

Abstract
Oncogenic transformation of postmitotic neurons triggers cell death, but the identity of genes critical for degeneration remain unclear. The antitumor antibiotic mithramycin prolongs survival of mouse models of Huntington's disease in vivo and inhibits oxidative stress-induced death in cortical neurons in vitro. We had correlated protection by mithramycin with its ability to bind to GC-rich DNA and globally displace Sp1 family transcription factors. To understand how antitumor drugs prevent neurodegeneration, here we use structure–activity relationships of mithramycin analogs to discover that selective DNA-binding inhibition of the drug is necessary for its neuroprotective effect. We identify several genes (Myc, c-Src, Hif1α, and p21waf1/cip1) involved in neoplastic transformation, whose altered expression correlates with protective doses of mithramycin or its analogs. Most interestingly, inhibition of one these genes, Myc, is neuroprotective, whereas forced expression of Myc induces Rattus norvegicus neuronal cell death. These results support a model in which cancer cell transformation shares key genetic components with neurodegeneration.
Introduction
Developmental and pathological cell deaths are highly regulated decisions in postmitotic neurons. Among the molecules that have been shown to modulate neuronal loss in physiology and disease, cell cycle mediators have garnered significant attention (Heintz, 1993; Freeman et al., 1994; Liu and Greene, 2001; Herrup, 2010). Early studies involving the simian virus 40 T-antigen, which promotes cell cycle progression via its ability to bind tumor suppressors, showed that transformation is simultaneously sufficient to drive transformation in dividing neuroblasts and, surprisingly, cell death in postmitotic neurons (Park et al., 2007). Since then, a role for cell cycle proteins in cell death in neurological disorders has been confirmed, but key regulators regulating this transition in postmitotic neurons are unknown, in part because of the absence of specific tools to elucidate the convergence between oncogenesis and neurodegeneration; such tools are essential because transformation is likely not the product of a single gene but rather a cooperative interactome culminating in a multifaceted phenotype (McMurray et al., 2008).
Over the past decade, the DNA binding drug mithramycin (MTM) has emerged as a useful chemical tool for studying the convergence between tumor transformation and neurodegeneration. Fifty years ago, Curreri and Ansfield (1960) demonstrated that MTM produced remissions in cases of embryonal carcinoma and choriocarcinoma of the testis. In vitro footprinting studies showed that MTM binds with highest affinity to GC-rich DNA (Van Dyke and Dervan, 1983). Footprinting and gel shift analyses were also used to show that MTM and Sp1 competitively bind to a GC-rich motif in the Myc (Snyder et al., 1991) and c-Src (Remsing et al., 2003) promoters. Indeed, the ability of MTM to bind to GC-rich DNA and displace the Sp1 family proteins from binding sites in numerous genes has been a model supported by >100 publications.
Interest in understanding the mechanism of action of MTM was further stimulated by two unexpected but converging preclinical studies. Torrance et al. (2001) screened 30,000 compounds and identified four, including MTM, that selectively suppressed growth in DLD-1 colon cancer cells but left their normal counterparts unaffected. The concentrations used matched studies that showed that MTM completely suppressed apoptosis of neurons as a result of oxidative stress or DNA damage without affecting global protein synthesis (Chatterjee et al., 2001). These findings were consistent with a scheme in which oxidative stress robustly induces DNA binding of Sp1 and Sp3 to affect proapoptotic gene expression (Ryu et al., 2003). Support for the model came from studies in which MTM improved behavior and extended the survival of Huntington's disease (HD) mice significantly (Ferrante et al., 2004; Voisine et al., 2007). Together, these data show that nanomolar concentrations of MTM could suppress tumor growth, enhance neuronal survival, and have no immediate effect on normal cell division.
We have used MTM analogs to probe transcriptional pathways necessary for oxidative neuronal death in vitro and in a fly model of HD. Our studies indicate a remarkable convergence between genes involved in transformation and those necessary for some types of neurodegeneration.
Materials and Methods
Cell culture
Immature primary cortical neurons were obtained from fetal Sprague Dawley rats [embryonic day 17 (E17)] as described previously (Ratan et al., 1994). Transformed SH-SY5Y neuroblastoma cells were cultured in DMEM/F-12 plus GlutaMAXmedium and 10% fetal bovine serum (Invitrogen).
Drosophila stocks, crosses, and survival assay
The polyglutamine-expressing transgenic stock used in this study is w; P{UAS-Httex1p Q93}4F1. These flies were mated with the pan-neuronal elav driver w; P{w+mW.hs = GawB}elavC155. Cultures were raised at 25°C. Virgin female progeny from the elav-Gal4 males × Httex1p Q93 females cross were collected and transferred to vials containing standard Drosophila food supplemented with 10 μm mithramycin A (MTM), 10 μm mithramycin SDK (SDK), 10 μm mithramycin SK (SK), 10 μm premithrmycin B (PreB), and water. The survival of at least 30–100 animals was monitored. In separate experiments, the flies were decapitated, and the heads were mounted on microscopic slides with nail polish after 10 d of feeding with MTM and its analogs. The rhabdomeres were analyzed under oil emersion at 63× to visualize individual rhabdomeres. The scoring (counting the number of rhabdomeres per ommatidium) was done by a blinded investigator. Rescue was calculated compared with the control group by the formula 100 * (Rt − Rc)/7 − Rc, where Rc is the number of rhabdomeres per ommatidium in the control group and Rt is the number of rhabdomeres per ommatidium in the treatment groups. RNA was isolated from the Drosophila heads, and expression of the mutant Huntington (mHtt) was assessed using reverse transcription-PCR followed by real-time PCR using the SYBR Green PCR Master Mix (Applied Biosystems) and the following primer pairs (huntingtin, 5′-CCGCTCAGGTTCTGCTTTTA-3′ and 5′ AAGGACTTGAGGGACTCGAA-3′; Act5C, 5′-ATCATCGTTTTGGGCGCATG-3′ and 5′-CATAGTATATCAATATCATATCTCATG-3′).
Cell viability
For cytotoxicity studies, immature primary cortical neurons (E17) were isolated as described above and plated at a density of 106 cells/ml in 96-well plates. The next day, three sets of experiments were performed. Cells were rinsed with warm PBS and then placed in medium containing the glutamate analog homocysteic acid (HCA) (5 mm). HCA was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5. In the first set of experiments, increasing concentrations of MTM or its analogs (SDK, SK, and PreB) were added at the time of HCA treatment and were present throughout the experiment. MTM and its analogs were prepared as 10 mm solutions, and serial dilutions were used to arrive to the desired concentration. In the second set of experiments, MTM and its analogs were added at various time points after HCA addition (0, 2, 3, 4, 5, 6, and 7 h after HCA addition). In the third set of experiments, 40 μm Int–H1–S6A F8 c-myc inhibitor (cell permeable; Enzo Life Sciences) (Giorello et al., 1998) or the H1–S6A F8 c-myc inhibitor (non-cell-permeable negative control; Enzo Life Sciences) were added at the time of HCA treatment and were present throughout the experiment. The next day, cell viability was assessed by the MTT assay (Promega) (Mosmann, 1983). One-way or two-way ANOVA followed by the Dunnett's or Bonferroni's post hoc tests, respectively, were used to measure statistical significance. p < 0.05 was considered to be statistically significant. The effectiveness of the MTT assays in measuring cell viability was confirmed by using the Live/Dead assay (Invitrogen) and fluorescence microscopy.
RNA extraction and real-time PCR
Total RNA was prepared from immature primary cortical neurons (E17) using the NucleoSpin RNA II kit (MACHEREY-NAGEL) according to the protocol of the manufacturer. Real-time PCRs were performed as a duplex reaction using a Myc (Rn00561507_m1), p21waf1/cip1 (Rn01427989_s1), c-Src (Rn01418228_m1), and Hif1α (Rn00577560_m1) gene expression assay (Applied Biosystems), which use a 6-carboxyfluorescein-labeled probe, and a β-actin gene expression assay, which uses a VIC-labeled probe (Applied Biosystems) so that gene amplification could be normalized to β-actin. These experiments were performed using a 7500 Real-time PCR System (Applied Biosystems) using standard PCR protocol and amplification conditions. One-way ANOVA followed by the Dunnett's post hoc test were performed to measure statistical significance. p < 0.05 was considered to be statistically significant.
Microarray analysis
Total RNA was extracted from immature primary cortical neurons (E17), after 4 h treatments with HCA (5 mm) along with protective doses (300 nm MTM and 75 nm SDK) as well as non-protective doses (50 nm MTM and 7.5 nm SDK) of MTM and SDK. The PreB (300 nm) treatments were used as negative controls. Three replicates were run per sample category, for a total of 36 arrays. RNA quantity was assessed with Nanodrop (Nanodrop Technologies) and quality with the Agilent Bioanalyzer (Agilent Technologies). Total RNA (200 ng) was amplified, biotinylated, and hybridized on Illumina Chips. Slides were scanned using Illumina BeadStation and signal extracted using Illumina BeadStudio software. Raw data was analyzed using Bioconductor packages. Quality assessment was performed looking at the inter-array Pearson's correlation, and clustering based on top variant genes was used to assess overall data coherence. Contrast analysis of differential expression was performed using the LIMMA package (Smyth et al., 2005). After linear model fitting, a Bayesian estimate of differential expression was calculated, and the threshold for statistical significance was set at p < 0.005. Data analysis was aimed at assessing the effect of drug treatment on control cells and cells treated with HCA. Gene Ontology (GO) analysis was performed using DAVID (for Database for Annotation, Visualization, and Integrated Discovery), and pathway analysis was performed by using the Functional Analysis Annotation tool in the Ingenuity Pathways Analysis software (Ingenuity Systems).
Weighted gene coexpression network analysis.
The weighted gene coexpression network analysis was performed as described (Zhang and Horvath, 2005; Oldham and Geschwind, 2006; Oldham et al., 2008). Briefly, after selecting genes the 6000 most variant genes, the absolute Pearson's correlation coefficients between one gene and every other screened gene were computed, weighted, and used to determine the topological overlap (TO), a measure of connection strength, or “neighborhood sharing” in the network. A pair of nodes in a network is said to have high TO if they are both strongly connected to the same group of nodes. In gene networks, genes with high TO have been found to have an increased chance of being part of the same tissue, cell type, or biological pathway.
Promoter analysis.
Promoter analysis was performed using the Transcription Analysis Listening System (TeLiS) (Cole et al., 2005). Of 4026 annotated probes, 1703 genes were found in the TeLiS database for promoter analysis (42% of submitted). One hundred ninety-two transcription factor binding motif matrices were scanned using TRANSFAC 3.2 [promoter size, −600 bases; high stringency (0.90)]. Of these matrices, seven were significantly overrepresented at p < 0.001 and a 2% false discovery rate: Sp1 (two occurrences), GC box elements, AP2, ELK1 VMYB, ATF, and CREBP1 (Cole et al., 2005).
Promoter activity assays
Immature primary cortical neurons (E17) were transfected with the reporter expression vectors [p21::Luc, p21 (Sp1 mut)::Luc (Xiao et al., 1999), Myc::Luc (Chung et al., 2006), Myc (Del6)::Luc (kind gift from Drs. David Levens and Hye-Jung Chung, National Cancer Institute, Bethesda, MD), E-box::Luc (pMyc–TA–Luc; Clontech), and control::Luc (pTA–Luc; Clontech) firefly luciferase plasmids using Lipofectamine 2000 (Invitrogen)] in accordance with the protocols of the manufacturers. The next day, cells were treated with MTM (300 nm), SDK (75 nm), SK (300 nm), PreB (75 and 300 nm), or Int–H1–S6A F8 c-myc inhibitor (40 μm) overnight. Next, the cells were washed with PBS and then lysed with luciferase assay buffer (Promega). The protein concentration was measured using a protein assay kit (Bio-Rad). Firefly luciferase activities were measured using a luciferase reporter assay system (Promega) and an LMax II384 luminometer (Molecular Devices). Firefly luciferase values were standardized to total protein concentration.
Immunoblot analysis
Nuclear and cytoplasmic protein extracts were obtained using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) in the presence of protease inhibitors, the proteasome inhibitor MG-132, and phosphatase inhibitors according to the protocol of the manufacturer. Samples were boiled in Laemli's buffer and electrophoresed under reducing conditions on NuPAGE Novex 4–12% Bis-Tris Gel polyacrylamide gels (Invitrogen). Proteins were transferred to a nitrocellulose membrane (Bio-Rad) by electroblotting. Nonspecific binding was inhibited by incubation in Odyssey blocking buffer (LI-COR Biosciences). Antibodies against Sp1 (PEP2; Santa Cruz Biotechnology), c-Myc (1472-1; Epitomics), p21waf1/cip1 (OP-76; Calbiochem), and β-actin (AC-74; Sigma-Aldrich) were diluted 1:1000, 1:1000; 1:1000, and 1:10,000, respectively, in Odyssey blocking buffer, and the membranes were incubated overnight at 4°C. Fluorophore-conjugated Odyssey IRDye-680 or IRDye-800 secondary antibody (LI-COR Biosciences) was used at 1:10,000 dilution followed by incubation for 1 h at room temperature. Finally, proteins were detected using an Odyssey infrared imaging system (LI-COR Biosciences).
Chromatin immunoprecipitation
The Ez-Magna ChIP assay kit was used as directed by the manufacturer (Millipore). Briefly, primary cortical cells were crosslinked with 1% formaldehyde at 37°C for 7 min. Cells were then sonicated using the Bioruptor (Diagenode) and immunoprecipitated with primary antibodies (10 μg). The crosslinking was reversed, and the DNA was isolated on the columns provided by the kit. Shearing size was determined to be between 150 and 1000 bp. Real-time PCR was conducted with primers targeted to the Myc promoter (rat promoter primers pair, 5′-GAGCGAGAGGAGGAAAAAAATAGA-3′ and 5′-CAGACCCCCGGATTATAAAGG-3′) or the p21waf1/cip1 promoter (rat promoter primer pair, 5′-CCGATGCTATAAGACCGCAGATCT-3′ and 5′-CAATTCCCCTTAACTCGGGCACTGTAGCAGCTC-3′) and SYBR Green PCR Master mix (Applied Biosystems). Each experiment was conducted at least three times by crosslinking cells from different primary cortical neuron preparations.
Myc short hairpin RNA knockdown
Two Myc (NM_012603) short hairpin RNA (shRNA) clones (TRCN0000039641, 5′ AAACCCAGGGCTGCCTTGGAAAAG 3′ and TRCN0000039641, 5′ AAACCCAGGGCTGCCTTGGAAAAG 3′; Open Biosystems) and Non-Target shRNA Control Vector (Sigma) were introduced into immature primary cortical neurons (E17) using the Amaxa Rat Neuron Nucleofector kit as directed by the manufacturer (Lonza). The next day, Myc knockdown was confirmed by whole-cell lysate Western blots. In addition, cells were treated with 5 mm HCA overnight, and viability was assessed using the MTT assay.
Adenoviral infections
Immature (E17) rat primary cortical cells were infected with 80 multiplicity of infection of a Myc adenovirus (kind gift from Drs. Preeti Ahuja and Robb MacLellan, University of California Los Angeles, Los Angeles, CA) or enhanced green fluorescent protein (GFP) adenovirus for 2 h in HBSS, after which the normal growth media were restored. The next day, Myc overexpression was confirmed by whole-cell lysate Western blots. In addition, cells were treated with either HCA (5 mm) alone or with both HCA (5 mm) and MTM (300 nm).
Statistical analysis
One-way or two-way ANOVA followed by the Dunnett's or Bonferroni's post hoc tests, respectively, were used to measure statistical significance. p < 0.05 was considered to be statistically significant.
Results
MTM and its analogs increase the lifespan of an HD model of Drosophila melanogaster
HD is a dominantly inherited neurodegenerative disorder. It is caused by expansion of a polyglutamine tract in Htt. Like other neurodegenerative disorders, HD is characterized by oxidative stress, DNA damage, and apoptosis. Previously, we have shown that MTM is one of the most effective compounds in ameliorating the phenotypes in the R6/2 mouse model of HD (Ferrante et al., 2004). These findings have been replicated in other laboratories (Stack et al., 2007), but the limited availability of the drug and its potential toxicity have precluded its testing in HD patients. Our goal in the current study was to use the MTM chemical analogs (SDK, SK, and PreB) (Fig. 1) to (1) better understand the mechanism of action of MTM, (2) to identify a source of MTM analogs that could allow scalable production for human use, and (3) to identify compounds that are at least as efficacious as MTM but that have a higher threshold for toxicity.
Figure 1.

A, MTM and analog chemical structures. B, Description of MTM analog structure, DNA binding ability, inhibition of Sp1 and Sp3 function, and role in carcinogenesis compared with the parent MTM compound.
To begin to address these questions in an in vivo context, we examined the efficacy of MTM and its analogs in a Drosophila model of HD. The basic structure of MTM is that of a tricyclic chromophore with a unique hydrophilic side chain attached in 3-position. MTM analogs SDK and SK are obtained by targeted gene inactivation of enzymes involved in the biosynthetic pathway of MTM, differ from the parent compound only in the structure and length of the hydrophilic side chain, and exhibit lower DNA binding affinity (Fig. 1). In contrast, PreB has four rings compared with three in MTM, lacks the hydrophilic 3-side chain, and is unable to bind to DNA (Remsing et al., 2003). To determine whether the MTM analogs can extend lifespan of flies expressing mHtt in neurons in vivo as well as MTM, we examined the effect of MTM and the MTM analogs (SDK, SK, and PreB) on the lifespan of transgenic flies expressing Htt exon 1 containing 93 glutamines (Q93) in all neurons. Normally, these flies exhibit motor defects and have a median survival of 9.5 d (Fig. 2A). As expected, flies fed with MTM (10 μm) had an extended median survival of 13 d, a lifespan extension comparable with the most effective treatments tested in this model. Similarly, flies fed with both SDK (10 μm) and SK (10 μm) had an extended survival of 13 and 12.5 d, respectively. Moreover, feeding of MTM and its analogs (SDK and SK) enhanced the number of rhabdomeres per ommatidium in HD flies and protected against degeneration of the eye. Rescue by MTM, SDK, and SK was calculated at 40, 45, and 81%, respectively, compared with flies receiving no treatment (Fig. 2C,D). Interestingly, feeding HD flies with PreB (10 μm) did not extend their lifespan, nor did it rescue eye degeneration (Fig. 2A,C,D). The distinct effects of MTM analogs could not be attributed to their ability to differentially affect mutant Htt transgene expression levels (Fig. 2B). These results suggest that MTM analogs can be effective therapeutic agents in a fly model of HD. They also demonstrate a structure–activity relationship (SAR) with MTM, SDK, and SK being equally effective in prolonging survival, whereas SK is superior in stemming eye degeneration, and PreB being indistinguishable from control.
Figure 2.
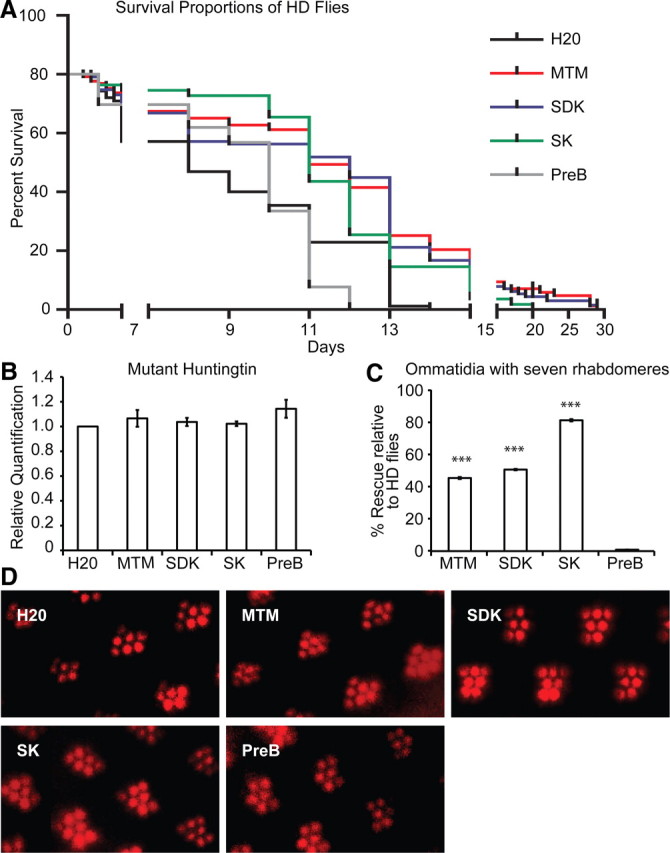
MTM and its analogs SDK and SK are protective in a Drosophila melanogaster model of HD. A, MTM or its analogs SDK and SK significantly extend the lifespan of HD flies expressing exon 1 of Htt with 93 CAG repeats (Q93). The median survival of flies receiving no treatment was 9.5 d compared with 13 d for flies receiving 10 μm MTM or SDK in their food or 12 d for flies receiving 10 μm SK in their food. For the MTM and SDK feeding survival curves, p < 0.0001 was calculated by the Mantel–Cox and Gehan–Beslow–Wilcoxon tests, whereas for the SK feeding survival curves, p < 0.001 or p < 0.0005 were calculated by the Mantel–Cox and Gehan–Beslow–Wilcoxon tests, respectively. Feeding of PreB (10 μm) had no statistically significant effect on the survival of the HD flies. The median survival of these flies was 10 d. These results indicate that the DNA binding ability of MTM and its analogs is necessary for their protective effect in vivo. B, MTM and its analogs had no significant effect on mHtt transgene expression as measured by real-time PCR. This indicates that the protective effect of MTM, SDK, and SK is independent of modulation of mHtt levels in the flies. C, D, MTM (10 μm), SDK (10 μm), and SK (10 μm) feeding enhanced the number of rhabdomeres per ommatidium in HD flies compared with ones receiving no treatment. Rescue was calculated at 40, 45, and 81% compared with the control group by the formula 100 * (Rt − Rc)/7 − Rc, where Rc is the number of rhabdomeres per ommatidium in the control group, and Rt is the number of rhabdomeres per ommatidium in the treatment groups. PreB (10 μm) feeding did not ameliorate the loss of rhabdomeres per ommatidium observed in the HD Q93 flies, indicating that the DNA binding ability of MTM and its analogs is necessary to suppress the neurodegeneration observed in the eyes of the HD flies. ***p < 0.0001.
The SAR for MTM or its analogs in vivo in affecting survival in a fly model of HD is similar to their SAR affecting oxidative death in cortical neurons in vitro
The in vivo data suggest a SAR that can be used to further elucidate the targets of MTM. However, they do not exclude the possibility that differences in the efficacies of MTM analogs reflect pharmacokinetic properties of the compounds or, alternatively, deleterious effects in non-neuronal cells that compromise overall efficacy. Furthermore, we examined all of the MTM analogs at a single dose. To further refine our understanding of the SAR of MTM and its analogs in neurodegeneration, we used an in vitro model of neuronal oxidative death. Early in their development in culture, cortical neurons exposed continuously to glutamate (or a glutamate analog, e.g., HCA) succumb through a mechanism dependent on competitive inhibition of cystine transport (Ratan et al., 1994). Reduced intracellular cyst(e)ine leads to depletion of the antioxidant glutathione. Cell death attributable to glutathione depletion has features of apoptosis and can be completely prevented by classical antioxidants (Ratan et al., 1994). Because oxidative stress resulting from impaired cysteine uptake and in turn glutathione depletion is a putative mediator of dysfunction and death in HD (140Q/140Q) mice (Li et al., 2010) as well as in a host of other acute and chronic neurodegenerative conditions and because many agents protective in our in vitro model are effective in rodent HD models, we used this model to explore the SAR of MTM analogs identified in HD flies. We treated primary cortical neurons with HCA (5 mm) alone or with varying concentrations of MTM, SDK, SK, and PreB and then quantified cell death using MTT reduction and Live/Dead staining. We observed that, like MTM, both SK and SDK potently protect immature neurons from oxidative stress-induced cell death (Fig. 3). The increased potency of SDK in protecting postmitotic neurons correlates with its rapid uptake (Albertini et al., 2006) and increased potency in killing of SH-SY5Y neuroblastoma cells (Fig. 4). In contrast, and consistent with the relative DNA binding affinity and cellular uptake in cancer cells (Albertini et al., 2006), SK exhibited the same potency as MTM in neurons. However, unlike MTM, SK did not lead to significant loss in viability in cells not undergoing oxidative stress. Moreover, in accordance with the fly studies, PreB did not protect the neurons from oxidative stress-induced cell death. Together, these findings suggest that the SAR for MTM analogs defined in flies could be related to distinct differences in the cell-autonomous mechanism of action of these drugs in degenerating postmitotic neurons (Chatterjee et al., 2001). The lack of a perfect congruence in SAR between the fly model in vivo and the neuron in vitro systems is likely related to the fact that exquisitely detailed concentration–response curves were generated in vitro, whereas only a limited number of doses were tested in the fly model and the pharmacodynamics is likely different in vivo.
Figure 3.
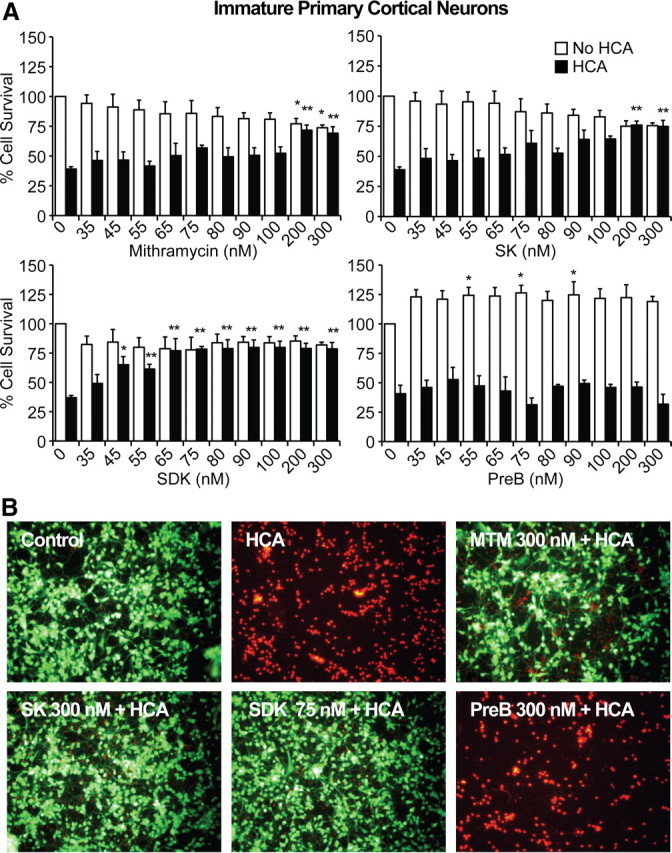
MTM, SK, and SDK protect immature primary cortical neurons (E17) from oxidative stress in a DNA binding-dependent manner. A, MTM, SK, and SDK abrogate neuronal cell death induced by HCA (5 mm) in a dose-dependent manner. SDK was protective at lower doses compared with MTM and SK. In addition, SK and SDK treatment induced no significant toxicity compared with non-HCA control, whereas MTM treatment did. *, Significant death compared with non-HCA control; **, significant protection compared with HCA treatment alone. PreB did not protect primary neurons from HCA-induced cell death. B, Live/Dead staining of treated neuron cultures. Live cells are identified by green, whereas dead cells are identified by red fluorescence.
Figure 4.
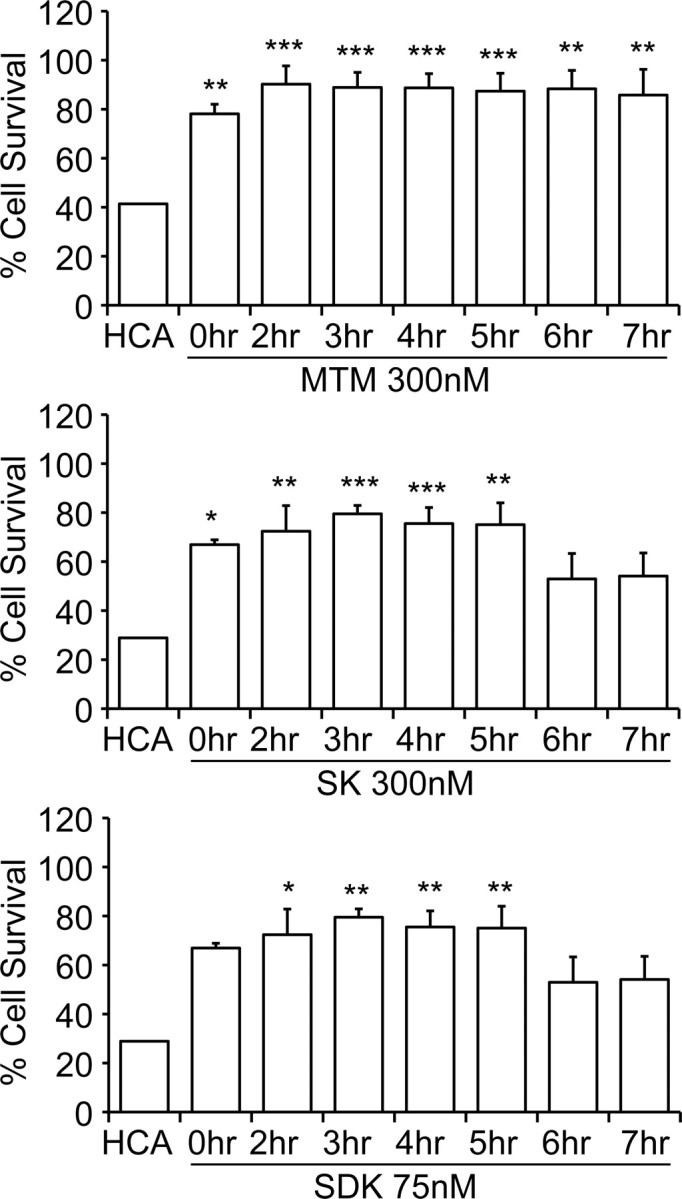
MTM and its analogs SK and SDK protect primary neurons from oxidative stress when administered up to several hours after HCA (5 mm) treatment. MTM (300 nm) significantly protects neurons from oxidative stress when administered up to 7 h after HCA treatment, whereas the SK (300 nm) and SDK (75 nm) analogs continue to significantly protect up to 5 h after HCA treatment. Statistical analysis was conducted by one-way ANOVA followed by the Dunnett's post hoc test. Significant protection compared with HCA treatment alone: *p < 0.05, **p < 0.01, and ***p < 0.0001. Untreated controls are calculated at 100% survival.
To harness the distinct effects of the analogs on protection from oxidative death for better understanding of the mechanism of action of MTM, we next focused on determining the kinetics of MTM protection from oxidative death. We reasoned that the optimal time to analyze gene changes induced by MTM, SDK, and SK that are relevant to their respective biological effects would require an understanding of the latest time the drugs can be added and still protect 100% of neurons. To determine the “commitment point,” we exposed cells to HCA and then added MTM, SK, or SDK at different time points. We observed a decline in protection from 100% when the analogs were added >5 h after exposure to HCA (Fig. 5). Because MTM diffuses rapidly into cells to bind to DNA (Albertini et al., 2006), we believe that the commitment point reflects the actual time after which MTM ceases to affect viability. From this, we conclude that 5 h is the point after which MTM or its analogs cease to be effective in stemming the apoptotic response; as a result, any transcriptional responses observed beyond this time point are likely contaminated by secondary changes rather than a primary result of MTM or analog treatment. Accordingly, we performed a gene microarray 4 h after exposure to HCA with or without protective or nonprotective doses of the analogs.
Figure 5.

Doses of MTM and its analogs (SDK and SK) that promote protection in immature primary cortical neurons induce cell death in transformed SH-SY5Y neuroblastoma cells. As expected, SDK was more effective in inducing cell death than MTM or SK. PreB did not induce cell death in neuroblastoma cells, further suggesting that the DNA binding ability of these compounds is necessary for their function. **p < 0.01; ***p < 0.0001.
A battery of genes covaries with protective doses of MTM and its analogs versus nonprotective doses
To identify the transcriptional networks associated with protection by MTM and its effective analogs, we used Illumina array technology to simultaneously query ∼20,000 reference–sequence-curated transcripts. As an initial step, we compared the levels of gene expression from neuronal cells treated with protective doses of MTM (300 nm) or SDK (75 nm) versus nonprotective doses (50 nm MTM or 7.5 nm SDK) or versus HCA (5 mm) alone. We also examined gene expression from cells treated with the non-neuroprotective analog PreB (300 nm) as an additional control. As expected, treatment with effective doses of MTM and SDK induced robust changes in gene expression, whereas nonprotective doses induced little or no change in gene expression at the chosen statistical threshold (p < 0.005) (Fig. 6A). Differentially expressed (DE) genes after treatment with MTM and SDK were highly overlapping (84% of the DE genes after MTM treatment are also DE after SDK treatment), supporting the idea that these two compounds act on the same pathways (Fig. 6B). GO analysis showed over-representation of a number of functional categories, including apoptosis, mitochondria, protein dimerization, and S-adenosyl-methionine-dependent methyltransferase activity. Pathway analysis confirmed those categories and suggested a main effect on transcriptional regulation. Interestingly, one of the networks identified by our pathway analysis was centered on Myc (Fig. 6C), which is downregulated by protective doses of MTM and SDK and unaffected by the nonprotective doses or PreB at both the mRNA and protein levels (Fig. 7C,G).
Figure 6.
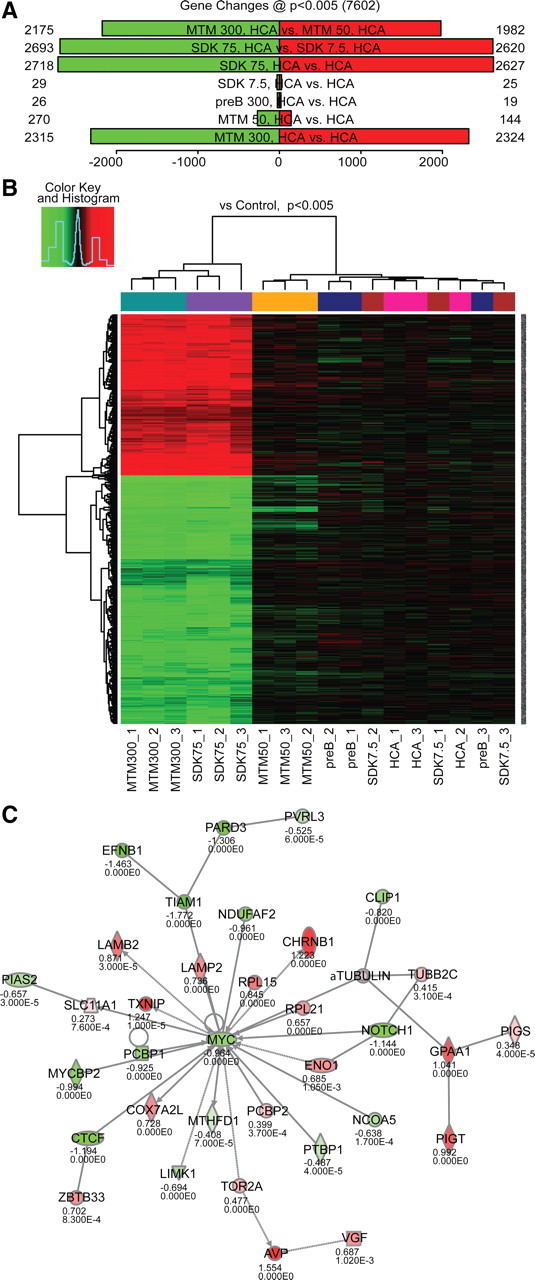
A network of genes centered on Myc covaries with MTM-mediated neuroprotection in immature primary cortical neurons (E17). A, Protective doses of MTM (300 nm) and SDK (75 nm) induce significant changes in gene expression compared with HCA (5 mm) alone or with nonprotective doses of MTM (50 nm), SDK (7.5 nm), or PreB (300 nm). Cells were treated with the compounds for 4 h. B, DE genes are highly overlapping after treatment with protective doses of MTM and SDK. C, Ingenuity analysis reveals a network of genes centered on Myc and whose expression covaries with protection by MTM and its analogs.
Figure 7.

Expression of genes involved in carcinogenesis covaries with protective doses of MTM, SK, and SDK in immature cortical neurons (E17). Protective doses of MTM, SK, and SDK significantly inhibit expression of c-Src, Hif1α, and Myc (A–C) and upregulate the expression of p21waf1/cip1 within 4 h (D). PreB has no effect. Significant protection compared with HCA treatment alone: *p < 0.05, **p < 0.01, and ***p < 0.0001. E, MTM (300 nm), SDK (75 nm), and SK (300 nm) significantly inhibit the expression of full-length Myc promoter driving luciferase as well as a deletion fragment that lost the five Sp1 binding sites upstream of the SmaI site at −101 relative to P1 promoter and that retains the Sp1 site between P1 and P2 promoter that we studied in our ChIP experiments, indicating that MTM and its analogs mediate their function through these sites. PreB (300 nm) did not have any effect on any of the constructs. Untreated controls are calculated at 100% luciferase activity. F, MTM (300 nm), SDK (75 nm), and SK (300 nm) significantly induce the expression of a 60 bp p21cip1/waf1 promoter driving luciferase. Conversely, mutating the Sp1 binding sites present in this construct abolishes the induction by MTM and its analogs, indicating that MTM and its analogs mediate their function through these sites. PreB (300 nm) did not have any effect on any of the constructs. Statistical analysis was conducted by one-way ANOVA followed by the Dunnett's post hoc test. Significant protection compared with the respective control: *p < 0.05, **p < 0.01, and ***p < 0.0001. Untreated controls are calculated at 100% luciferase activity. G, MTM (300 nm) treatment significantly reduced the Myc protein levels. H, MTM (300 nm) treatment significantly induced p21waf1/cip1 protein levels.
To dissect the transcriptional changes induced by protective MTM and SDK treatment and to prioritize candidates for additional study, we performed an unbiased network analysis (Oldham et al., 2008) of the microarray expression profiles, which allows groups of tightly coexpressed genes (modules) to be identified across many experimental conditions. Because genes expressed by the same cells or involved in similar functions are often closely coexpressed in modules, large datasets enable such gene groups to be identified (Oldham et al., 2008). Three main modules showed strong correlation with MTM and SDK treatment. Although partially overlapping, the GO analysis of these modules showed some distinctive features, suggesting overrepresentation of mitochondrial, neurogenesis, and apoptosis genes, respectively. For example, members of the mitogen-activated protein kinase pathway, including Raf1, Mek, and Erk1/2, were clustered in these three modules. Interestingly, Myc (oncogene), Mycn (oncogene), Src (oncogene), and Hif1α (increased in many cancers) were all clustered in a single module. We confirmed by quantitative real-time PCR that c-Src, Hif1α, and Myc were significantly downregulated by protective doses but unaffected by nonprotective doses of MTM/SDK/SK or PreB (Fig. 7A–C). c-Src has the ability to induce Myc expression (Chang et al., 2008). Hif-1α has been postulated to associate with Myc to repress DNA repair genes, thus enhancing mutations in genetic hotspots (Yoo et al., 2009). This is interesting considering that DNA repair is among the top categories affected by MTM and SDK treatment. Of note, p21waf1/cip1 expression is increased by MTM and SDK treatment. Real-time PCR and Western blots confirmed that its mRNA and protein levels are increased (Fig. 7D,H). Previous studies from our laboratory and others have shown that c-Src inhibition (Khanna et al., 2002), Hif-1α deletion (Aminova et al., 2008), or p21waf1/cip1 overexpression (Langley et al., 2008) can protect neurons from oxidative death, but the SAR combined with the array are consistent with a model whereby oxidative stress requires the expression of genes involved in oncogenic transformation to mediate their deleterious effects on viability or lifespan.
To understand whether there is common promoter architecture for those genes that are downregulated that might explain their sensitivity to MTM or its analogs, we used the Telis database (www.telis.ucla.edu), which looks for overrepresentation of transcription factor binding sites in the promoters of genes in DE gene lists. We applied this analysis on the subset of probes affected by both MTM and SDK (n = 4026). The top overrepresented transcription factor was Sp1 (present in 50% of 1703 annotated genes vs. 40% in the control set; p = 1.00e-10). Nearly all of the genes that are downregulated contain phylogenetically conserved Sp1 consensus sites in their promoters. In contrast, p21waf1/cip1, a tumor suppressor gene, also has phylogenetically conserved Sp1 sites in its promoter, and yet its expression was increased. The remarkable ability of MTM or its bioactive analogs to reduce the expression in neurons of genes associated traditionally with oncogenesis and yet to increase the expression of a tumor suppressor gene, p21waf1/cip1, led us to directly examine the binding of Sp1 family members to distinct promoters of genes suppressed or enhanced by MTM or its analogs.
Mithramycin and its analogs only partially mediate their neuroprotective effect by inhibiting Sp1 family binding to target genes
MTM is believed to be a canonical inhibitor of Sp family members binding to their consensus DNA binding sites. To determine whether the protective effects of MTM and its analogs covary relative to their ability to inhibit Sp1 binding to target sites in the promoters of genes identified in our informatics analysis, we performed chromatin immunoprecipitation (ChIP) using both Sp1 and Sp3 antibodies. We chose Sp1 and Sp3 because we have shown previously by electrophoretic mobility shift assay (EMSA) that their DNA binding is robustly induced by oxidative stress in cortical neurons (Ryu et al., 2003). We also chose these two factors because their DNA binding domains are virtually identical (Fig. 8A,B). Based on the accepted mode of action of MTM, it should inhibit both of these proteins to a similar extent. For the ChIP assays, we focused on well-defined Sp1 consensus motifs in the Myc (Snyder et al., 1991) and p21waf1/cip1 (Xiao et al., 1999) promoters because nearly complete downregulation of Myc (oncogene) and upregulation of p21waf1/cip1 (tumor suppressor gene) occurred at protective concentrations of all the analogs. We validated these Sp1 consensus motifs in the Myc and p21waf1/cip1 promoters by conducting promoter reporter analysis. As expected, MTM, SDK, and SK, but not PreB, inhibited the expression of luciferase driven by the Myc promoter (Fig. 7E). In contrast, MTM, SDK, and SK, but not PreB, induced the expression of a luciferase driven by the proximal p21waf1/cip1 promoter (Fig. 7F). In each case, induction or repression was dependent on the presence of the Sp1 sites. These findings focused our ChIP experiments on those regions of the Myc or p21waf1/cip1 promoter with Sp1 sites necessary for MTM-mediated repression or induction. We exposed cortical neurons to oxidative stress with protective or nonprotective doses of MTM or SDK as well as with the nonprotective PreB and then conducted the ChIP experiments at the Myc or p21waf1/cip1 loci. Protective doses of MTM or SDK significantly inhibited Sp1 occupancy at the Myc promoter, whereas nonprotective doses had no effect (Fig. 8D). As expected, treatment of cells with PreB had no effect on Sp1 binding; moreover, the MTM/SDK-dependent decrease in Sp1 occupancy on the Myc promoter was not attributable to an effect of MTM/SDK treatment on Sp1 protein levels (Fig. 8C). Sp3 was also bound to the Myc promoter. Surprisingly, MTM or SDK at protective or nonprotective doses failed to displace Sp3 from its site on the Myc promoter (Fig. 8E). Together, these studies show that protective doses of MTM analogs inhibit Sp1, but not Sp3, occupancy at the proximal Myc promoter and argue against the previously held model that MTM binds to the minor groove of GC-rich DNA to displace the zinc finger DNA binding domain of Sp1 family transcription factors. Indeed, our data suggest that other factors outside of the DNA binding domain of Sp1 or Sp3 must explain the selectivity of MTM or its analogs in inhibiting Sp1, but not Sp3, binding to distinct promoters.
Figure 8.

Protective doses of MTM and SDK inhibit Sp1 binding to a subset of its targets but have no effect on Sp3 binding. A, Schematic depicting protein structures of Sp1 and Sp3. The glutamine-rich regions, serine/threonine regions, and the zinc fingers that make up the DNA binding domain (DBD) are depicted by dark gray, light gray, and black boxes, respectively. The region (+/−) is rich in charged amino acids. The four regions (A, B, C, and D) contribute to the transcriptional activity. Activation domains (AD) and inhibitory domains (ID) are shown. B, Protein sequence alignment of the DNA binding domain of Sp1 and Sp3. Cysteine and histidine residues that coordinate zinc ions are highlighted in light gray, and protein regions that contact the DNA are highlighted in dark gray. C, MTM (300 nm) treatment does not affect Sp1 protein levels in immature cortical (E17) neurons. D, E, Protective doses of MTM and SDK inhibit Sp1, but not Sp3, binding to their sites in the Myc promoter, whereas nonprotective doses or PreB do not. F, G, MTM and SDK do not inhibit Sp1 or Sp3 binding to their sites in the p21waf1/cip1 promoter. Immature cortical neurons (E17) were treated with the compounds for 4 h for both the Western blots and chromatin immunoprecipitation experiments. Statistical analysis was conducted by one-way ANOVA followed by the Dunnett's post hoc test. Significant protection compared with the respective control: *p < 0.05. Untreated controls are calculated at onefold.
Because MTM or its analogs increase the expression of p21waf1/cip1, we examined the effect of MTM and SDK treatment of cortical neurons exposed to oxidative stress on Sp1 and Sp3 occupancy of the p21waf1/cip1 promoter. As expected from our gene expression and protein data, we observed that MTM and SDK had no effect on both Sp1 and Sp3 occupancy at the p21waf1/cip1 promoter (Fig. 8F,G). These results are the first demonstration that MTM and its analogs do not uniformly inhibit Sp1 family binding to target sites in promoters. Indeed, in previous EMSAs, we found that MTM inhibited both Sp1 and Sp3 binding to a consensus DNA binding sequence (Chatterjee et al., 2001). These results suggest a model in which MTM inhibits Sp1 binding to a subset of genes associated with oncogenesis but leaves Sp3 unperturbed. In contrast, MTM has no effect of Sp1 binding to one tumor suppressor; again Sp3 is unperturbed. To exclude the possibility that our ChIP results reflect nonspecific cross-reactivity of our antibodies with other proteins, we reduced the expression of both Sp1 and Sp3 and then performed ChIPs. We observed a significant reduction in Sp1 and Sp3 occupancy on the Myc and p21waf1/cip1 promoters compared with control conditions, thus verifying the specificity of our antibodies (data not shown).
Mithramycin and its analogs only partially mediate their neuroprotective effect by inhibiting Myc expression
Our findings pointed to MTM-specific reductions in a gene network associated with Myc oncogenesis (Fig. 6C). To establish whether the decreased expression of the Myc gene in response to MTM and its analogs can contribute to a protective phenotype in neurons, we used an established peptide inhibitor to reduce Myc function. The Int–H1–S6A F8 c-myc inhibitor is cell permeable, affects Myc/Max dimer formation, and inhibits Myc-dependent transcription (Giorello et al., 1998). As such, both MTM and the Int–H1–S6A F8 c-myc inhibitor significantly affect the expression of an E-box driving luciferase (Fig. 9A). As expected, the Int–H1–S6A F8 c-myc inhibitor partially protects immature primary cortical neurons from oxidative stress-induced cell death, whereas a nonpermeable negative control peptide (H1–S6A F8 c-myc inhibitor) had no effect on oxidative stress-induced neuronal cell death (Fig. 9B,C). To further verify the specificity of our peptide inhibitor, we directly reduced Myc expression using shRNA. Selective reduction in Myc (Fig. 9D) significantly protects immature cortical neurons from oxidative death (Fig. 9E). In contrast, overexpression of Myc in immature cortical neurons (Fig. 9F) significantly reduced the survival of these cells (Fig. 9G), although it did not sensitize them to oxidative death (Fig. 9E). In addition, Myc overexpression did not abolish the protective effect of MTM.
Figure 9.
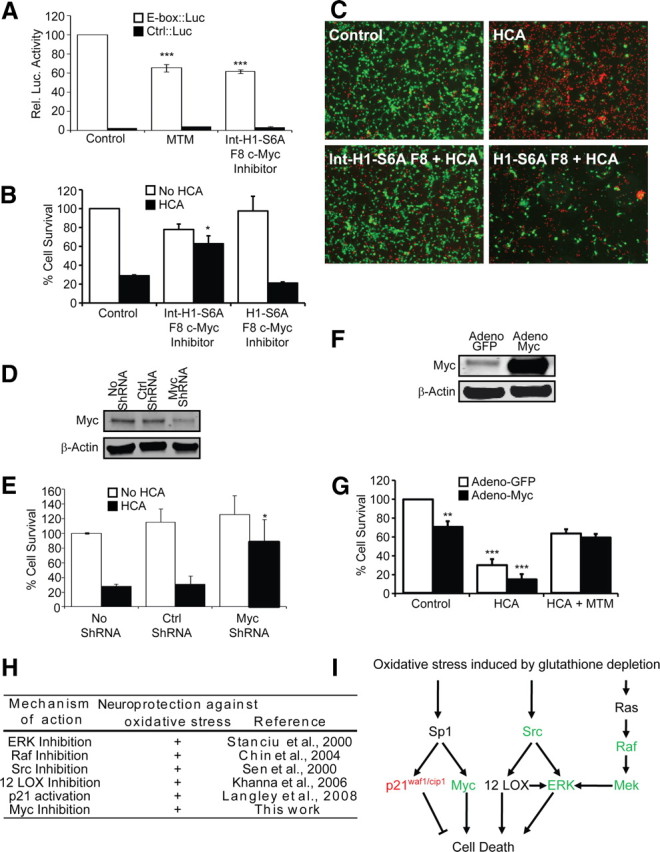
Myc suppression is sufficient but not necessary to mediate the protective effects of MTM in immature cortical neurons exposed to oxidative stress. A, Protective doses of both MTM (300 nm) and Int–H1–S6A F8 c-myc inhibitor (40 μm) significantly inhibit the expression of an E-box driving luciferase construct (Clontech). ***p < 0.0001. B, Treatment with Int–H1–S6A F8 c-myc inhibitor (40 μm) significantly protects cells from oxidative stress-induced death. The negative control peptide (H1–S6A F8 c-myc inhibitor, 40 μm) has no effect. *p < 0.05. C, Live/Dead staining of treated neurons. D, Western blots showing a significant reduction in Myc protein levels in cells expressing Myc shRNA compared with control shRNA. E, Inhibition of Myc expression using shRNA protects immature primary cortical neurons (E17) from oxidative stress-induced death. *p < 0.05. F, Western blots showing that Myc protein is increased in immature primary cortical neurons (E17) infected with the Myc2 adenovirus. G, Overexpression of Myc2 in immature primary cortical neurons (E17) significantly affects their survival but is not sufficient to block the ability of MTM to protect them from oxidative stress. Statistical analysis was conducted by one-way ANOVA followed by the Dunnett's post hoc test. Significant protection compared with control cells infected with Adeno-GFP: **p < 0.01 and ***p < 0.0001. H, Table highlighting the multiple signaling molecules targeted by protective doses of MTM and its analogs and shown to modulate neuroprotection in our model of oxidative stress. I, Model depicting pathways affecting oxidative death in neurons and that are targeted by MTM, SDK, and SK. Expression of genes depicted in green is reduced, whereas expression of genes depicted in red is induced by protective doses of MTM.
A distinctive feature of Myc is that it encodes two translational forms, Myc1 and Myc2, that differ at their N terminal. Myc1 is generated by a non-AUG translational start site. There is evidence showing that Myc1 and Myc2 are functionally distinct (Hann et al., 1994). Although in these experiments, we overexpressed the Myc2 translational isoform of the MYC gene, overexpression of Myc1 yields a similar outcome (data not shown). These results suggest that downregulation of Myc is sufficient, but not necessary, for the protective response induced by MTM. Altogether, these studies support the notion that MTM-mediated repression of expression of a host of genes associated with oncogenesis, including Myc, Src, and Hif-1α, can protect postmitotic neurons from oxidative death (Fig. 9H,I). Furthermore, a striking correlation exists between protective doses of structurally diverse MTM analogs and the reduction of expression of these genes. Although the simple model has been that MTM inhibits Sp1 binding to many GC-rich binding sites in promoters, our findings suggests that the protective effects of MTM in neurons derive from reduction in Sp1 binding to a small set of promoters of genes whose expression is classically associated with oncogenesis (Table 1) and its ability to selectively ignore Sp1 binding sites in other promoters such as p21cip1/waf1 that are classically linked with tumor suppression.
Table 1.
List of genes that are downregulated by protective doses of MTM (300 nm) and SDK (75 nm), involved in carcinogenesis, and obtained from two networks identified in the microarray analysis: cell cycle and molecular mechanism of cancer
| Gene | Mediate carcinogenesis |
|---|---|
| ADCY3 | + |
| ADCY8 | + |
| ADCY9 | + |
| BMPR1A | + |
| HIF1A | + |
| LRP5 | + |
| LRP6 | + |
| MAP2K4 | + |
| MAPK14 | + |
| NFKBIB | + |
| PIK3C3 | + |
| PRKAG2 | + |
| RAP1B | + |
| RRAS2 | + |
| SMAD1 | + |
| SMAD6 | + |
| SRC | + |
| TCF3 | + |
| AATF | + |
| ACVR1 | + |
| BCAR3 | + |
| CCNH | + |
| CCT2 | + |
| CDC2A | + |
| CHES1 | + |
| CRK | + |
| CRKL | + |
| CSPG6 | + |
| CTCF | + |
| CUL1 | + |
| CUL3 | + |
| DLG7 | + |
| DNAJA2 | + |
| DNM2 | + |
| FOXG1 | + |
| FRAP1 | + |
| GSPT1 | + |
| ID3 | + |
| JAK2 | + |
| MAP2K1 | + |
| MAP2K6 | + |
| MAPK1 | + |
| MYC | + |
| PBEF1 | + |
| PCNA | + |
| RAF1 | + |
| RALBP1 | + |
| RUVBL1 | + |
| SIAH1A | + |
| STRN3 | + |
| TNFSF5IP1 | + |
| TPX2 | + |
| TSG101 | + |
| TUBB3 | + |
| NOTCH1 | − |
| SMAD2 | − |
| SMAD4 | − |
| BCCIP | − |
| BRINP2 | − |
| COMMD5 | − |
| DAB2IP | − |
| DLGH1 | − |
| GAK | − |
| LZTS1 | − |
| MAPK6 | − |
| MSH2 | − |
| PTEN | − |
| RAD1 | − |
| RAP1A | − |
| RB1 | − |
| SMPD3 | − |
| TBRG1 | − |
| UBE1C | − |
The majority of these downregulated genes normally promote carcinogenesis as opposed to a small number that inhibit carcinogenesis.
Discussion
MTM is a prototypic Sp1 inhibitor that was engaged as a chemotherapeutic agent and found to have efficacy long before its putative mechanism of action was understood. Using various methods, several groups developed a model whereby MTM displaces Sp1 from its sites in aberrantly expressed oncogenes such as Myc to mediate its antitumor actions. Here, we provide data using careful SAR analysis of MTM analogs in vitro and in vivo along with promoter analysis and chromatin immunoprecipitations to support aspects of the previous model in an unexpected context, neurodegeneration. Indeed, we have found that MTM displaces Sp1 from its sites in the promoters of select oncogenes such as Myc (Fig. 8D) and that the reduction in expression of such genes not only covaries with biological phenotype of neuroprotection in neurons (Fig. 6A,C, 7C) but also inhibition of each of these targets individually [Myc (Fig. 9B,C,E), c-Src (Sen et al., 2000; Khanna et al., 2002, 2006), Erk (Stanciu et al., 2000), and Raf (Chin et al., 2004)] protects neurons. Interestingly, although oxidative stress does not increase Myc protein levels, we speculate that it may induce a posttranslational change in the Myc protein that alters its ability to bind DNA or its transactivation potential (data not shown). Additionally, our analysis also provides unexpected insights that may require a complete refinement in our understanding of the mode of action of MTM. First, MTM fails to displace Sp1 or Sp3 from all promoters in neurons (Fig. 8E–G). This suggests that MTM must interact with DNA in a manner that allows it to selectively displace Sp1 from some sites and not others. We have found that Sp1 levels are much lower than Sp3 levels, so the differences may simply relate to differences in capacity or affinity of Sp3 versus Sp1, a feature not easily queried using ChIP analysis. It is also possible that adjacent sequences in the promoters play an important role in establishing whether MTM can competitively inhibit Sp1 occupancy. The concentration dependence of protection and reductions in gene expression or DNA binding does suggest a simple competitive interaction between Sp1 and MTM.
MTM was identified in a screen for compounds effective in inhibiting tumor cell growth without affecting normal cells (Torrance et al., 2001). It is also one of the most effective agents tested to date in a worm model (Voisine et al., 2007) and mouse model (Chatterjee et al., 2001; Ferrante et al., 2004; Qiu et al., 2006; Ryu et al., 2006) of HD. Moreover, MTM protects against dopaminergic neurotoxicity in the mouse brain after administration of methamphetamine, suggesting that it may be effective in treating methamphetamine users (Hagiwara et al., 2009). Together, these studies highlight its potential therapeutic potential for many cancers as well as CNS diseases characterized by neurodegeneration or acute insults. Indeed, MTM is approved by the Food and Drug Administration for leukemia and testicular cancer treatment; however, the side effects associated with its administration in humans have limited its use. Translation of MTM to the bedside will be facilitated by the development of analogs that are less toxic and more efficacious. These analogs should also be available in quantities sufficient to ultimately complete a human clinical trial. We chose to characterize two previously identified MTM analogs (SDK and SK) that are obtained by genetic manipulation of the MTM biosynthetic pathway because they have improved transcriptional and anti-proliferative activity in cancer cell lines (Fig. 1) with minimal effects on normal cell growth (Albertini et al., 2006) as well as in human ovarian xenogafts in vivo (Previdi et al., 2010). Although SDK and SK have a lower DNA binding affinity than the parent compound, the doses of SDK and SK required for the inhibition of tumor growth in orthotopic xenografts and for mice survival were lower than those required for MTM (Previdi et al., 2010). This could be in part attributable to their increased cellular uptake and bioavailability (Albertini et al., 2006); moreover, no toxicity was associated with SDK and SK treatment in vivo (Previdi et al., 2010). We found that they, like MTM, are protective in a fly model of HD (Fig. 2) as well as in a neuronal model of oxidative stress (Fig. 3). Because of their reduced toxicity, we speculate that their use may help circumvent the side effects of MTM.
We have identified a battery of genes that are regulated in response to protective doses of MTM (Figs. 6, 7). During examination of the top networks, we observe that a subset of these genes mediate cancer-related processes and that inhibition of at least one of them, namely Myc, is sufficient to protect against oxidative stress (Fig. 9B,C,E). Indeed, mice exclusively overexpressing Myc in neurons exhibit a neurodegenerative phenotype (Lee et al., 2009); in addition, Myc overexpression in neurons is sufficient to induce significant cell death (Fig. 9F,G). We speculate that, when cells are challenged with oxidative stress, oncogenic signaling pathways become critical mediators of the apoptotic response and that MTM functions to prevent these pathways from tipping the balance toward death (Fig. 9I). We find that MTM and its analogs target several key pathways (Erk, c-Src, Hif1α, and p21waf1/cip1) involved in carcinogenesis and previously described to affect oxidative stress-mediated neuronal cell death (Fig. 9H,I) (Sen et al., 2000; Khanna et al., 2002; Chin et al., 2004; Aminova et al., 2008; Langley et al., 2008; Siddiq et al., 2009). Indeed, the list of compounds that are effective in inhibiting both cancer cell growth and protecting neurons is growing and includes histone deacetylase inhibitors (Sleiman et al., 2009), mammalian target of rapamycin inhibitors (Ravikumar et al., 2004; Shor et al., 2009), and transglutaminase inhibitors (Caccamo et al., 2010; McConoughey et al., 2010).
In agreement with our results, previous studies reveal that MTM extends the survival of the R6/2 HD mice by normalizing the expression of a subset the genes dysregulated in the presence of mHtt (Stack et al., 2007), and careful analysis of this list reveals that all of these genes are involved in carcinogenesis. Indeed, an increasing body of evidence from cell culture studies and postmortem tissue from human disease hint at a mechanistic connection between oncogenic pathways and neurodegeneration/acute insults to the CNS. For example, ectopic expression of oncogenes such as simian virus 40 T antigen in Purkinje neurons induces their degeneration (Feddersen et al., 1992). Moreover, expression of cell cycle markers is induced in postmitotic neurons in response to spinal cord injury (Di Giovanni et al., 2003), in striatal neurons in response to excitotoxic injury (Liang et al., 2005), and in neurodegenerative diseases such as Alzheimer's disease (AD) (Herrup et al., 2004; Yang and Herrup, 2007), and inhibition of cyclin-dependent kinases blocks this neuronal cell death (Park et al., 1997, 1998; Giardina and Beart, 2002; Rideout et al., 2003). Additionally, dysregulation of multiple genes that mediate postmitotic degeneration is also observed in carcinogenesis (Staropoli, 2008). For example, mutations in the tumor suppressor Park2 lead to cancer and Parkinson's disease (Mehdi et al., 2010; Poulogiannis et al., 2010; Tay et al., 2010; Veeriah et al., 2010). Loss-of-function mutations in ataxia telangiectasia gene, ATM, lead to degeneration of the nigrostriatal neurons (Eilam et al., 1998, 2003) and increase the risk of breast cancer and leukemia (Morrell et al., 1986; Thompson et al., 2005a,b), whereas gain-of-function mutations in the amyloid precursor protein (APP) lead to rare forms of AD as well as leukemia (Baldus et al., 2004). In addition to APP, multiple oncogenic pathways known to be involved in the G1 transition, such as Cdc42/Rac (Zhu et al., 2000) and retinoblastoma p130 (Previll et al., 2007), contribute to AD.
Our findings suggest that MTM functions at the crossroad between transformation and neurodegeneration, fit the notion that transformation in cancer and neurodegeneration share pathways, and are in agreement with the proposed notion that, in postmitotic neurons, clonal expansion has been supplanted by clonal deletion to avoid tumors from developing all over the CNS during aging (Heintz, 1993). In conclusion, it would be of interest to determine whether aging or disease creates mutation hotspots in oncogenes that would drive the neurons toward death.
Footnotes
This work was funded by a Dr. Miriam and Sheldon G. Adelson Medical Research Foundation grant (R.R.R., D.H.G., G.C.), New York State Department of Health Grant C0199771 (R.R.R.), and The Burke Foundation. We thank Drs. David Levens and Hye-Jung Chung for the Myc–Luc constructs and Dr. David Levens and Vicki Brandt for critical review of this manuscript.
References
- Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I, Nur-e-Alam M, Bergant J, Bertoni F, Carbone GM, Rohr J, Catapano CV. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucleic Acids Res. 2006;34:1721–1734. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminova LR, Siddiq A, Ratan RR. Antioxidants, HIF prolyl hydroxylase inhibitors or short interfering RNAs to BNIP3 or PUMA, can prevent prodeath effects of the transcriptional activator, HIF-1alpha, in a mouse hippocampal neuronal line. Antioxid Redox Signal. 2008;10:1989–1998. doi: 10.1089/ars.2008.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldus CD, Liyanarachchi S, Mrózek K, Auer H, Tanner SM, Guimond M, Ruppert AS, Mohamed N, Davuluri RV, Caligiuri MA, Bloomfield CD, de la Chapelle A. Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: amplification discloses overexpression of APP, ETS2, and ERG genes. Proc Natl Acad Sci U S A. 2004;101:3915–3920. doi: 10.1073/pnas.0400272101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo D, Currò M, Ientile R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin Ther Targets. 2010;14:989–1003. doi: 10.1517/14728222.2010.510134. [DOI] [PubMed] [Google Scholar]
- Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–6375. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol. 2001;49:345–354. [PubMed] [Google Scholar]
- Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D'Mello SR. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Liu J, Dundr M, Nie Z, Sanford S, Levens D. FBPs are calibrated molecular tools to adjust gene expression. Mol Cell Biol. 2006;26:6584–6597. doi: 10.1128/MCB.00754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, Yan W, Galic Z, Arevalo J, Zack JA. Expression-based monitoring of transcription factor activity: the TELiS database. Bioinformatics. 2005;21:803–810. doi: 10.1093/bioinformatics/bti038. [DOI] [PubMed] [Google Scholar]
- Curreri AR, Ansfield FJ. Mithramycin-human toxicology and preliminary therapeutic investigation. Cancer Chemother Rep. 1960;8:18–22. [PubMed] [Google Scholar]
- Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP, Faden AI. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol. 2003;53:454–468. doi: 10.1002/ana.10472. [DOI] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Elson A, Rotman G, Shiloh Y, Groner Y, Segal M. Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc Natl Acad Sci U S A. 1998;95:12653–12656. doi: 10.1073/pnas.95.21.12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Groner Y, Segal M. Late degeneration of nigro-striatal neurons in ATM−/− mice. Neuroscience. 2003;121:83–98. doi: 10.1016/s0306-4522(03)00322-1. [DOI] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Ryu H, Kubilus JK, D'Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington's disease. J Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Giardina SF, Beart PM. Kainate receptor-mediated apoptosis in primary cultures of cerebellar granule cells is attenuated by mitogen-activated protein and cyclin-dependent kinase inhibitors. Br J Pharmacol. 2002;135:1733–1742. doi: 10.1038/sj.bjp.0704636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorello L, Clerico L, Pescarolo MP, Vikhanskaya F, Salmona M, Colella G, Bruno S, Mancuso T, Bagnasco L, Russo P, Parodi S. Inhibition of cancer cell growth and c-Myc transcriptional activity by a c-Myc helix 1-type peptide fused to an internalization sequence. Cancer Res. 1998;58:3654–3659. [PubMed] [Google Scholar]
- Hagiwara H, Iyo M, Hashimoto K. Mithramycin protects against dopaminergic neurotoxicity in the mouse brain after administration of methamphetamine. Brain Res. 2009;1301:189–196. doi: 10.1016/j.brainres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Hann SR, Dixit M, Sears RC, Sealy L. The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 1994;8:2441–2452. doi: 10.1101/gad.8.20.2441. [DOI] [PubMed] [Google Scholar]
- Heintz N. Cell death and the cell cycle: a relationship between transformation and neurodegeneration? Trends Biochem Sci. 1993;18:157–159. doi: 10.1016/0968-0004(93)90103-t. [DOI] [PubMed] [Google Scholar]
- Herrup K. The involvement of cell cycle events in the pathogenesis of Alzheimer's disease. Alzheimers Res Ther. 2010;2:13. doi: 10.1186/alzrt37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Venojarvi M, Roy S, Sen CK. Glutamate-induced c-Src activation in neuronal cells. Methods Enzymol. 2002;352:191–198. doi: 10.1016/s0076-6879(02)52019-x. [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK. Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem. 2006;98:1474–1486. doi: 10.1111/j.1471-4159.2006.04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, Richardson SL, Perry G, Felsher DW, Petersen RB, Smith MA. The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol. 2009;174:891–897. doi: 10.2353/ajpath.2009.080583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P, Kegel KB, Aronin N, Difiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington's disease. J Neurosci. 2010;30:4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang ZQ, Wang XX, Wang Y, Chuang DM, DiFiglia M, Chase TN, Qin ZH. Susceptibility of striatal neurons to excitotoxic injury correlates with basal levels of Bcl-2 and the induction of P53 and c-Myc immunoreactivity. Neurobiol Dis. 2005;20:562–573. doi: 10.1016/j.nbd.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001;32:425–438. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- McConoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, Mahishi L, Cooper AJ, Antonyak MA, Cerione RA, Li B, Starkov A, Chaturvedi RK, Beal MF, Coppola G, Geschwind DH, Ryu H, Xia L, Iismaa SE, Pallos J, Pasternack R, Hils M, Fan J, Raymond LA, Marsh JL, Thompson LM, Ratan RR. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2010;2:349–370. doi: 10.1002/emmm.201000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, Smith B, Chen SR, Klebanov L, Salzman P, Yakovlev A, Land H. Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature. 2008;453:1112–1116. doi: 10.1038/nature06973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdi SJ, Alam MS, Batra S, Rizvi MM. Allelic loss of 6q25–27, the PARKIN tumor suppressor gene locus, in cervical carcinoma. Med Oncol. 2010 doi: 10.1007/s12032-010-9633-x. [DOI] [PubMed] [Google Scholar]
- Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986;77:89–92. [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Geschwind DH. Deconstructing language by comparative gene expression: from neurobiology to microarray. Genes Brain Behav [5 Suppl] 2006;1:54–63. doi: 10.1111/j.1601-183X.2006.00195.x. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I. Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J Neurosci. 2007;27:2969–2978. doi: 10.1523/JNEUROSCI.0186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previdi S, Malek A, Albertini V, Riva C, Capella C, Broggini M, Carbone GM, Rohr J, Catapano CV. Inhibition of Sp1-dependent transcription and antitumor activity of the new aureolic acid analogues mithramycin SDK and SK in human ovarian cancer xenografts. Gynecol Oncol. 2010;118:182–188. doi: 10.1016/j.ygyno.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previll LA, Crosby ME, Castellani RJ, Bowser R, Perry G, Smith MA, Zhu X. Increased expression of p130 in Alzheimer disease. Neurochem Res. 2007;32:639–644. doi: 10.1007/s11064-006-9146-3. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Norflus F, Singh B, Swindell MK, Buzescu R, Bejarano M, Chopra R, Zucker B, Benn CL, DiRocco DP, Cha JH, Ferrante RJ, Hersch SM. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem. 2006;281:16672–16680. doi: 10.1074/jbc.M511648200. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Wang Q, Park DS, Stefanis L. Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J Neurosci. 2003;23:1237–1245. doi: 10.1523/JNEUROSCI.23-04-01237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci U S A. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem. 2000;275:13049–13055. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–3837. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the “HAT” back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- Snyder RC, Ray R, Blume S, Miller DM. Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry. 1991;30:4290–4297. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- Stack EC, Del Signore SJ, Luthi-Carter R, Soh BY, Goldstein DR, Matson S, Goodrich S, Markey AL, Cormier K, Hagerty SW, Smith K, Ryu H, Ferrante RJ. Modulation of nucleosome dynamics in Huntington's disease. Hum Mol Genet. 2007;16:1164–1175. doi: 10.1093/hmg/ddm064. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Staropoli JF. Tumorigenesis and neurodegeneration: two sides of the same coin? Bioessays. 2008;30:719–727. doi: 10.1002/bies.20784. [DOI] [PubMed] [Google Scholar]
- Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX, Yip DL, Sung JX, Tan PH, Bay BH, Wong SH, Tang C, Tan JM, Lim KL. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem. 2010;285:29231–29238. doi: 10.1074/jbc.M110.108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005a;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- Thompson D, Antoniou AC, Jenkins M, Marsh A, Chen X, Wayne T, Tesoriero A, Milne R, Spurdle A, Thorstenson Y, Southey M, Giles GG, Khanna KK, Sambrook J, Oefner P, Goldgar D, Hopper JL, Easton D, Chenevix-Trench G. Two ATM variants and breast cancer risk. Hum Mutat. 2005b;25:594–595. doi: 10.1002/humu.9344. [DOI] [PubMed] [Google Scholar]
- Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- Van Dyke MW, Dervan PB. Chromomycin, mithramycin, and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II) Biochemistry. 1983;22:2373–2377. doi: 10.1021/bi00279a011. [DOI] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA. Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2010;42:77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC. Identification of potential therapeutic drugs for huntington's disease using Caenorhabditis elegans. PLoS One. 2007;2:e504. doi: 10.1371/journal.pone.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Hasegawa T, Isobe K. Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J Cell Biochem. 1999;73:291–302. [PubMed] [Google Scholar]
- Yang Y, Herrup K. Cell division in the CNS: protective response or lethal event in post-mitotic neurons? Biochim Biophys Acta. 2007;1772:457–466. doi: 10.1016/j.bbadis.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo YG, Hayashi M, Christensen J, Huang LE. An essential role of the HIF-1alpha-c-Myc axis in malignant progression. Ann N Y Acad Sci. 2009;1177:198–204. doi: 10.1111/j.1749-6632.2009.05043.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Boux H, Simmons ZL, Takeda A, Smith MA. Activation of oncogenic pathways in degenerating neurons in Alzheimer disease. Int J Dev Neurosci. 2000;18:433–437. doi: 10.1016/s0736-5748(00)00010-1. [DOI] [PubMed] [Google Scholar]