

Abstract
BACKGROUND
Sativex®, a cannabis extract oromucosal spray containing Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD), is currently in phase III trials as an adjunct to opioids for cancer pain treatment, and recently received United Kingdom approval for treatment of spasticity. There are indications that CBD modulates THC’s effects, but it is unclear if this is due to a pharmacokinetic and/or pharmacodynamic interaction.
METHODS
Cannabis smokers provided written informed consent to participate in this randomized, controlled, double-blind, double-dummy institutional review board–approved study. Participants received 5 and 15 mg synthetic oral THC, low-dose (5.4 mg THC and 5.0 mg CBD) and high-dose (16.2 mg THC and 15.0 mg CBD) Sativex, and placebo over 5 sessions. CBD, THC, 11-hydroxy-THC, and 11-nor-9-carboxy-THC were quantified in plasma by 2-dimensional GC-MS. Lower limits of quantification were ≤0.25 μg/L.
RESULTS
Nine cannabis smokers completed all 5 dosing sessions. Significant differences (P < 0.05) in maximum plasma concentrations (Cmax) and areas under the curve from 0–10.5 h postdose (AUC0→10.5) for all analytes were found between low and high doses of synthetic THC and Sativex. There were no statistically significant differences in Cmax, time to maximum concentration or in the AUC0→10.5 between similar oral THC and Sativex doses. Relative bioavailability was calculated to determine the relative rate and extent of THC absorption; 5 and 15 mg oral THC bioavailability was 92.6% (13.1%) and 98.8% (11.0%) of low- and high-dose Sativex, respectively.
CONCLUSION
These data suggest that CBD modulation of THC’s effects is not due to a pharmacokinetic interaction at these therapeutic doses.
Cannabis has been used medicinally for thousands of years. The US Food and Drug Administration questioned the safety of smoked cannabis, citing its high abuse potential; cardiovascular, reproductive, and pulmonary effects; and lack of efficacy compared to the approved oral synthetic Δ9-tetrahydrocannabinol (THC,3 dronabinol, Marinol®) (1). Dronabinol has proven effective in treating chemotherapy-induced nausea and emesis and AIDS anorexia.
In 1999, the Office of National Drug Control Policy funded a study by the Institute of Medicine to evaluate medicinal cannabis (2). The Institute of Medicine recommended testing alternative cannabinoid delivery systems to smoking and clinical trials to assess efficacy of synthetic and plant-derived cannabinoids for treatment of spasticity, movement disorders, glaucoma, and other indications. Sativex, a whole-plant cannabis extract, is approved in Canada to treat neuropathic pain in multiple sclerosis and as an adjunctive analgesic in patients with cancer pain not adequately relieved by opiates (3). In the US, Sativex is in phase III trials for the latter indication. Most recently, Sativex was approved for multiple sclerosis spasticity in the United Kingdom (4). Each 100-μL Sativex actuation delivers 2.7 mg THC and 2.5 mg cannabidiol (CBD), a nonpsychotropic cannabinoid (5). In addition to THC and CBD, Sativex contains potentially synergistic cannabinoids, terpenes, and flavonoids, which also may contribute to overall therapeutic effects (6).
THC oral absorption is slow and unpredictable, with peak concentrations occurring 1–5 h postdose (7). THC bioavailability was 6% when orally administered, compared to up to 27% (8) when inhaled. Plasma THC maximum concentrations were 4.4–11.0 μg/L after a 20 mg dose of oral THC (7). Once absorbed, THC is oxidized by the cytochrome P450 hepatic mixed-function oxidase system to equipotent 11-hydroxy-THC (11-OH-THC), and further metabolized to inactive 11-nor-9-carboxy-THC (THCCOOH).
CBD has similar oral absorption and bioavailability to THC (9). Following doses of 10 mg CBD + 10 mg THC in an oral capsule, mean (SD) peak plasma concentrations were 2.5 (2.2) μg/L CBD and 6.4 (3.1) μg/L THC, and after buccal Sativex (10 mg THC + 10 mg CBD) were 3.0 (3.1) μg/L CBD and 6.1 (5.4) μg/L THC (10). Time to maximum concentration (Tmax) was 1.3 (0.8) h for CBD and 1.0 (0.6) h for THC after oral administration, and 2.8 (1.3) h for CBD and 2.4 (1.1) h for THC after buccal administration.
Although CBD administered alone has beneficial effects (11-12), it also may attenuate the euphoria produced by THC (13). What remains unclear is whether this effect is attributable to a pharmacokinetic and/or pharmacodynamic interaction. When human liver microsomes were coincubated with THC (130 μmol/L) and CBD (up to 300 μmol/L), in vitro metabolism of THC to 11-OH-THC was reduced, although statistical significance was not described (14). Although some investigators have reported no significant pharmacokinetic interactions when these cannabinoids were coadministered to humans in vivo (15), others have suggested that CBD inhibits cytochrome P450 (CYP) 2C (16) and 3A (17) enzymes, reducing first-pass metabolism to 11-OH-THC.
Sativex is being considered as a treatment for a number of different therapeutic indications, including neuropathic pain (18-19), spasticity from multiple sclerosis (20), urinary incontinence (21), increased intraocular pressure (22), and pain from rheumatoid arthritis (23) and cancer (24). Elucidating potential pharmacokinetic interactions is essential for understanding the efficacy of this new cannabinoid medication. In this study, experienced cannabis smokers were randomly administered placebo, 5 and 15 mg oral THC, and low-dose (5.4 mg THC and 5.0 mg CBD) and high-dose (16.2 mg THC and 15.0 mg CBD) Sativex over 5 sessions. The aim of this research study was to contrast CBD, THC, 11-OH-THC, and THCCOOH pharmacokinetics after controlled administration of oromucosal Sativex and oral THC, specifically to determine if CBD modulates THC disposition.
Materials and Methods
Volunteers gave written informed consent to participate in this double-blind, double-dummy, within- and between-subject study, which was approved by the institutional review board of the National Institute on Drug Abuse Intramural Research Program. Study participants (age 18–45 years) had smoked cannabis at least once, but less than daily, during the 3 months before study entry. Women who participated in the study could not be pregnant or nursing and were directed to use a medically accepted form of birth control or abstain from vaginal sexual intercourse during the study and for 3 months thereafter. Men who participated in the study and used cannabis were instructed to use barrier-method contraception during the period of their study participation and for 3 months thereafter.
Individuals were screened for the presence of any clinically significant illness, as detected by history, physical examination, and/or clinical laboratory tests, that might put the individual at increased risk of adverse events or that might interfere with absorption, distribution, metabolism, or excretion of study medications. Criteria for exclusion from study participation included history of psychosis or current Diagnostic and Statistical Manual—Version IV axis I disorder, other than caffeine or nicotine dependence or simple phobia, as well as any history of adverse events associated with cannabis intoxication or dependence. Individuals who ingested ≥5 standard drinks per day 4 or more times per week and those who had donated >0.5 L blood within 30 days of study drug administration were excluded from study participation. Blood pressure and heart rate measured while the study participant was sitting after resting 5 min were required to be ≤140 mmHg systolic and ≤90 mmHg diastolic blood pressure, and ≤100 bpm heart rate. A 12-lead electrocardiogram and 3-min rhythm strip with normal results also were required. Lastly, if participants were allergic to sesame seed oil (an ingredient in dronabinol capsules), propylene glycol, ethanol, or peppermint oil (ingredients in Sativex), they were excluded from the study.
STUDY DESIGN
Participants spent the night before each session at the Johns Hopkins Bayview Behavioral Pharmacology Research Unit under 24-h medical surveillance to rule out acute intoxication. Women were required to have a negative urine pregnancy test result before each dosing session. Participants were provided with a standard breakfast the morning of each study session prior to dosing.
Each participant received, in random order, 1 of 5 treatments: oral synthetic THC, 5 and 15 mg; 2 Sativex actuations (low dose: 5.4 mg THC and 5.0 mg CBD) and 4 Sativex placebo actuations; 6 Sativex actuations (high dose: 16.2 mg THC and 15.0 mg CBD); or placebo oral THC and 6 Sativex placebo actuations. Placebo THC capsules formulated by the National Institute on Drug Abuse pharmacy contained only lactose, whereas placebo Sativex, provided by G.W. Pharma, contained propylene glycol, ethanol, and peppermint oil. Individuals swallowed 2 capsules with water and were subsequently administered 6 actuations of Sativex and/or Sativex placebo within 1–2 min by the study physician. Sativex and Sativex placebo actuations were directed sublingually and at the buccal mucosa.
SPECIMEN COLLECTION AND ANALYSIS
A peripheral venous catheter was placed the morning of each study session and was removed before discharge. Whole blood was collected in 7-mL green-top Vacutainer tubes containing sodium heparin. Samples were stored on ice, centrifuged, and plasma separated within 2 h. Plasma was transferred to cryotubes and frozen at −20 °C until analysis.
Plasma samples were analyzed with a previously described 2-dimensional GC-MS method with cryofocusing for CBD, THC, 11-OH-THC, and THCCOOH (25). Plasma (1.0 mL) was treated with 0.5-mL increments of cold acetonitrile (total 3 mL) to precipitate proteins. Supernatants were decanted into tubes containing 5 mL water, mixed, and subjected to solid-phase extraction with UCT SSTHC06Z columns. Analytes were eluted with 3 mL hexane/ethyl acetate/acetic acid (49:49:2 vol/vol), dried under nitrogen, and derivatized with N,O-bis-(trimethylsilyl)trifluoroacetamide + 1% trimethylchlorosilane for 30 min at 70 °C.
Extracts were analyzed by 2-dimensional GC-MS in the selected ion–monitoring mode. Low calibration curves were from 0.25 to 25 μg/L for CBD and THC, 0.125–25 μg/L for 11-OH-THC, and 0.25–50 μg/L for THCCOOH. High calibration curves from 5–100 μg/L for THC, 11-OH-THC, and THCCOOH used a smaller 2-μL injection volume. Analytes were quantified on the high calibration curve only if concentrations exceeded the low linear dynamic range upper limit of quantification (LOQ). Specimens quantifying >100 μg/L were diluted with blank human plasma and reextracted. Analytical method intraassay, interassay, and total imprecision were ≤9.5% and recovery/bias was ≤9.2%.
PHARMACOKINETIC AND STATISTICAL ANALYSES
The area under the curve from 0–10.5 h postdose (AUC0→10.5 h) was determined by linear trapezoidal noncompartmental analysis (WinNonlin; Pharsight). Main outcome measures were Cmax, Tmax, and AUC0→10.5 h for 4 analytes, examined by planned comparisons between 5 and 15 mg oral THC, 5 mg oral THC and low-dose Sativex, 15 mg oral THC and high-dose Sativex, and low- and high-dose Sativex. Because treatment conditions were assessed on different days for each study participant, and study participants may have smoked cannabis between sessions, most analytes were present at baseline. We made an a priori decision to control for this with multilevel models (SAS Proc Mixed; SAS Institute), which are functionally similar to repeated-measures ANOVA, but permit the inclusion of time-varying covariates. For example, to test treatment effects on Tmax for 11-OH-THC, we ran a multi-level model with 2 predictors: treatment (5 mg oral THC, 15 mg oral THC, low-dose Sativex, and high-dose Sativex) and baseline 11-OH-THC concentration (a time-varying covariate with a different value for each session). Twelve models were run, assessing Cmax, Tmax, or AUC0→10.5 h for each analyte, always controlling for the baseline concentration of the relevant analyte. In each model, the contrast statement in Proc Mixed was used to conduct planned comparisons between different conditions (4 planned comparisons in each model), not correcting for multiple tests. The models used a compound-symmetry error structure, which, based on the Akaike Information Criterion, provided a better fit to the data than several other error structures examined. As a sensitivity analysis, we reran the models without controlling for baseline concentrations. Results were considered statistically significant if the 2-tailed P value was <0.05. All participants were included in mean (SE), median, and range data. If analytes were not detected, a 0 was reported for descriptive statistical analyses. Relative bioavailability was calculated by normalizing for dose:
Relative bioavailability and mean peak ratios across treatments were analyzed with 1-way ANOVA. Two-way ANOVA was used to compare peak metabolite ratios between participants with no detectable plasma THC and those with measurable THC at the time of study entry.
Results
Six male and 3 female cannabis smokers, ages 19–43 years, each completed 5 study sessions (Table 1). Participants were predominantly (66.7%) black, and 3 were identified as white. Mean (SE) height and weight were 170.5 (4.8) cm and 74.9 (5.4) kg, respectively. Cannabis smoking frequency varied between participants. Mean age of first experience with cannabis was 15.6 (0.9) years, with the duration of longest use ranging from 2 to 10 years. Most participants reported drinking alcohol (88.9%) and smoking tobacco (66.7%) within the last year. One participant reported use of cocaine, and 2 reported consumption of psilocybin mushrooms in the year preceding the study. The median (range) of cannabinoid baseline concentrations 0.5 h before dosing were 0.0 [not detected (ND)–0.4], 1.6 (ND–6.5), 0.5 (ND–2.4), and 51.0 (ND–269.7) μg/L for CBD, THC, 11-OH-THC, and THCCOOH, respectively, across all sessions (n = 45).
Table 1.
Demographic and self-reported drug use histories for 9 cannabis smokers.
| Study participant | Sex | Age, years | Race | Weight, kg | Height, cm | Average use | Duration of longest use, years | Age of 1st use, years | Other drugs used in last year |
|---|---|---|---|---|---|---|---|---|---|
| A | M | 19 | Wa | 70 | 175 | 1×/month | 2 | 15 | T, A |
| B | F | 25 | W | 66 | 173 | 3×/week | 5 | 16 | T, A, Coc, H |
| C | M | 22 | B | 95 | 178 | 2×/month | 1 | 21 | A |
| D | M | 28 | B | 64 | 168 | 1×/week | 9 | 16 | T, A |
| E | M | 27 | B | 73 | 178 | 2×/month | 7 | 12 | T, A |
| F | M | 43 | B | 109 | 196 | 30×/week | 10 | 16 | — |
| G | F | 20 | B | 69 | 145 | 2×/week | 2 | 13 | T, A |
| H | F | 21 | W | 64 | 165 | 5–6×/week | 2 | 17 | T, A, H |
| I | M | 23 | B | 64 | 157 | 9×/week | 5 | 14 | A |
W, white; B, black; T, tobacco; A, alcohol; Coc, cocaine; H, hallucinogen (psilocybin mushrooms).
We report descriptive statistical data (Table 2) and median cannabinoid concentrations (Fig. 1). CBD mean (SE) time to first detection was 2.5 (0.4) h (n = 8) for low and 2.0 (0.3) h (n = 9) for high-dose Sativex. One participant had plasma CBD greater than the LOQ at baseline for the low Sativex dose, precluding determination of time to first detection for this individual. CBD mean plasma Cmax after low-dose (Fig. 1) and high-dose Sativex (Figs. 1 and 2) were 1.6 (0.4) and 6.7 (2.0) μg/L [F(1,7) = 7.56, P = 0.029] and occurred 3.7 (0.5) and 4.0 (0.5) h postdose, respectively. CBD AUC0→10.5 h also was significantly different between low and high doses [F(1,7) = 16.11, P = 0.005]. CBD was <LOQ by 10.5 h in 7 participants after low-dose and 2 participants after high-dose Sativex.
Table 2.
Pharmacokinetics following 5 and 15 mg oral THC and low-dose (5.4 mg THC + CBD) and high-dose (16.2 mg THC + 15 mg CBD) oromucosal Sativex (n = 9).
| Dose | Mean (SE) | Median | Range | |
|---|---|---|---|---|
| CBD Cmax, μg/L | Low Sativex | 1.6 (0.4) | 1.2 | 0.6–3.9 |
| High Sativex | 6.7 (2.0) | 3.7 | 2.0–20.5 | |
| CBD Tmax, h | Low Sativex | 3.7 (0.5) | 3.6 | 1.0–5.5 |
| High Sativex | 4.0 (0.5) | 4.5 | 1.2–5.6 | |
| CBD AUC0→10.5, h · μg/L | Low Sativex | 4.5 (0.8) | 4.1 | 2.0–8.5 |
| High Sativex | 18.1 (3.6) | 12.6 | 7.4–41.6 | |
| THC, Cmax, μg/L | 5 mg oral THC | 4.7 (0.9) | 4.6 | 1.4–10.4 |
| 15 mg oral THC | 14.3 (2.7) | 11.2 | 3.3–28.5 | |
| Low Sativex | 5.1 (1.0) | 5.1 | 1.2–9.6 | |
| High Sativex | 15.3 (3.4) | 14.5 | 3.2–38.2 | |
| THC, Tmax, h | 5 mg oral THC | 3.2 (0.3) | 3.1 | 1.5–4.5 |
| 15 mg oral THC | 3.4 (0.5) | 3.4 | 1.2–5.5 | |
| Low Sativex | 3.3 (0.3) | 3.5 | 1.2–4.5 | |
| High Sativex | 4.0 (0.5) | 4.5 | 1.2–5.6 | |
| THC, AUC0→10.5, h · μg/L | 5 mg oral THC | 30.6 (9.0) | 21.2 | 2.6–76.8 |
| 15 mg oral THC | 50.2 (9.2) | 43.5 | 9.9–97.9 | |
| Low Sativex | 32.3 (7.1) | 30.6 | 6.3–67.8 | |
| High Sativex | 58.8 (9.7) | 68.6 | 10.2–95.3 | |
| 11-OH-THC, Cmax, μg/L | 5 mg oral THC | 3.0 (0.4) | 2.6 | 1.8–5.9 |
| 15 mg oral THC | 11.1 (2.0) | 9.3 | 3.6–19.5 | |
| Low Sativex | 4.2 (0.7) | 3.7 | 2.1–7.5 | |
| High Sativex | 8.4 (1.2) | 7.6 | 3.8–13.7 | |
| 11-OH-THC, Tmax, h | 5 mg oral THC | 3.3 (0.4) | 3.3 | 1.5–5.6 |
| 15 mg oral THC | 3.4 (0.4) | 3.6 | 1.0–5.5 | |
| Low Sativex | 3.6 (0.6) | 3.3 | 1.0–7.5 | |
| High Sativex | 3.9 (0.5) | 3.7 | 1.2–5.6 | |
| 11-OH-THC, AUC0→10.5, h · μg/L | 5 mg oral THC | 14.8 (3.2) | 12.5 | 4.2–38.0 |
| 15 mg oral THC | 42.4 (6.3) | 40.1 | 15.8–71.2 | |
| Low Sativex | 21.0 (3.4) | 19.9 | 10.0–41.6 | |
| High Sativex | 34.7 (4.2) | 32.6 | 17.1–51.8 | |
| THCCOOH, Cmax, μg/L | 5 mg oral THC | 69.3 (17.6) | 57.1 | 15.9–179.7 |
| 15 mg oral THC | 133.6 (36.3) | 102.1 | 44.5–409.0 | |
| Low Sativex | 108.0 (30.5) | 79.8 | 19.1–281.6 | |
| High Sativex | 126.6 (25.9) | 92.4 | 55.9–304.1 | |
| THCCOOH, Tmax, h | 5 mg oral THC | 4.4 (0.5) | 4.3 | 2.7–7.5 |
| 15 mg oral THC | 4.9 (0.5) | 5.5 | 2.4–7.5 | |
| Low Sativex | 4.4 (0.7) | 4.5 | 1.2–7.5 | |
| High Sativex | 4.8 (0.3) | 5.0 | 2.6–5.6 | |
| THCCOOH, AUC0→10.5, h · μg/L | 5 mg oral THC | 581.6 (172.6) | 366.8 | 104.2–1671.8 |
| 15 mg oral THC | 1015.9 (331.9) | 663.0 | 298.1–3539.5 | |
| Low Sativex | 849.5 (257.7) | 680.3 | 116.6–2176.8 | |
| High Sativex | 921.8 (221.6) | 692.4 | 338.2–2451.7 |
Fig. 1.
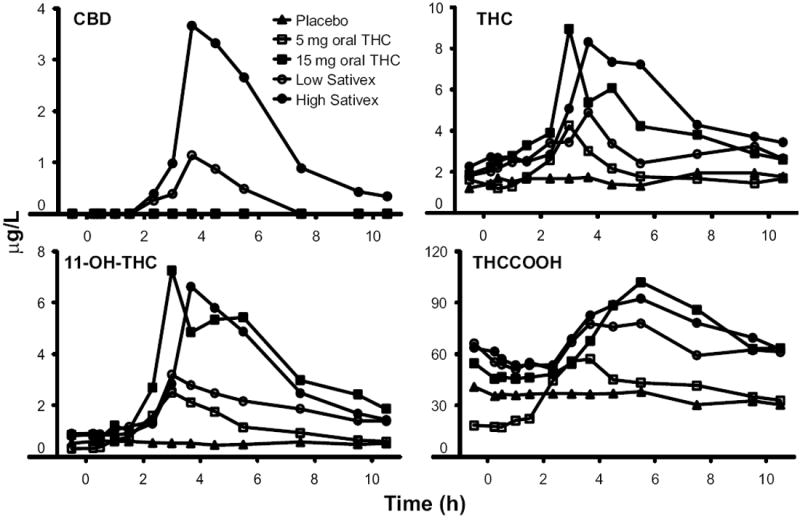
Median (n = 9) CBD, THC, 11-OH-THC, and THCCOOH concentrations following controlled administration of placebo, 5 and 15 mg oral THC and low-dose (5.4 mg THC + 5 mg CBD) and high-dose (16.2 mg THC + 15 mg CBD) oromucosal Sativex.
Fig. 2.
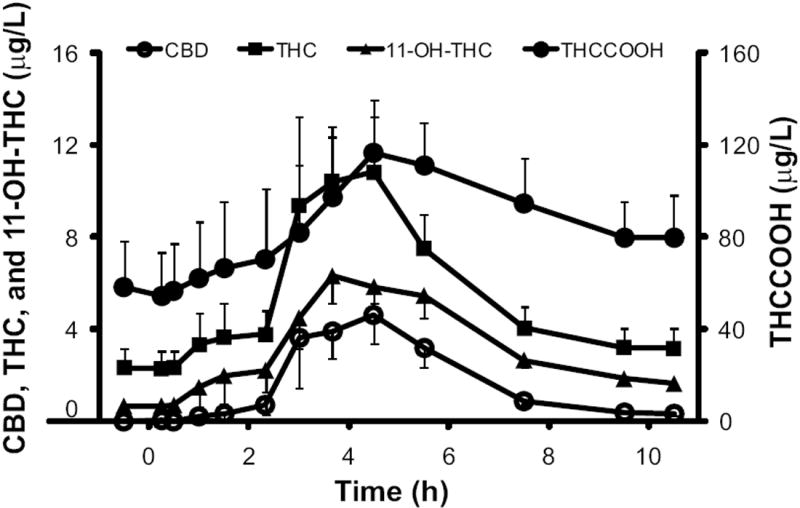
Mean (SE) CBD, THC, 11-OH-THC, and THCCOOH concentrations following high-dose oromucosal Sativex (16.2 mg THC + 15 mg CBD).
Mean THC Cmax values were not significantly different [F(1,23) = 0.01, P = 0.93] for 5 mg oral THC [4.7 (0.9) μg/L] and low-dose Sativex [5.1 (1.0) μg/L]. Similarly, mean THC plasma Cmax values were not significantly different for 15 mg oral THC [14.3 (2.7) μg/L] and high-dose Sativex [15.3 (3.4) μg/L]. THC Tmax values were not significantly different across all doses. Significant Cmax differences were observed between the low and high doses of oral THC [F(1, 23) = 14.15, P = 0.001] and low and high doses of Sativex [F(1,23) = 13.34, P = 0.0013]. Also, THC AUC0→10.5 h was significantly larger for 15 mg oral THC [F(1,23) = 28.12, P < 0.0001] and high-dose Sativex [F(1,23) = 20.47, P = 0.0002] compared to lower doses. We calculated relative bioavailability to determine the relative rate and extent of THC absorption; 5 mg oral THC bioavailability was 92.6% (13.1%) of low-dose Sativex, whereas 15 mg oral THC was 98.8% (11.0%) of high-dose Sativex. No significant differences were found in relative bioavailabilities.
11-OH-THC mean Cmax values were 3.0 (0.4) μg/L for 5 mg oral THC and 4.2 (0.7) μg/L for low-dose Sativex. At the higher doses, the mean 11-OH-THC Cmax values were 11.1 (2.0) (oral) and 8.4 (1.2) μg/L (Sativex), respectively. Mean 11-OH-THC Cmax was significantly higher for 15 mg [F(1,23) = 11.30, P = 0.0027] compared with 5 mg oral THC and for high- vs low-dose Sativex [F(1,23) = 31.38, P < 0.0001]. 11-OH-THC Cmax occurred 1.0–5.6 h after oral THC and 1.0–7.5 h after Sativex doses. Mean 11-OH-THC AUC0→10.5 h also significantly increased following 15 mg oral THC [F(1, 23) = 19.60, P = 0.0002] and high-dose Sativex [F(1,23) = 48.93, P < 0.0001] compared to the lower doses. Although statistically significant differences were not achieved, 11-OH-THC Cmax and AUC0→10.5 h were lower following high-dose Sativex compared with 15 mg oral THC [F(1,23) = 3.18, P = 0.09; F(1,23) = 3.25, P = 0.085].
THCCOOH Cmax was attained 1.2–7.5 h postdose. THCCOOH Cmax values were 69.3 (17.6), 133.6 (36.3), 108.0 (30.5), and 126.6 (25.9) μg/L after 5 and 15 mg oral THC and low- and high-dose Sativex, respectively. Both THCCOOH Cmax and AUC0→10.5 h were significantly larger after high-dose Sativex [F(1,23) = 11.58, P = 0.002; F(1,23) = 13.66, P = 0.0012] and 15 mg oral THC [F(1,23) = 14.82, P = 0.0008; F(1,23) = 11.88, P = 0.0022] in relation to the low doses administered by similar routes.
To illustrate the observed interindividual variability in cannabinoid concentrations, 5 dosing conditions are shown in Fig. 3 for participants A and G. Participant A reported smoking cannabis once monthly, and baseline samples contained no measurable CBD, THC, or 11-OH-THC, but THCCOOH concentrations <5.0 μg/L were observed. Participant G, who self-reported smoking cannabis twice per week, was admitted with plasma THC concentrations ≥3.9 μg/L, 11-OH-THC ≥1.5 μg/L, and THCCOOH ≥142.9 μg/L at each of the 5 drug administration sessions. After receiving placebo, participant G’s CBD concentrations fluctuated around the LOQ multiple times during a 10.5-h period. Participant A’s CBD Tmax were 5.5 and 4.5 h for low- and high-dose Sativex, respectively, whereas participant G’s CBD Tmax occurred earlier, at 1.0 and 1.2 h, respectively. THC and 11-OH-THC Tmax also were different for these participants, occurring at 2.8–4.5 h for participant A and 1–5.6 h for participant G. 11-OH-THC and THCCOOH Cmax were higher after 15 mg oral THC compared with high-dose Sativex in both participants. THC AUC0→10.5 h was substantially larger in participant G (97.9 h · μg/L) in relation to participant A (9.9 h · μg/L) following 15 mg oral THC. In fact, participant G had consistently larger THC, 11-OH-THC, and THCCOOH AUC0→10.5 h for all active doses.
Fig. 3.
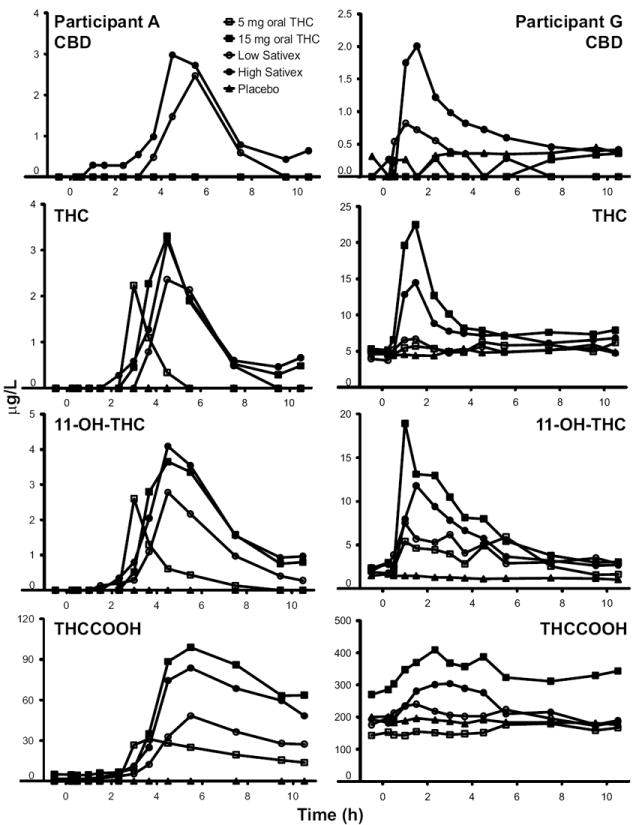
Participants’ A and G plasma CBD, THC, 11-OH-THC, and THCCOOH concentrations after placebo, 5 and 15 mg oral THC and low-dose (5.4 mg THC + 5 mg CBD) and high-dose (16.2 mg THC + 15 mg CBD) oromucosal Sativex.
Plasma cannabinoid ratios were examined in all participants and for all dosing conditions (Fig. 4). During the placebo session, which reflected previously self-administered smoked cannabis, 11-OH-THC/THC ratios ranged from 0.0 to 0.5 through 10.5 h. Ratios increased after Sativex and THC administration. Mean peak 11-OH-THC/THC ratios were 1.1 (0.2), 2.1 (0.6), 1.5 (0.3), and 1.4 (0.3) after 5 and 15 mg oral THC and low- and high-dose Sativex, respectively. Mean peak ratios were not significantly different between doses; however, there were individual differences in peak 11-OH-THC/THC ratios. Participant A’s peak ratios after 5 and 15 mg oral THC were 1.8 and 3.0, respectively, and 2.0 and 2.6 after low- and high-dose Sativex, respectively. Peak 11-OH-THC/THC ratios were 1.0 for both 5 and 15 mg oral THC and 1.3 and 1.0 for low- and high-dose Sativex, respectively, for participant G. Though not significant (P = 0.095), consistently lower 11-OH-THC/THC peak ratios were observed for participants with measurable baseline THC concentrations (n = 6) vs those without detectable THC (n = 3). No consistent THCCOOH/THC ratio pattern was evident.
Fig. 4.
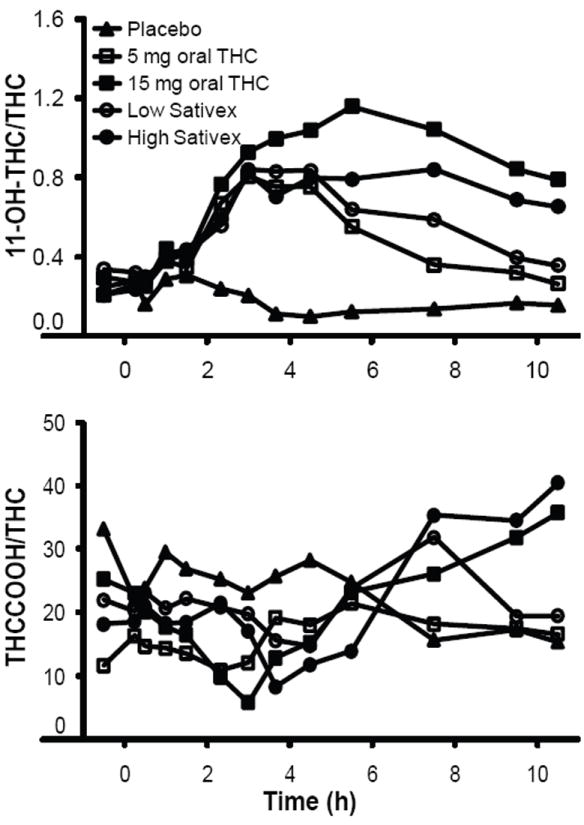
Median 11-OH-THC/THC and THCCOOH/THC ratios in 9 participants administered placebo, 5 and 15 mg oral THC and low-dose (5.4 mg THC + 5 mg CBD) and high-dose (16.2 mg THC + 15 mg CBD) oromucosal Sativex.
THC and THCCOOH LOQs were 0.25 μg/L and 11-OH-THC LOQ was 0.125 μg/L.
Discussion
Although CBD reportedly alters THC’s pharmacokinetics (26), in the present study there were no significant pharmacokinetic differences in similar oral THC and Sativex doses administered to 9 cannabis smokers. We did, however, observe lower, but not significant, 11-OH-THC Cmax (P = 0.09) and AUC0→10.5 h (P = 0.085) after high-dose Sativex in relation to 15 mg oral THC. The same changes were not noted after low-dose oral THC and Sativex. Decreases in mean 11-OH-THC Cmax and AUC0→10.5 h after only high-dose Sativex might indicate that CBD does not interact with THC at lower doses (5 mg), but could alter THC metabolism at higher CBD doses (≥15 mg). However, in vitro cytochrome P450 inhibition studies have revealed that CBD inhibits microsomal CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, but at much higher concentrations (≥2201 μg/L) than typically achieved in plasma (27). After high-dose Sativex in our study, maximum CBD concentrations were ≤20.5 μg/L. Thus, a marked pharmacokinetic interaction in vivo as a result of microsomal enzyme inhibition is unlikely after Sativex administration. Other possible factors contributing to observed lower mean 11-OH-THC Cmax and AUC0→10.5 h after high-dose Sativex were facilitated THC absorption through the oral mucosa and reduced first-pass metabolism. In the present study, THC bioavailability was enhanced when THC was administered by oromucosal spray, compared with oral administration, although differences did not reach statistical significance. Similarly, increased THC relative bioavailability was observed in a phase I open-label study, in which 10 mg oral THC had 93.9% relative bioavailability of 10 mg CBD + THC administered buccally, and 87.2% of 10 mg CBD + THC administered sublingually (10).
Metabolite ratios were examined to evaluate THC and metabolite disposition after multiple-dosing conditions. Residual THC, 11-OH-THC, and THCCOOH concentrations from self-administered cannabis smoking before study admission were present in multiple placebo-session samples. 11-OH-THC/THC ratios are low after smoked cannabis (28). THC enters the blood-stream directly from the alveoli, yielding approximately 5%–10% 11-OH-THC. Conversely, after oral THC administration, first-pass metabolism yields an 11-OH-THC/THC ratio of approximately 1 (29). Median 11-OH-THC/THC ratios increased after all active doses as THC was metabolized to 11-OH-THC (Fig. 4). If THC in Sativex was primarily absorbed through the oral mucosa, bypassing first pass metabolism, we might expect to see a difference between oral THC and Sativex 11-OH-THC/THC ratios; however, no statistical difference was found. Ratio differences observed between participants with no detectable THC vs those with THC at baseline were likely due to more frequent cannabis smoking and increased THC adipose stores, yielding decreased 11-OH-THC/THC ratios.
There were several limitations of the study, including the small sample size, potential underreporting of cannabis smoking, and the need to correct for baseline cannabinoid concentrations. Baseline cannabinoid concentrations suggest that self-report may have underestimated cannabis exposure; thus, we were unable to utilize self-report to compare light vs heavy cannabis smokers on the basis of self-report. It was necessary to correct for baseline cannabinoid concentrations, especially for THCCOOH, to adequately characterize the pharmacokinetics of our oromucosal and oral THC doses. Significant differences in THCCOOH Cmax and AUC0→10.5 h were not observed following low- and high-dose oral THC if baseline concentrations were not corrected. We did not adjust data for multiple comparisons; if Bonferroni-corrected, acceptable P values would be ≤.0013. Eight of 15 significant comparisons would survive this stringent correction. Another limitation of this study was the inability to evaluate oral THC and Sativex pharmacokinetics in cannabis naïve participants. Also, the presence of other cannabis plant components may have contributed to the observed effects.
Variable cannabinoid concentrations between study participants could be due to multiple factors. Shorter THC Tmax in participant G following high- dose oral THC and low- and high-dose Sativex could be due to saturated THC fat stores, resulting in a more rapid increase in blood THC and earlier Cmax. In participant A, THC in blood may be rapidly distributed into tissues, leading to a later Cmax. Because THC Tmax was similar after both oral THC and Sativex in participant G, CBD did not appear to influence THC absorption. Furthermore, although participants were encouraged to allow Sativex to absorb through their oral mucosa, a portion of the drug was inevitably swallowed, contributing to overall variability. Also, Sativex may have variable absorption in different areas of the oral mucosa (10). Oral THC is subject to degradation in the gut, first-pass metabolism, and enterohepatic reabsorption (7).
Several advantages exist for oromucosal cannabis plant extracts over single oral synthetic cannabinoids. A combination of cannabinoids and other plant compounds provide additional therapeutic possibilities for treating a variety of medical conditions (18-24). Oromucosal administration also is more desirable than the oral route for treating nausea and for increasing appetite. In addition, self-titration often is necessary in this population to control pain and spasticity, and to reduce unwanted subjective effects. To our knowledge, this is the first time the pharmacokinetics of 2 oral THC and 2 Sativex doses were compared. These data will improve interpretation of CBD, THC, and metabolite concentrations. There were no clinically significant pharmacokinetic differences between Sativex and oral administration of similar THC doses in this preliminary study, suggesting that modulation of THC’s physiological or behavioral effects is not due to a pharmacokinetic interaction.
Acknowledgments
The authors acknowledge G.W. Pharma for generously providing Sativex and placebo Sativex, Drs. G. Guy and C. Stott for design assistance, D. Epstein for data analysis support, and S. Baskin, K. Demuth, J. Nichels, and J. Etter for clinical research assistance.
Research Funding: E.L. Karschner, W.D. Darwin, R.S. Goodwin, and M.A. Huestis, Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Nonstandard abbreviations: THC, Δ9-tetrahydrocannabinol; CBD, cannabidiol; 11-OH-THC, 11-hydroxy-THC; THCCOOH, 11-nor-9-carboxy-THC; Tmax, time to maximum concentration; CYP, cytochrome P450; Cmax, maximum plasma concentrations; LOQ, limit of quantification; AUC0→10.5 h, area under the curve from 0–10.5 h postdose.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: S. Wright, G.W. Pharma.
Consultant or Advisory Role: None declared.
Stock Ownership: S. Wright, G.W. Pharma.
Honoraria: None declared.
Expert Testimony: None declared.
References
- 1.US Food and Drug Administration. Inter-agency advisory regarding claims that smoked marijuana is a medicine. Rockville (MD): Department of Health and Human Services; 2006. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108643.htm. [Google Scholar]
- 2.Watson SJ, Benson JA, Jr, Joy JE. Marijuana and medicine: assessing the science base: a summary of the 1999 Institute of Medicine report. Arch Gen Psychiatry. 2000;57:547–52. doi: 10.1001/archpsyc.57.6.547. [DOI] [PubMed] [Google Scholar]
- 3.GW Pharmaceuticals. Sativex receives qualifying notice for approval in Canada for the relief of cancer pain. 2007 [Press Release]. Available at: http://www.gwpharm.com/relief%20of%20Cancer%20Pain.aspx.
- 4.GW Pharmaceuticals. GW announces UK launch of world’s first prescription cannabis medicine. 2010 [Press Release]. Available at: http://www.gwpharm.com/release-sativex-launch.aspx.
- 5.Dalton WS, Martz R, Lemberger L, Rodda BE, Forney RB. Influence of cannabidiol on delta-9-tetrahydrocannabinol effects. Clin Pharmacol Ther. 1976;19:300–9. doi: 10.1002/cpt1976193300. [DOI] [PubMed] [Google Scholar]
- 6.McPartland JM, Russo EB. Cannabis and cannabis extracts: greater than the sum of their parts? J Cannabis Ther. 2001;1:103–32. [Google Scholar]
- 7.Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. Plasma delta-9-tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clin Pharmacol Ther. 1980;28:409–16. doi: 10.1038/clpt.1980.181. [DOI] [PubMed] [Google Scholar]
- 8.Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. Single dose kinetics of deuterium labelled delta-1-tetrahydrocannabinol in heavy and light cannabis users. Biomed Environ Mass Spectrom. 1982;9:6–10. doi: 10.1002/bms.1200090103. [DOI] [PubMed] [Google Scholar]
- 9.Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, Hollister LE. Single-dose kinetics of deuterium-labelled cannabidol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom. 1986;13:77–83. doi: 10.1002/bms.1200130206. [DOI] [PubMed] [Google Scholar]
- 10.Guy GW, Robson P. A phase I, open label, four-way crossover study to compare the pharmacokinetic profiles of a single dose of 20 mg of a cannabis based medicine extract (CBME) administered on 3 difference areas of the buccal mucosa and to investigate the pharmacokinetics of CBME per oral in healthy male and female volunteers. J Cannabis Ther. 2003;3:79–120. [Google Scholar]
- 11.Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-) delta-9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. 1998;95:8268–73. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuardi AW, Morais SL, Guimaraes FS, Mechoulam R. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56:485–6. [PubMed] [Google Scholar]
- 13.Karniol IG, Shirakawa I, Kasinski N, Pfeferman A, Carlini EA. Cannabidiol interferes with the effects of delta 9-tetrahydrocannabinol in man. Eur J Pharmacol. 1974;28:172–7. doi: 10.1016/0014-2999(74)90129-0. [DOI] [PubMed] [Google Scholar]
- 14.McArdle K, Mackie P, Pertwee R, Guy G, Whittle B, Hawksworth G. Selective inhibition of delta-9-tetrahydrocannabinol metabolite formation by cannabidiol in vitro. Toxicology; Proceedings of the BTS Annual Congress; 2001. pp. 133–4. Abstract. [Google Scholar]
- 15.Hunt CA, Jones RT, Herning RI, Bachman J. Evidence that cannabidiol does not significantly alter the pharmacokinetics of tetrahydrocannabinol in man. J Pharmacokinet Biopharm. 1981;9:245–60. doi: 10.1007/BF01059266. [DOI] [PubMed] [Google Scholar]
- 16.Bornheim LM, Correia MA. Purification and characterization of the major hepatic cannabinoid hydroxylase in the mouse: a possible member of the cytochrome P-450IIC subfamily. Mol Pharmacol. 1991;40:228–34. [PubMed] [Google Scholar]
- 17.Bornheim LM, Grillo MP. Characterization of cytochrome P450 3A inactivation by cannabidiol-hydrozyquinone as a P450 inactivator. Chem Res Toxicol. 1998;11:1209–16. doi: 10.1021/tx9800598. [DOI] [PubMed] [Google Scholar]
- 18.Selvarajah D, Gandhi R, Emery CJ, Tesfaye S. Randomized placebo-controlled double-blind clinical trial of cannabis-based medicinal product (Sativex) in painful diabetic neuropathy: depression is a major confounding factor. Diabetes Care. 2010;33:128–30. doi: 10.2337/dc09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berman JS, Symonds C, Birch R. Efficacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain. 2004;112:299–306. doi: 10.1016/j.pain.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Wade DT, Makela PM, House H, Bateman C, Robson P. Long-term use of a cannabis-based medicine in the treatment of spasticity and other symptoms in multiple sclerosis. Multiple Sclerosis. 2006;12:639–45. doi: 10.1177/1352458505070618. [DOI] [PubMed] [Google Scholar]
- 21.Brady CM, DasGupta R, Dalton C, Wiseman OJ, Berkley KJ, Fowler CJ. An open-label pilot study of cannabis-based extracts for bladder dysfunction in advanced multiple sclerosis. Multiple Sclerosis. 2004;10:425–33. doi: 10.1191/1352458504ms1063oa. [DOI] [PubMed] [Google Scholar]
- 22.Tomida I, Azuara-Blanco A, House H, Flint M, Pertwee RG, Robson PJ. Effect of sublingual application of cannabinoids on intraocular pressure: a pilot study. J Glaucoma. 2006;15:349–53. doi: 10.1097/01.ijg.0000212260.04488.60. [DOI] [PubMed] [Google Scholar]
- 23.Blake DR, Robson P, Ho M, Jubb RW, McCabe CS. Preliminary assessment of the efficacy, tolerability and safety of a cannabis-based medicine (Sativex) in the treatment of pain caused by rheumatoid arthritis. Rheumatology (Oxford) 2006;45:50–2. doi: 10.1093/rheumatology/kei183. [DOI] [PubMed] [Google Scholar]
- 24.Johnson JR, Burnell-Nugent M, Lossignol D, Ganae-Motan ED, Potts R, Fallon MT. Multi-center, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage. 2010;39:167–79. doi: 10.1016/j.jpainsymman.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Karschner E, Barnes A, Lowe R, Scheidweiler K, Huestis M. Validation of a two-dimensional gas chromatography mass spectrometry method for the simultaneous quantification of cannabidiol, Delta (9)-tetrahydrocannabinol (THC), 11-hydroxy-THC, and 11-nor-9-carboxy-THC in plasma. Anal Bioanal Chem. 2010;397:603–11. doi: 10.1007/s00216-010-3599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guy GW, Robson PJ. A phase I, double blind, three-way crossover study to assess the pharmacokinetic profile of cannabis based medicine extract (CBME) administered sublingually in variant cannabinoid ratios in normal healthy male volunteers (GWPK0215) J Cannabis Ther. 2004;3:121–52. [Google Scholar]
- 27.Barnes MP. Sativex: clinical efficacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain. Expert Opin Pharmacother. 2006;7:607–15. doi: 10.1517/14656566.7.5.607. [DOI] [PubMed] [Google Scholar]
- 28.Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids, I: absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol. 1992;16:276–82. doi: 10.1093/jat/16.5.276. [DOI] [PubMed] [Google Scholar]
- 29.Wall ME, Sadler BM, Brine D, Taylor H, Perez-Reyes M. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin Pharmacol Ther. 1983;34:352–63. doi: 10.1038/clpt.1983.179. [DOI] [PubMed] [Google Scholar]