

Abstract
Background
The endocannabinoid system modulates the hypothalamic–pituitary–adrenal (HPA) axis, but the effect of cannabinoid type 1 (CB1) receptor antagonism following chronic CB1 receptor stimulation in humans is unknown.
Objectives
To evaluate effects of the CB1 receptor antagonist rimonabant on the HPA axis in cannabis-dependent individuals.
Methods
Fourteen daily cannabis smokers received increasingly frequent 20 mg oral Δ9-tetrahydrocannabinol (THC) doses (60–120 mg/day) over 8 days to standardize cannabis tolerance. Concurrent with the last THC dose, double-blind placebo or rimonabant (20 or 40 mg) was administered. Cannabinoid, rimonabant, and cortisol plasma concentrations were measured 1.5 hours prior to rimonabant administration and 2.0, 5.5, and 12.5 hours post-dose.
Results
Ten participants completed before premature study termination due to rimonabant’s withdrawal from development. Five participants received 20 mg, three received 40 mg, and two placebo. There was a significant positive association between rimonabant concentration and change in cortisol concentration from baseline (r = .53, p < .01). There also was a borderline significant association between rimonabant dose and cortisol concentrations when the dose-by-time interaction was included. Four of eight participants receiving rimonabant (none of two receiving placebo) had greater cortisol concentrations 2 hours after dosing (at 11:30) than at 08:00, while normal diurnal variation should have peak concentrations at 08:00.
Conclusion
Rimonabant 20 or 40 mg did not significantly increase plasma cortisol concentrations, consistent with an absence of antagonist-elicited cannabis withdrawal.
Scientific Significance
Rimonabant doses >40 mg might elicit cortisol changes, confirming a role for CB1 receptors in modulating the HPA axis in humans.
Keywords: cortisol, rimonabant, cannabis, antagonist, withdrawal
INTRODUCTION
Cannabis sativa (marijuana) is the most widely used illicit drug in the world (1). Its primary psychoactive constituent, Δ9-tetrahydrocannabinol (THC) (2), acts at cannabinoid type 1 (CB1) receptors (3) to engage the endocannabinoid system (CB receptors and their endogenous ligands).
The endocannabinoid system maintains brain homoeostasis. Endocannabinoids inhibit the presynaptic release of neurotransmitters, thereby controlling the activation of neuronal circuits, including those involved in neuroendocrine regulation. Exogenous cannabinoids such as THC activate the hypothalamic–pituitary–adrenal (HPA) axis, the major neuroendocrine stress response system of mammals. However, the function of the endocannabinoid system in the regulation of stress hormone secretion remains unclear (4).
Cannabinoids alter HPA axis function by modulating corticotropin-releasing hormone (CRH) concentrations, either directly through CB1 receptor-mediated effects on CRH neurons in the paraventricular nucleus (5) or indirectly through other pathways (6). Preclinical studies show that acute THC administration dose-dependently increases adrenocorticotropic hormone, CRH, and cortisol concentrations (7,8). There is rapid development of tolerance following chronic exposure to cannabinoid agonists (8), blunting the cortisol response to subsequent acute exposure (6,7).
Human studies showed variable cannabinoid effects on the HPA axis. Similar to animals, acute cannabis administration increased cortisol levels (9). However, no change in the diurnal rhythm of cortisol secretion was observed during THC ingestion in chronic smokers (10). Chronic, frequent cannabis smokers have normal levels of cortisol and prolactin (11), suggesting tolerance development.
Tolerance to the stimulatory effects of THC on plasma adrenocorticotropic hormone (ACTH) and cortisol occurs in humans (12). Frequent cannabis smokers had blunted increases in cortisol release compared to healthy controls in response to intravenous THC administration (13).
Drug withdrawal stimulates the HPA axis, generally increasing corticosteroid concentrations (14-18). This is the case in rodents during CB1 antagonist-elicited cannabis withdrawal. In rats treated for 2 weeks with the synthetic CB1 agonist HU-210, withdrawal elicited by the selective CB1 antagonist rimonabant was associated with a 2.5-fold increase in CRH concentration in the amygdala and a one-third increase in plasma corticosterone (rodent equivalent of human cortisol) concentration (19).
To our knowledge, there was only one human study that examined effects of cannabis withdrawal on the HPA system. In 30 healthy male cannabis smokers who smoked cannabis cigarettes (2.2% THC) ad lib for 9 weeks while residing in a hospital unit, cortisol concentrations after 6 days of abstinence were unchanged compared to concentrations during daily cannabis smoking (20).
As the HPA axis is integral to the hormonal stress response, it is important to investigate its interaction with the endocannabinoid system, and specifically the impact of cannabis withdrawal on the CRH system. CB1 antagonists were developed as potential treatments for obesity, metabolic syndrome, and substance use disorders (21-23). As this drug class may eventually have widespread use for one or more of these indications, it would be prudent to assess the effects of antagonist-elicited withdrawal on the HPA axis. We report the effects of rimonabant administration on plasma cortisol concentration in male cannabis smokers following multiple daily oral THC doses to standardize cannabis tolerance.
METHODS
Participants
Participants provided written informed consent when not acutely intoxicated or in withdrawal. The study was approved by the Institutional Review Boards of the National Institute on Drug Abuse (NIDA) Intramural Research Program (IRP), the University of Maryland School of Medicine, and the Maryland Department of Health and Mental Hygiene. Inclusion criteria included 18–45 years old, smoked cannabis ≥1 year with daily use for at least 3 months prior to admission, urine specimen positive for cannabinoids in the 30 days prior to study entry, normal cardiac function, and IQ ≥ 85 (Wechsler Abbreviated Scale of Intelligence). Exclusion criteria included past or present clinically significant medical disease that might interfere with safe study participation; history of psychosis or any current Diagnostic & Statistical Manual, 4th edition (DSM-IV) axis I disorder (other than cannabis, caffeine, or nicotine dependence, or simple phobia); current physical dependence on substances other than cannabis, nicotine, or caffeine; history of clinically significant adverse events associated with cannabis intoxication or withdrawal, for example, psychosis or seizure; ≥6 alcohol drinks/day ≥4 times/week in the month prior to study entry; sesame oil allergy; or interest in drug abuse treatment. Participants were admitted to a secure research unit the evening of Day 1, 20 hours before the first THC dose, and discharged 22 hours after the last dose. The unit had 24-hour staffing, ensuring that subjects had no access to drugs except those provided in the study.
Study Design
A randomized, placebo-controlled, double-blind, escalating dose design was utilized. A total of 14 participants enrolled in the study and 10 completed. Subjects were enrolled in groups of 6. Five participants in the first group received rimonabant 20 mg; 1 was randomly assigned to receive placebo. The second dosing group received rimonabant 40 mg. Three participants received this dose of rimonabant; 1 was randomly assigned to receive placebo. The protocol was terminated prematurely during the second (40 mg) dosing group when worldwide clinical trials were stopped by the manufacturer, ending all rimonabant clinical development efforts (24).
Oral THC Administration
Oral synthetic THC (dronabinol, Marinol®, Unimed Pharmaceuticals, Marietta, GA, USA) was administered in 20 mg capsules with increasing frequency (every 4–8 hours) for total daily doses as follows: 40 mg on Day 2; 100 mg on Days 3–5; 120 mg on Days 6–8; and 40 mg on Day 9. The first dose was administered at 15:00 on Day 2 and the last at 09:30 on Day 9. This regimen standardized cannabis tolerance across participants while minimizing adverse events previously reported with 30 mg THC doses (25).
Rimonabant Administration
Subjects received a single oral dose of active or placebo rimonabant (Sanofi-aventis, Malvern, PA, USA) at 09:30 on Day 9.
Specimen Collection
Blood for cortisol, cannabinoids, and rimonabant was drawn from a peripheral vein on Day 9 at 08:00, 11:30, 15:00, and 22:00. For participants A, B, E, and I, an intravenous catheter placed at least 16 hours prior to collecting the 08:00 specimen was used to obtain all specimens. For the remaining subjects, the 08:00 specimen was drawn by venipuncture. An intravenous catheter was then placed within 90 minutes and was used to collect subsequent specimens.
Specimens for cannabinoids and rimonabant were collected in Vacutainer® tubes (BD Diagnostics, Franklin Lakes, NJ, USA) with sodium heparin, stored on ice no more than 2 hours prior to centrifugation, and separated plasma stored refrigerated at 4°C (cannabinoid specimens) or frozen at −20°C (rimonabant specimens) until analysis. Specimens for cortisol analysis were collected in Vacutainers® containing ethylenediamine tetraacetic acid, stored on ice no more than 2 hours prior to centrifugation, and separated plasma frozen at −70°C until analysis.
Cannabinoid Analysis
Free plasma cannabinoids were determined by a fully validated, previously published analytical method (26). Split calibration curves were employed for quantification of .25–100 ng/mL THC and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (THCCOOH) and .5–75 ng/mL 11-hydroxy-tetrahydrocannabinol (11-OH-THC). Intra- and inter-assay imprecision were <11% and <14%, respectively, and analytical recoveries were 86–113%.
Cortisol Radioimmunoassay and Rimonabant Assay
Blood specimens for cortisol analysis were spun in a refrigerated centrifuge at 495 g for 10 minutes Plasma (25 μl) was assayed with a commercially available antibody coated-tube [125I] cortisol RIA kit (MP Biomedicals, Irvine, CA, USA). All specimens were assayed in a single run, and average intra-assay coefficient of variability was 6.7%. The lower limit of assay sensitivity was 1.0 μg/dL.
Rimonabant plasma concentrations were measured by a validated liquid chromatography-tandem mass spectrometry method with a 1.0 μg/L limit of quantification (Sanofi-aventis).
Statistical Methods
Baseline cortisol concentrations in the venipuncture and intravenous catheter groups were compared using the nonparametric Wilcoxon two-sample test. Nonparametric methods were used due to non-normality of data distributions. Because venipuncture might be expected to elevate (but not depress) cortisol concentration compared to an indwelling catheter (27), a one-tailed p-value was used.
The influence of rimonabant on plasma cortisol concentrations was evaluated by repeated measures linear regression, with rimonabant dose as the independent variable and cortisol concentration (expressed as change from pre-rimonabant baseline) as the dependent variable. Change scores were employed to evaluate cortisol concentrations, as these were fairly normally distributed, whereas raw concentration values were not. Planned single-df contrasts evaluated the effect of each rimonabant dose relative to placebo. Because the rimonabant dose groups appeared to have different cortisol trajectories over time, a second repeated measures regression model was fit, including dose, time, and a dose-by-time interaction term. Because of the greater than twofold intersubject variability in plasma rimonabant concentrations (Table 1), this regression model also was evaluated with plasma rimonabant concentrations or the ratio of rimonabant/(THC + 11-OH-THC) plasma concentrations as independent variables, rather than rimonabant dose. The bivariate association between change in cortisol concentration and rimonabant concentration at each time point was evaluated using Pearson’s correlation coefficient.
TABLE 1.
Plasma rimonabant concentrations following a single 20 or 40 mg rimonabant dose in eight daily cannabis smokers.
| Rimonabant concentration (ng/mL)
| ||||
|---|---|---|---|---|
| Subject | Dose | Time of sample
|
||
| 11:30 | 15:00 | 22:00 | ||
| A | 20 | 89.5 | 76.2 | 49.1 |
| B | 20 | 181 | 67.0 | 23.6 |
| C | 20 | 71.8 | 33.2 | 10.7 |
| D | 20 | 172 | 54.7 | 27.6 |
| E | 20 | 168 | 61.4 | 44.8 |
| F | 40 | 378 | 111 | 45.0 |
| G | 40 | 143 | 67.7 | 23.5 |
| H | 40 | 187 | 57.2 | 30.3 |
Note: Daily cannabis smokers were administered Δ9-tetrahydrocannabinol (THC) 20 mg orally every 4–8 hours over 8 days (120 mg THC/day for last 3 days prior to rimonabant). At 9:30 on the ninth day, five subjects received rimonabant 20 mg and three subjects received rimonabant 40 mg concurrently with the last dose of THC.
The associations between cortisol concentrations and (1) cannabinoid concentrations or (2) rimonabant Cmax and AUC were evaluated using Spearman correlation coefficients (due to non-normal distribution of the data). Correlations were calculated separately for each time point because time also could affect the nature of the correlation.
Power calculations were performed using NCSS/PASS software 2004 release (Number Cruncher Statistical Systems, Kaysville, UT, USA).
RESULTS
Baseline (08:00) cortisol concentrations (median ± SD [range]) did not differ significantly in the venipuncture and intravenous catheter groups (12.9 ± 7.6 [7.2 –26.2] μg/dL and 9.4 ± 3.9 [6.1 – 13.7] μg/dL, respectively) (z = 1.17, p = .12), so the two groups were combined for all further analyses.
Cortisol concentrations were generally highest at 08:00 (mean ± SE = 13.3 ± 2.5 for 8 rimonabant subjects, 10.4 ± 3.3 for 2 placebo subjects), followed by decreasing concentrations later in the day (Figure 1), the expected normal diurnal rhythm. Four of eight subjects (A, B, C, and F) receiving rimonabant had greater concentrations of cortisol at 11:30 than at 08:00, while neither of the two subjects receiving placebo did so.
FIGURE 1.
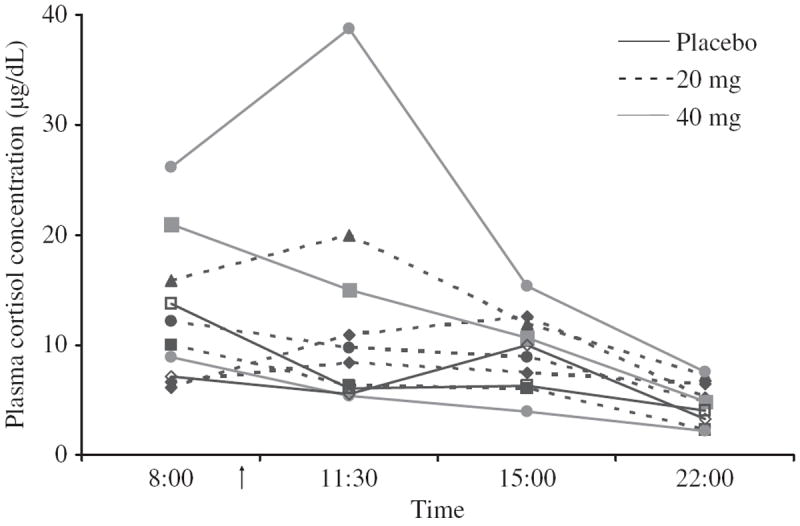
Plasma cortisol concentrations (μg/dL) before and after single placebo, 20, or 40 mg rimonabant doses in daily cannabis smokers. Ten cannabis smokers were administered Δ9-tetrahydrocannabinol (THC) 20 mg orally every 4–8 hours in escalating daily doses for 8 days to maintain and standardize cannabis tolerance (120 mg/day for 3 days prior to rimonabant). At 9:30 on the ninth day (arrow), subjects A, B, C, D, and E (dashed lines) received oral rimonabant 20 mg concurrently with the last dose of THC; subjects F, G, and H (gray lines) received oral rimonabant 40 mg concurrently with the last dose of THC. Subjects I and J (black lines) received placebo rimonabant.
Cortisol Concentrations and Rimonabant
There was no statistically significant effect of rimonabant dose on plasma cortisol concentration (20 mg vs. placebo: F1,20 = .89, p = .36; 40 mg vs. placebo: F1,20 = .56, p = .46). The model that included rimonabant dose, plus time, plus the dose-by-time interaction, found the effect of dose to be nonsignificant (F2,17 = 1.17, p = .33), the effect of time to be highly significant (F1,17 = 13.03, p = .002), and the effect of dose-by-time interaction term to be borderline significant (F2,17 = 3.00, p = .08).
Table 1 lists plasma rimonabant concentrations for each participant. There was a statistically significant positive association between change from baseline in plasma cortisol concentration and rimonabant concentrations (Figure 2; F1,15 = 30.3, p < .0001). The bivariate correlation also was significant (r = .53, p < .01). When time and interaction terms were added to the regression model, none of the terms were statistically significant (F1,13 = 1.21, p = .29 for rimonabant; F1,13 = .39, p = .54 for time; F1,13 = .04, p = .85 for rimonabant × time). There was no statistically significant association between plasma cortisol concentration and the ratio of rimonabant/(THC + 11-OH-THC) plasma concentrations, whether this was evaluated by itself (F1,15 = 2.06, p = .17) or including time and interaction terms in the regression model (F1,13 = 2.44, p = .14 for rimonabant ratio; F1,13 = .00, p = .95 for time; F1,13 = 2.05, p = .18 for rimonabant ratio × time). There were no statistically significant correlations between plasma cortisol concentrations at any time point and rimonabant Cmax or AUC (data not shown).
FIGURE 2.
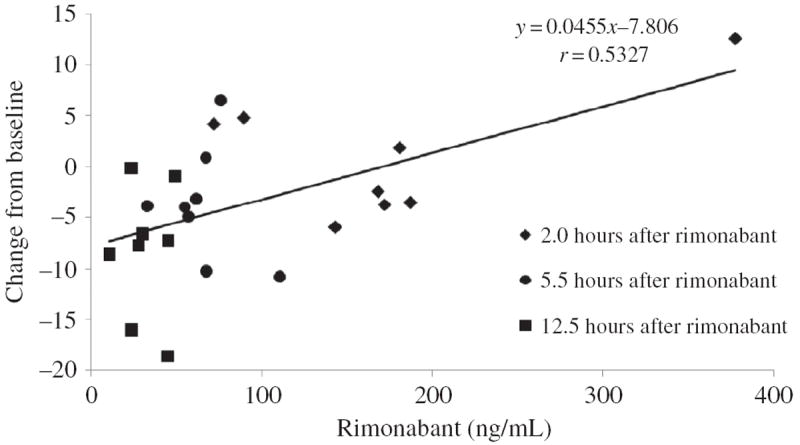
The association between plasma rimonabant concentrations and change from baseline plasma cortisol concentrations up to 12.5 hours after 20 or 40 mg rimonabant. Eight daily cannabis smokers were administered 20 mg Δ9-tetrahydrocannabinol (THC) orally every 4–8 hours for 8 days. On the ninth day, baseline cortisol concentrations were obtained at 08:00 and subjects received oral rimonabant at 09:30 concurrently with the last dose of THC. Line shows least squares linear regression best fit to all rimonabant concentrations.
Cortisol Concentrations and Cannabinoid Concentrations
There was a significant negative correlation between plasma cortisol and THCCOOH concentrations (r = −.64, p = .05). There were no other significant correlations between cortisol concentration and any cannabinoid at any time point (data not shown).
DISCUSSION
Single doses of 20 or 40 mg rimonabant did not significantly increase plasma cortisol concentrations in cannabis-dependent men. This is clinically relevant because 20 mg was the recommended therapeutic dose for this first-in-class agent for the treatment of obesity, metabolic syndrome, and substance use disorders. The lack of a significant effect on plasma cortisol is possibly due to the fact that these doses did not robustly elicit other measures of cannabis withdrawal (e.g., subjective symptoms, cardiovascular changes) in these cannabis-dependent individuals (Gorelick et al., J Clin Psychopharmacol, in press). Higher rimonabant doses might elicit withdrawal and increase cortisol concentrations.
Doses of rimonabant greater than the 20 or 40 mg single doses administered in this study are required to block the acute subjective and physiological effects of smoked cannabis. A single 90 mg rimonabant dose was required to significantly attenuate psychological and cardiovascular effects of smoked cannabis (28). A second study replicated the significant attenuation by a 90 mg single dose, and also found significant attenuation after 8 or 15 days of 40 mg daily doses (29). The plasma rimonabant Cmax achieved in those blockade studies was substantially higher (478.6 ng/mL following a single 90 mg dose and 307.8 and 334.6 ng/mL after 8 or 15 days, respectively, of daily 40 mg doses) than the Cmax achieved in this study.
Another explanation for the failure to observe the expected increase in cortisol concentrations may be the low power of the study as a result of premature termination. Based on the mean differences actually observed, 19 subjects would need to receive rimonabant to have 80% power to detect significant effects of rimonabant dose and time, as well as a significant dose-by-time interaction.
A methodological limitation of this study was the collection of baseline plasma samples by venipuncture (rather than indwelling catheter) in six subjects. Venipuncture is known to acutely and transiently increase plasma cortisol concentration as part of a stress response (27). However, this is unlikely to have substantially affected our results. There was no statistically significant difference in cortisol concentrations between the venipuncture and indwelling catheter groups. If venipuncture increased baseline cortisol concentrations, less of a change from baseline would be expected with the cortisol measurement at 11:00. Furthermore, the statistically significant positive association between cortisol and rimonabant concentrations was based on change from baseline concentration, thus minimizing the influence of baseline levels.
Precautions were taken to minimize the risk of adverse events which could spuriously elevate cortisol levels. Increasingly frequent doses of THC 20 mg were administered during the 8 days of continuous dosing to standardize cannabis tolerance across participants. The dosing regimen utilized in this study was well tolerated by all participants.
Three aspects of the findings suggest that a true phenomenon may have been missed due to early termination. First, there was a significant positive association between cortisol change from baseline and rimonabant concentrations, albeit only when time and interaction terms were ignored. Second, the statistical model that included rimonabant dose, plus time, plus the dose-by-time interaction showed a trend toward statistical significance for the effect of dose-by-time. These results suggest possible group differences in cortisol trends over time which might have achieved statistical significance with a larger sample size.
Finally, four of eight participants receiving rimonabant had greater cortisol concentrations at 11:30 (2 hours after dosing) than at 8:00, including one participant with a concentration of 38.7 μg/dL, well above the upper limit of normal. This is not consistent with the normal diurnal rhythm of cortisol, in which concentrations peak in the morning (typical values up to 24 μg/dL at 08:00), followed by a progressive decline throughout the day. Concentrations at 16:00 in healthy adults are in the range of 3–12 μg/dL (30). This concentration range was exceeded (at 15:00) by only one subject who achieved a peak plasma rimonabant concentration which was likely needed to elicit cannabis withdrawal (see Gorelick et al., J Clin Psychopharmacol, in press). Both participants receiving placebo showed the expected diurnal rhythm of cortisol concentration.
These changes are of potential clinical significance, given the possible effects of elevated cortisol concentrations on psychological and physiological function. Further studies with larger sample sizes and adequate power are warranted to evaluate the effect of CB1 antagonists on the HPA axis.
Acknowledgments
We acknowledge the clinical research teams of the NIDA IRP, Johns Hopkins Behavioral Pharmacology Research Unit, and Maryland Psychiatric Research Center (MPRC) Treatment Research Program for their support. This study was funded by the IRP, NIDA, National Institutes of Health, NIDA Residential Research Support Services Contract HHSN271200599091CADB (D. Kelly, PI), and Sanofi-aventis, and conducted under a Cooperative Research and Development Agreement (CRADA) between the National Institutes of Health and Sanofi-aventis.
Footnotes
Declaration of Interest
Drs. Ortemann-Renon and Bonnet are employees of Sanofi-aventis. No other authors have any conflicts of interest to declare. The authors alone are responsible for the content and writing of the paper.
References
- 1.United Nations Office on Drugs and Crime. 2008 World Drug Report. Vol. 310 Vienna: United Nations Office on Drugs and Crime; [Google Scholar]
- 2.Hawks RL. The constituents of cannabis and the disposition and metabolism of cannabinoids. In: Hawks R, editor. The Analysis of Cannabinoids in Biological Fluids. Research Monograph. Vol. 42. Rockville: National Institute on Drug Abuse; 1982. pp. 125–137. [PubMed] [Google Scholar]
- 3.Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34(5):605–613. [PubMed] [Google Scholar]
- 4.Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432. doi: 10.1016/S0079-6123(08)00433-0. [DOI] [PubMed] [Google Scholar]
- 5.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J Neurosci. 1991;11(2):563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy LL, Munoz RM, Adrian BA, Villanua MA. Function of cannabinoid receptors in the neuroendocrine regulation of hormone secretion. Neurobiol Dis. 1998;5:432–446. doi: 10.1006/nbdi.1998.0224. [DOI] [PubMed] [Google Scholar]
- 7.Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27(1):73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- 8.Brown TT, Dobs AS. Endocrine effects of marijuana. J Clin Pharmacol. 2002;42:90S–96S. doi: 10.1002/j.1552-4604.2002.tb06008.x. [DOI] [PubMed] [Google Scholar]
- 9.Cone EJ, Johnson RE, Moore JD, Roache JD. Acute effects of smoking marijuana on hormones, subjective effects and performance in male human subjects. Pharmacol Biochem Behav. 1986;24:1749–1754. doi: 10.1016/0091-3057(86)90515-0. [DOI] [PubMed] [Google Scholar]
- 10.Dax EM, Pilotte NS, Adler WH, Nagel JE, Lange WR. The effects of 9-ene-tetrahydrocannabinol on hormone release and immune function. J Steroid Biochem. 1989;34:1–6. doi: 10.1016/0022-4731(89)90090-3. [DOI] [PubMed] [Google Scholar]
- 11.Block RI, Farinpour R, Schlechte JA. Effects of chronic marijuana use on testosterone, luteinizing hormone, follicle stimulating hormone, prolactin and cortisol in men and women. Drug Alcohol Depend. 1991;28:121–128. doi: 10.1016/0376-8716(91)90068-a. [DOI] [PubMed] [Google Scholar]
- 12.D’Souza DC, Ranganathan M, Braley G, Gueorguieva R, Zimolo Z, Cooper T, Perry E, Krystal J. Blunted psychotomimetic and amnestic effects of delta-9-tetrahydrocannabinol in frequent users of cannabis. Neuropsychopharmacology. 2008;33(10):2505–2516. doi: 10.1038/sj.npp.1301643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranganathan M, Braley G, Pittman B, Cooper T, Perry E, Krystal J, D’Souza DC. The effects of cannabinoids on serum cortisol and prolactin in humans. Psychopharmacology (Berl) 2009;203(4):737–744. doi: 10.1007/s00213-008-1422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickworth WB, Fant RV. Endocrine effects of nicotine administration, tobacco and other drug withdrawal in humans. Psychoneuroendocrinology. 1998;23(2):131–141. doi: 10.1016/s0306-4530(97)00075-9. [DOI] [PubMed] [Google Scholar]
- 15.Adinoff B, Iranmanesh A, Veldhuis J, Fisher L. Disturbances of the stress response: The role of the HPA axis during alcohol withdrawal and abstinence. Alcohol Health Res World. 1998;22(1):67–72. [PMC free article] [PubMed] [Google Scholar]
- 16.Koob GF. Drug addiction: The yin and yang of hedonic homeostasis. Neuron. 1996;16(5):893–896. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- 17.Menzaghi F, Rassnick S, Heinrichs S, Baldwin H, Pich EM, Weiss F, Koob GF. The role of corticotropin-releasing factor in the anxiogenic effects of ethanol withdrawal. Ann N Y Acad Sci. 1994;739:176–184. doi: 10.1111/j.1749-6632.1994.tb19819.x. [DOI] [PubMed] [Google Scholar]
- 18.Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extra-cellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15(8):5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Fonseca FR, Carrera MRA, Navarro M, Koob GF, Weiss F. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 1997;276:2050–2053. doi: 10.1126/science.276.5321.2050. [DOI] [PubMed] [Google Scholar]
- 20.Cohen S. The 94-day cannabis study. Ann N Y Acad Sci. 1976;282:211–220. doi: 10.1111/j.1749-6632.1976.tb49900.x. [DOI] [PubMed] [Google Scholar]
- 21.Scheen A, Paquot N. Inhibitors of cannabinoid receptors and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2008;11(4):505–511. doi: 10.1097/MCO.0b013e3282fcea11. [DOI] [PubMed] [Google Scholar]
- 22.Xie S, Furjanic MA, Ferrara JJ, McAndrew NR, Ardino EL, Ngondara A, Bernstein Y, Thomas KJ, Kim E, Walker JM, Nagar S, Ward SJ, Raffa RB. The endocannabinoid system and rimonabant: A new drug with a novel mechanism of action involving cannabinoid CB1 receptor antagonism–or inverse agonism–as potential obesity treatment and other therapeutic use. J Clin Pharm Therapeut. 2007;32(3):209–231. doi: 10.1111/j.1365-2710.2007.00817.x. [DOI] [PubMed] [Google Scholar]
- 23.Tucci SA, Halford JC, Harrold JA, Kirkham TC. Therapeutic potential of targeting the endocannabinoids: Implications for the treatment of obesity, metabolic syndrome, drug abuse and smoking cessation. Curr Med Chem. 2006;13(22):2669–2680. doi: 10.2174/092986706778201512. [DOI] [PubMed] [Google Scholar]
- 24.Le Foll B, Gorelick D, Goldberg S. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology. 2009;205(1):171–174. doi: 10.1007/s00213-009-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW. Abstinence symptoms following oral THC administration to humans. Psychopharmacology. 1999;141:385–394. doi: 10.1007/s002130050848. [DOI] [PubMed] [Google Scholar]
- 26.Lowe RH, Karschner EL, Schwilke EW, Barnes AJ, Huestis MA. Simultaneous quantification of delta-9-tetrahydrocannabinol (THC), 11-hydroxy-delta-9-tetrahydrocannabinol (11-OH-THC), and 11-nor-delta-9-tetrahydrocannabinol-9-carboxylic acid (THCCOOH) in human plasma using two-dimensional gas chromatography, cryofocusing, and electron impact-mass spectrometry. J Chromatogr A. 2007;1163:318–327. doi: 10.1016/j.chroma.2007.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine A, Zagoory-Sharon O, Feldman R, Lewis JG, Weller A. Measuring cortisol in human psychobiological studies. Physiol Behav. 2007;90(1):43–53. doi: 10.1016/j.physbeh.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 28.Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, Frank RA. Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry. 2001;58:322–330. doi: 10.1001/archpsyc.58.4.322. [DOI] [PubMed] [Google Scholar]
- 29.Huestis MA, Boyd SJ, Heishman SJ, Preston KL, Bonnet D, Le Fur G, Gorelick DA. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology (Berl) 2007;194(4):505–515. doi: 10.1007/s00213-007-0861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fauci AS, et al., editors. Harrison’s Principles of Internal Medicine. 17. New York: McGraw-Hill, Health Professions Division; 2008. [Google Scholar]