-Cα bonds with the formation of c/z ion pairs (see Figure 4). As labile PTMs are preserved, the glycosylation site can be identified. As such, combinations of IRMPD/ECD have been used to characterize N-linked glycopeptides [76], and combinations of CID/ETD have been even incorporated in on-line LC-MS/MS glycoproteomic workflows [77]. Using such techniques, Perdivara et al. have performed LC-CID-MS/MS followed by LC-ETD-MS/MS to elucidate the structure and linkages of O-linked sialylated glycans in the β-amyloid protein [69]. Alternatively, Alley et al. have used a combination of CID/ETD, alternating within one single LC-MS/MS analysis, to characterize and identify the glycosylation sites in model N-linked and sialylated glycoproteins. CID generated glycan sequence information, while ETD generated amino acid sequence information [78]. To expand on this strategy, Wu et al. developed an on-line LC-MSn method involving CID (MS2) / ETD(MS2) / CID(MS3) that was used for the low fmol sequencing of both N- and O-linked glycopeptides. The method enabled the identification of phosphorylated sites in model proteins (α-casein, epidermal growth factor receptor and tissue plasminogen activator) [45]. As yet another MS development for the analysis of glycoproteins, Clemmer and co-workers have used ion mobility spectrometry (IMS) to obtain information about glycan conformational and isomeric composition. Without a front-end separation system, IMS-MS was able to distinguish between 19 different glycan structures and 42 distinct isomers/conformers of ovalbumin [79], and between the ion mobility distribution of specific m/z ions derived from serum glycoproteins from liver cancer and cirrhosis patients [80]. While these examples highlight the progress that was made by mass spectrometry in analyzing glycoproteomic samples, the full capabilities of all these technologies have yet to be harvested.
7.3. Enzymatic sequencing for glycan sequence identification
An alternative method to tandem MS, that helps clarifying the sequence of specific carbohydrates in a glycan moiety, relies on enzymatic sequencing. While the method is laborious, and requires glycan purification by chromatographic techniques prior to enzymatic treatment and MALDI-MS detection, it enables the identification of specific carbohydrate isomers. An example of how enzymatic sequencing with MS detection can be implemented in a laboratory is provided in the followings (unpublished work, Lazar, A.C.). A purified glycan sample is divided in aliquots (1, 2, 3, 4 and 5) and subjected to sequential exoglycosidase treatments with a series of enzymes such as Sialidase A, β (1-4) Galactosidase, β-N-Acetylhexosaminidase, α-Mannosidase Jack Bean, and α-Mannosidase X. manihotis (Figure 7). Removal of specific sugar residues will occur from the non-reducing end. The first aliquot (1) will not be treated with any enzyme, aliquot 2 may be treated with Sialidase A, aliquot 3 with Sialidase A + β (1-4) Galactosidase, aliquot 4 with Sialidase A + β (1-4) Galactosidase + β-N-Acetylhexosaminidase, and aliquot 5 with Sialidase A + β (1-4) Galactosidase + β-N-Acetylhexosaminidase + α-Mannosidase Jack Bean + α-Mannosidase X. manihotis. The MALDI-MS analysis of each aliquot reveals that: aliquot 1 corresponds to the undigested glycan; digestion of aliquot 2 does not change the mass of the glycan, which indicates that no sialic acid is present; digestion of aliquot 3 with Sialidase A and β (1-4) Galactosidase decreases the mass of the glycan, and the mass difference corresponds to the mass of two Gal residues; digestion of aliquot 4 reveals that besides the removal of the Gal residues by β (1-4) Galactosidase, two GlcNAc residues are also removed by the β-N-Acetylhexosaminidase enzyme; and, ultimately, the digestion of aliquot 5 with the mixture of all five enzymes indicates the removal of additional two Man residues from the tri-mannose structure by the two Mannosidase enzymes.
Figure 7.
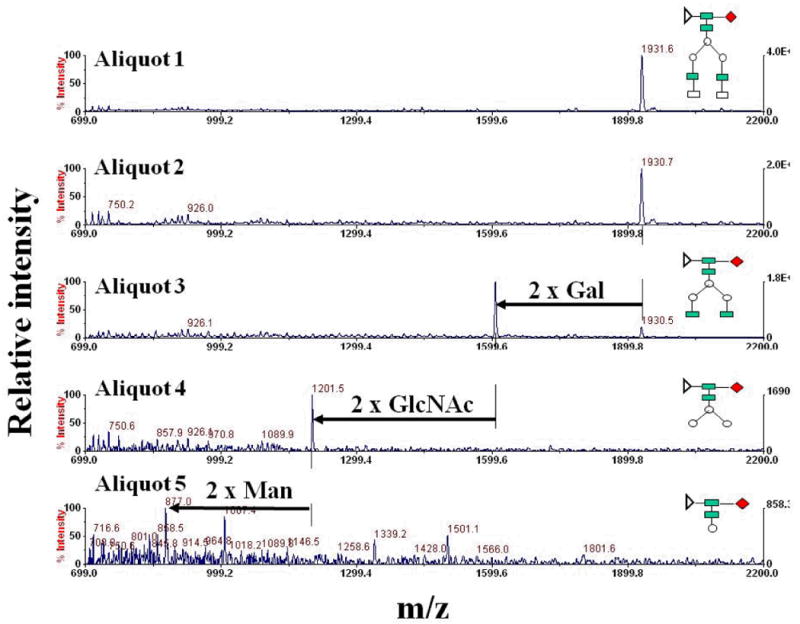
Set of MALDI mass spectra illustrating the principle of glycan enzymatic sequencing. Enzymatic treatments: Aliquot 1: sample + no enzyme; Aliquot 2: sample + Sialidase A; Aliquot 3: sample + Sialidase A + β (1-4) Galactosidase; Aliquot 4: sample + Sialidase A + β (1-4) Galactosidase + β-N-Acetylhexosaminidase; and Aliquot 5: sample + Sialidase A + β (1-4) Galactosidase + β-N-Acetylhexosaminidase + α-Mannosidase Jack Bean + α-Mannosidase X. manihotis.
7.4. Identification of the glycosylation site
With advancements in glycomic and proteomic methodologies, as well as in MS instrumentation, “glycosylation site-specific analysis” for correlating the glycosylation site on a peptide with the attached glycan has become possible. The identification of the glycosylation site is performed by enzymatic or chemical cleavage of the glycan moiety, with possible incorporation of tags of a specific mass, or by MS fragmentation of glycopeptides.
Enzymatic methods rely on the release of glycans with enzymes that will tag the glycosylation site on the parent peptide. PNGase F, the most commonly used enzyme for releasing N-linked glycans, has additional amidase activity and will convert Asn to Asp. This conversion will result in an amino acid residue mass shift (Δm) of 1 Da that is detectable by mass spectrometers with superior mass accuracy [81]. Assignment of a glycosylation site is performed by taking into account the Asn-to-Asp conversion as long as the Asn residue is part of the N-glycosylation consensus sequence Asn-Xxx-Ser/Thr. However, as Asn deamidation may frequently occur as a result of both in vivo and in vitro processes, to avoid false positive assignments, the PNGase F digestion is performed in H218O to incorporate the 18O isotope in the newly formed Asp residue (Δm=3 Da) [38, 82]. PNGase F treatment can be complemented with PNGase A to enhance specificity vs. α1,3 core fucosylated N-linked glycoproteins [37], or with Endo H and Endo D to preserve the innermost GlcNAc of the conserved N-glycan core on the Asn residues. The Endo H/Endo D strategy is applicable to the identification of O-glycosylation sites, as well [38]. The use of additional exoglycosidases (β-galactosidase, neuraminidase, N-acetyl-β-glucosaminidase) may be included in the analysis to facilitate the identification of glycosylation sites in complex and hybrid type N-glycans [38].
Mass spectrometry based methods for the identification of glycosylation sites rely, as described above, on the use of precursor ion activation/fragmentation strategies that preserve the intact glycan on the peptide, i.e., ECD, ETD and photodissociation. Alternatively, carefully selected fragmentation energies in the ion source and the MS analyzer, may enable the preservation of sugar residues at the glycosylation site (see discussion of Figure 4).
8. Concluding remarks
While the analysis of glycoproteins continues to remain a laborious task, significant progress has been accomplished as a result of novel developments in sample enrichment, processing, separation and MS detection. Sample processing times in various stages of analysis have been reduced from hours to minutes. The detection of glycoproteins with putative biomarker attributes has become feasible from sample volume as small as 10 μL of serum. Improvements in separation column technology and MS instrumentation have pushed the detection limits to sub-fmol levels. The capability to perform advanced MSn sequencing with a combination of various ion activation strategies has enabled unprecedented glycopeptide structural characterizations. The combined benefits of the technologies that have been summarized in this manuscript have fused synergistically to facilitate the analysis of not only a few glycoproteins, but of the entire glycoproteome as a whole. The workflows that have emerged for the large-scale analysis of glycoproteins in complex biological samples are the subject of our next review.
Acknowledgments
This work was supported in part by grants from NSF (Career BES-0448840) and NIH/NCI (R21CA126669).
References
- 1.Vanderschaeghe D, Festjens N, Delanghe J, Callewaert N. Biol Chem. 391:149–161. doi: 10.1515/bc.2010.031. [DOI] [PubMed] [Google Scholar]
- 2.Kim YS, Yoo HS, Ko JH. Protein and Peptide Letters. 2009;16:499–507. doi: 10.2174/092986609788167798. [DOI] [PubMed] [Google Scholar]
- 3.Brooks SA. Molecular Biotechnology. 2009;43:76–88. doi: 10.1007/s12033-009-9184-6. [DOI] [PubMed] [Google Scholar]
- 4.Marklova E, Albahri Z. Clinica Chimica Acta. 2007;385:6–20. doi: 10.1016/j.cca.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Drake PM, Cho W, Li BS, Prakobphol A, et al. Clinical Chemistry. 56:223–236. doi: 10.1373/clinchem.2009.136333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Temporini C, Callerli E, Massolini G, Caccialanza G. Mass Spectrometry Reviews. 2008;27:207–236. doi: 10.1002/mas.20164. [DOI] [PubMed] [Google Scholar]
- 7.Morelle W. Current Analytical Chemistry. 2009;5:144–165. [Google Scholar]
- 8.Morelle W, Canis K, Chirat F, Faid V, Michalski JC. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- 9.Gesslbauer B, Rek A, Falsone F, Rajkovic E, Kungl AJ. Proteomics. 2007;7:2870–2880. doi: 10.1002/pmic.200700176. [DOI] [PubMed] [Google Scholar]
- 10.Dai Z, Zhou J, Qiu SJ, Liu YK, Fan J. Electrophoresis. 2009;30:2957–2966. doi: 10.1002/elps.200900064. [DOI] [PubMed] [Google Scholar]
- 11.Wu AM, Lisowska E, Duk M, Yang Z. Glycoconj J. 2009;26:899–913. doi: 10.1007/s10719-008-9119-7. [DOI] [PubMed] [Google Scholar]
- 12.Abbott KL, Aoki K, Lim JM, Porterfield M, et al. J Proteome Res. 2008;7:1470–1480. doi: 10.1021/pr700792g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Harris LE, Palmer-Toy DE, Hancock WS. Clin Chem. 2006;52:1897–1905. doi: 10.1373/clinchem.2005.065862. [DOI] [PubMed] [Google Scholar]
- 14.Madera M, Mechref Y, Klouckova I, Novotny MV. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;845:121–137. doi: 10.1016/j.jchromb.2006.07.067. [DOI] [PubMed] [Google Scholar]
- 15.Jung K, Cho W, Regnier FE. J Proteome Res. 2009;8:643–650. doi: 10.1021/pr8007495. [DOI] [PubMed] [Google Scholar]
- 16.Plavina T, Wakshull E, Hancock WS, Hincapie M. J Proteome Res. 2007;6:662–671. doi: 10.1021/pr060413k. [DOI] [PubMed] [Google Scholar]
- 17.Zeng Z, Hincapie M, Haab BB, Hanash S, et al. J Chromatogr A. 1217:3307–3315. doi: 10.1016/j.chroma.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kullolli M, Hancock WS, Hincapie M. Anal Chem. 82:115–120. doi: 10.1021/ac9013308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Li XJ, Martin DB, Aebersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 20.Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Nat Protoc. 2007;2:334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arcinas A, Yen TY, Kebebew E, Macher BA. J Proteome Res. 2009;8:3958–3968. doi: 10.1021/pr900278c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian Y, Gurley K, Meany DL, Kemp CJ, Zhang H. J Proteome Res. 2009;8:1657–1662. doi: 10.1021/pr800952h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klement E, Lipinszki Z, Kupihar Z, Udvardy A, Medzihradszky KF. J Proteome Res. 9:2200–2206. doi: 10.1021/pr900984h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berven FS, Ahmad R, Clauser KR, Carr SA. J Proteome Res. 9:1706–1715. doi: 10.1021/pr900845m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee A, Kolarich D, Haynes PA, Jensen PH, et al. J Proteome Res. 2009;8:770–781. doi: 10.1021/pr800910w. [DOI] [PubMed] [Google Scholar]
- 26.Madera M, Mechref Y, Klouckova I, Novotny MV. J Proteome Res. 2006;5:2348–2363. doi: 10.1021/pr060169x. [DOI] [PubMed] [Google Scholar]
- 27.Feng S, Yang N, Pennathur S, Goodison S, Lubman DM. Anal Chem. 2009;81:3776–3783. doi: 10.1021/ac900085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang J, Liu Y, Yin P, Yao G, et al. Proteomics. 2010;10:2000–2014. doi: 10.1002/pmic.200900377. [DOI] [PubMed] [Google Scholar]
- 29.Abbott KL, Lim JM, Wells L, Benigno BB, et al. Proteomics. 2010;10:470–481. doi: 10.1002/pmic.200900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen MR, Hojrup P, Roepstorff P. Mol Cell Proteomics. 2005;4:107–119. doi: 10.1074/mcp.M400068-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez-Manilla G, Atwood J, Guo Y, Warren NL, et al. Journal of Proteome Research. 2006;5:701–708. doi: 10.1021/pr050275j. [DOI] [PubMed] [Google Scholar]
- 32.Kang D, Ji ES, Moon MH, Yoo JS. J Proteome Res. 2010 doi: 10.1021/pr900937w. [DOI] [PubMed] [Google Scholar]
- 33.Omaetxebarria MJ, Hagglund P, Elortza F, Hooper NM, et al. Anal Chem. 2006;78:3335–3341. doi: 10.1021/ac0517949. [DOI] [PubMed] [Google Scholar]
- 34.Luo Q, Rejtar T, Wu SL, Karger BL. J Chromatogr A. 2009;1216:1223–1231. doi: 10.1016/j.chroma.2008.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wuhrer M, de Boer AR, Deelder AM. Mass Spectrom Rev. 2009;28:192–206. doi: 10.1002/mas.20195. [DOI] [PubMed] [Google Scholar]
- 36.Goetz JA, Novotny MV, Mechref Y. Anal Chem. 2009;81:9546–9552. doi: 10.1021/ac901363h. [DOI] [PubMed] [Google Scholar]
- 37.Lim JM, Aoki K, Angel P, Garrison D, et al. J Proteome Res. 2009;8:673–680. doi: 10.1021/pr800855f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagglund P, Matthiesen R, Elortza F, Hojrup P, et al. J Proteome Res. 2007;6:3021–3031. doi: 10.1021/pr0700605. [DOI] [PubMed] [Google Scholar]
- 39.Sandoval WN, Arellano F, Arnott D, Raab H, et al. Int J Mass Spectrom. 2007;259:117–123. [Google Scholar]
- 40.Szabo Z, Guttman A, Karger BL. Anal Chem. 82:2588–2593. doi: 10.1021/ac100098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanisch FG, Teitz S, Schwientek T, Muller S. Proteomics. 2009;9:710–719. doi: 10.1002/pmic.200800492. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y, Konse T, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2002;16:1199–1204. doi: 10.1002/rcm.701. [DOI] [PubMed] [Google Scholar]
- 43.Hanisch FG, Jovanovic M, Peter-Katalinic J. Anal Biochem. 2001;290:47–59. doi: 10.1006/abio.2000.4955. [DOI] [PubMed] [Google Scholar]
- 44.Chen R, Jiang X, Sun D, Han G, et al. J Proteome Res. 2009;8:651–661. doi: 10.1021/pr8008012. [DOI] [PubMed] [Google Scholar]
- 45.Wu SL, Huhmer AF, Hao Z, Karger BL. J Proteome Res. 2007;6:4230–4244. doi: 10.1021/pr070313u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Anal Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 47.Liu X, McNally DJ, Nothaft H, Szymanski CM, et al. Anal Chem. 2006;78:6081–6087. doi: 10.1021/ac060516m. [DOI] [PubMed] [Google Scholar]
- 48.Clowers BH, Dodds ED, Seipert RR, Lebrilla CB. J Proteome Res. 2007;6:4032–4040. doi: 10.1021/pr070317z. [DOI] [PubMed] [Google Scholar]
- 49.Dodds ED, Seipert RR, Clowers BH, German JB, Lebrilla CB. J Proteome Res. 2009;8:502–512. doi: 10.1021/pr800708h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mechref Y, Novotny MV. Mass Spectrometry Reviews. 2009;28:207–222. doi: 10.1002/mas.20196. [DOI] [PubMed] [Google Scholar]
- 51.Bateman KP, White RL, Thibault P. J Mass Spectrom. 1998;33:1109–1123. doi: 10.1002/(SICI)1096-9888(199811)33:11<1109::AID-JMS728>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 52.Balaguer E, Neususs C. Anal Chem. 2006;78:5384–5393. doi: 10.1021/ac060376g. [DOI] [PubMed] [Google Scholar]
- 53.Hitchcock AM, Bowman MJ, Staples GO, Zaia J. Electrophoresis. 2008;29:4538–4548. doi: 10.1002/elps.200800335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thakur D, Rejtar T, Karger BL, Washburn NJ, et al. Anal Chem. 2009;81:8900–8907. doi: 10.1021/ac901506p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabo Z, Guttman A, Rejtar T, Karger BL. Electrophoresis. 31:1389–1395. doi: 10.1002/elps.201000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong H, El Rassi Z. J Sep Sci. 2009;32:1642–1653. doi: 10.1002/jssc.200900127. [DOI] [PubMed] [Google Scholar]
- 57.Zhong H, El Rassi Z. J Sep Sci. 2009;32:10–20. doi: 10.1002/jssc.200800546. [DOI] [PubMed] [Google Scholar]
- 58.Harvey DJ. Mass Spectrom Rev. 1999;18:349–450. doi: 10.1002/(SICI)1098-2787(1999)18:6<349::AID-MAS1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 59.Harvey DJ. Mass Spectrom Rev. 2006;25:595–662. doi: 10.1002/mas.20080. [DOI] [PubMed] [Google Scholar]
- 60.Harvey DJ. Mass Spectrom Rev. 2008;27:125–201. doi: 10.1002/mas.20157. [DOI] [PubMed] [Google Scholar]
- 61.Harvey DJ. Mass Spectrom Rev. 2009;28:273–361. doi: 10.1002/mas.20192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaia J. Mass Spectrom Rev. 2004;23:161–227. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- 63.Zaia J. Mass Spectrom Rev. 2009;28:254–272. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ciucanu I, Kerek F. Carbohydrate Research. 1984;131:209–217. [Google Scholar]
- 65.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zaia J. Chemistry & Biology. 2008;15:881–892. doi: 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tajiri M, Kadoya M, Wada Y. J Proteome Res. 2009;8:688–693. doi: 10.1021/pr800727w. [DOI] [PubMed] [Google Scholar]
- 68.Papayannopoulos IA. Mass Spectrom Rev. 1995;14:49–73. [Google Scholar]
- 69.Perdivara I, Petrovich R, Allinquant B, Deterding LJ, et al. J Proteome Res. 2009;8:631–642. doi: 10.1021/pr800758g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2007;849:115–128. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 71.Seipert RR, Dodds ED, Lebrilla CB. J Proteome Res. 2009;8:493–501. doi: 10.1021/pr8007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L, Reilly JP. J Proteome Res. 2009;8:734–742. doi: 10.1021/pr800766f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zubarev RA, Demirev PA. J Am Soc Mass Spectrom. 1998;9:149–156. [Google Scholar]
- 74.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coon JJ, Ueberheide B, Syka JE, Dryhurst DD, et al. Proc Natl Acad Sci U S A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adamson JT, Hakansson K. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 77.Wuhrer M, Stam JC, van de Geijn FE, Koeleman CA, et al. Proteomics. 2007;7:4070–4081. doi: 10.1002/pmic.200700289. [DOI] [PubMed] [Google Scholar]
- 78.Alley WR, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2009;23:495–505. doi: 10.1002/rcm.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Plasencia MD, Isailovic D, Merenbloom SI, Mechref Y, et al. J Am Soc Mass Spectrom. 2008;19:1706–1715. doi: 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Isailovic D, Kurulugama RT, Plasencia MD, Stokes ST, et al. J Proteome Res. 2008;7:1109–1117. doi: 10.1021/pr700702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cao J, Shen C, Wang H, Shen H, et al. J Proteome Res. 2009;8:662–672. doi: 10.1021/pr800826u. [DOI] [PubMed] [Google Scholar]
- 82.Kuster B, Mann M. Anal Chem. 1999;71:1431–1440. doi: 10.1021/ac981012u. [DOI] [PubMed] [Google Scholar]
- 83.Lazar IM. Electrophoresis. 2009;30:262–275. doi: 10.1002/elps.200800427. [DOI] [PubMed] [Google Scholar]