

Abstract
The Sec machinery constitutes the major pathway for protein translocation in bacteria. SecA is thought to act as a molecular motor driving preprotein translocation across the membrane by repeated ATP-driven cycles of insertion and retraction at the translocon channel. SecA is predominately a dimer under physiological conditions; however, its oligomeric state during active protein translocation is still unresolved. Five SecA crystal structures have been determined, each displaying a different dimer interface, suggesting that SecA may adopt different dimer configurations. In this study, a Förster resonance energy transfer (FRET) approach was utilized with nine functional monocysteine SecA mutants labeled with appropriate dyes to determine the predominant solution state dimer. Three different dye pairs allowed interprotomer distances ranging from 20-140 Å to be investigated. Comparison of 15 experimentally determined distances with those predicted from X-ray structures showed the greatest agreement with the B. subtilis SecA anti-parallel dimer structure (Hunt, J., Weinkauf, S., Henry, L., Fak, J.J., McNicholas, P., Oliver, D.B., and Deisenhfer, J. (2002) Science 297, 2018-2026). The binding of two signal peptides to SecA was also examined to determine their effect on SecA dimer structure. We found that the SecA dimer is maintained upon peptide binding; however, the preprotein cross-linking domain (PPXD) and helical wing domain (HWD) regions experience significant conformational changes, and the PPXD movement is greatly enhanced by binding of an extended signal peptide containing an additional 19 residues. Modeling of an ‘open’ antiparallel dimer structure suggests that binding of preprotein to SecA induces an activated open conformation suitable for binding to SecYEG.
Nearly one third of the proteins synthesized in the cytoplasm of bacteria must either insert into or cross the plasma membrane in order to reach their functional location within the cell. In Escherichia coli, the General Secretion pathway (Sec pathway) is the major pathway for secretory protein translocation (reviewed in du Plessis et al.1). The Sec machinery consists of two major components, a heterotrimeric translocon complex, composed of SecY, SecE, and SecG, that is located within the plasma membrane, and a large soluble ATPase protein, SecA, that binds preproteins and associates with SecYEG. At this latter location, SecA utilizes ATP-dependent conformational changes to drive transport of preproteins through the protein-conducting channel of SecYEG. The oligomeric form of both SecA and SecYEG in their functional state has remained a matter of considerable controversy during the past decade, as has the precise structural form of the different dimer states observed in vitro (for a recent review, see Rusch et al.2).
E. coli SecA protein was originally described as a homodimer in solution that is subject to a concentration-dependent monomer-dimer equilibrium which can be modulated by both temperature and salt concentration 3-5. This observation was followed up by numerous reports that described how key SecA ligands such as nucleotides, signal peptides, phospholipids, and SecYEG, all affect its dynamic monomer-dimer equilibrium (for a recent review, see 6). Such findings have been used to argue that SecA functions as either a monomer or a dimer or in some type of monomer-dimer cycle during the normal protein translocation cycle. Deficiencies with the current equivocal literature fall along several main lines: (i) certain studies utilized non-equilibrium techniques that can distort the normal monomer-dimer equilibrium, such as protein crosslinking, to assess SecA monomer-dimer status 7, 8, (ii) other studies utilized monomer-biased SecA or preactivated SecY (prlA4) mutants, duplicated and linked gene copies (e.g. SecAA or SecYY), or artificially crosslinked dimers that can give rise to artificial situations 8-15, (iii) the presence of detergents or high salt in biochemical or structural studies of the SecA-SecYEG complex would induce SecA monomerization 16, 17, (iv) certain biochemical assays were not sensitive enough to detect residual SecA dimer, while other assays utilized potentially non-physiological levels of ligands such as signal peptides that could have resulted in nonspecific effects 7, 18, 19. Clearly the use of additional in vivo or equilibrium in vitro methodologies is needed in order to resolve this complex but critical question of SecA oligomer function. In that regard, recent fluorescence burst analyses, an equilibrium technique, indicated that SecA bound to SecYEG as a dimer 20.
Not only has the oligomeric functional state of SecA been a matter of dispute, but also the precise structural form of the SecA dimer has remained unclear given the different SecA dimer crystal forms that have been observed recently. SecA from four bacterial species has been crystallized as both monomers and dimers 21-26. While all of the SecA crystal structures displayed a similar protomer building block, strikingly different dimer interfaces were observed (Figure 1). The initial B. subtilis SecA crystal structure (1M6N) predicted an anti-parallel dimer orientation with extensive interactions between the various N-terminal and C-terminal domains of SecA on opposing protomers, which was supported experimentally by a study employing FRET 21, 27. However since then, four additional SecA dimer structures have been published, all with different dimer interfaces. The M. tuberculosis SecA dimer structure (1NL3) also has the two protomers arranged in an antiparallel fashion, but it contains the smallest dimer interface formed by symmetrical interactions between two α-helices at the junction of NBF21 and HSD of one protomer with the tip of the two-helix finger sub-domain and a β-strand of PPXD of the other protomer 23. An alternative antiparallel B. subtilis SecA dimer structure (2IBM) has the two protomers arranged in an orthogonal fashion utilizing a dimer interface that is located at the prominent groove formed by the junction of NBF2 and PPXD 25. The E. coli SecA dimer structure (2FSF) also has the two protomers arranged in an antiparallel fashion, but the dimer interface consists solely of contacts between NBF1 and NBF2 of the respective protomers 22. Finally, the T. thermophilus SecA dimer structure (2IPC) is the only one presently that has the two protomers arranged in a parallel fashion in which an α-helix from NBF2 and the most N-terminal portion of HSD of one protomer insert into the NBF1-NBF2 interface of the other protomer to form a hinge-like structure that gives the dimer an overall open scissors-like configuration along its long axis 24. Whether any of these SecA dimers is physiologically relevant or is solely the result of its crystallization conditions remains presently unclear. In that regard, a recent cryo-electron microscopy study supported an antiparallel orientation for the E. coli SecA dimer, but it found a more extensive dimer interface that was incompatible with the E. coli SecA crystal structure 28.
Figure 1.

SecA dimer crystal structures with monocysteine residues colored. The color assignments for the residues are as follows: 59-dark green, 340-yellow, 402-blue, 427-hot pink, 458-brown, 470-purple, 506-red, 696-green, and 734-orange. E. coli amino acid residues are given and homologous residues in other species are depicted based on sequence alignment. For each dimer, the individual protomers are shown in grey or cyan. (A) Hunt et al. B. subtilis structure (PDB ID: 1M6N) 21, (B) Zimmer et al. B. subtilis structure (PDB ID: 2IBM) 25, (C) Vassylyev et al. T. thermophilus structure (PDB ID: 2IPC) 24, (D) Papanikolau et al. E. coli structure (PDB ID: 2FSF)22, and (E) Sharma et al. M. tuberculosis structure (PDB ID: 1NL3) 23.
The existence of additional SecA dimer structures has led to problems in interpreting the relevant literature. For example, the small data set obtained in the FRET study of Ding et al. 27 that was originally utilized to support the 1M6N dimer orientation is now inadequate given its potential compatibility with other SecA dimers. Likewise, the crosslinking study of Jilaveanu et al. 13 was consistent with the 1M6N dimer, but it is compatible with the 2IPC dimer as well 10. Finally, the behavior of the monomer-biased SecAΔ11 mutant that is missing N-terminal residues 2-11 was consistent with the 1M6N dimer, since this region is an important subunit contact site for this structure 8, 11; however this region also plays a similar role for the 2IPC dimer 24. Given these ambiguities, additional studies are now needed in order to clearly distinguish between the different SecA dimer forms and determine which one(s) is physiologically relevant for SecA in solution or bound to its different ligands.
Considerable information on the structure and function of SecA has been gained through the use of fluorescence techniques. Previous fluorescence studies of SecA have utilized intrinsic tryptophan fluorescence along with collisional quenchers to study SecA conformational changes mediated by temperature or binding of signal peptides, nucleotides, model membranes, or SecYEG-containing proteoliposomes 18, 29-31. FRET approaches have been used successfully to study conformational changes between PPXD and HSD of SecA that are induced by the binding of nucleotide or model membranes, as well as to assess the dimeric form of SecA in solution and to study its stability in the presence of phospholipids, signal peptides, and nucleotides 7, 27, 32. More recently, FRET has been used to map the signal peptide-binding site on SecA to the PPXD-NBF1-HSD multi-domain interface 33, which was found to be consistent with earlier biochemical and NMR studies 34-36. Collectively, these studies demonstrate the utility of fluorescence and particularly the FRET methodology for detailed elucidation of SecA structure-function relationships under biochemically relevant and more physiological conditions.
In order to begin to resolve the dominant structural form of the E. coli SecA dimer in its ground solution state, we undertook a FRET approach in the present study. Nine different positions on SecA were chosen for dye labeling in order to distinguish between the previously observed SecA dimers, and three different dye pairs with widely different R0 values were utilized to make accurate interprotomer measurements from 20-120 Å. Our data strongly support the existence of a 1M6N-like dimer as the dominant form of SecA protein in solution 21. Furthermore, we have also investigated structural changes of the SecA dimer upon signal peptide binding. Our data indicate that SecA dimer structure is maintained upon signal peptide binding, but the PPXD and HWD regions experience significant conformational changes during this interaction. Intriguingly, movement of the PPXD is greatly enhanced by the use of an elongated signal peptide containing an additional 19 residues from the early mature region of the preprotein.
Experimental Procedures
Materials
SP-Sepharose, spectroscopic grade Tris-HCl, phenylmethanesulfonyl fluoride, isopropyl-β-D thiogalactopyranoside, and most other reagent quality chemicals were purchased from Sigma. The fluorescent probes IAEDANS (IAE), IANBD-Ester (IAN), Alexa Fluor 488 (AF488), Alexa Fluor 568 (AF568), and Alexa Fluor 647 (AF647) in the maleimide form were obtained from Invitrogen (Eugene, OR). The E. coli alkaline phosphatase signal peptides, SP2, MCKQSTIALALLPLLFTPVTKA-NH2, SP22, MKQSTIALALLPLLFTPVTKAC-NH2, and SP41, MCKQSTIALALLPLLYTPVTKARTPEMPVLENRAAQGDITA-NH2, were synthesized by Biomolecules Midwest Inc. (Waterloo, IL). The cysteine residue incorporated into the peptides at position 2 or 22 was used for IAN-labeling, while the carboxyl-termini of the peptides was capped with an amide to prevent an unnatural negative charge 37. Peptides were purified by reverse-phase HPLC on a C18 or C4 column, and their identity was verified with Electrospray Ionization Mass Spectrometry at the Keck Biotechnology Resource Laboratory at Yale University.
Monocysteine SecA Mutant Protein Expression and Purification
Escherichia coli BL21.19 (secA13(Am) supF(Ts) trp(Am) zch∷Tn10 recA∷CAT clpA∷KAN) is derived from BL21(λDE3) 38 and was used as the host for all secA-containing plasmids. Plasmid pT7secA-Cys-0, a derivative of pT7secA2 that has all four cysteine codons within secA changed to serine, has been described previously 39, and it was used to create the monocysteine secA mutants described in this study. Mutants were generated with a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and appropriate oligonucleotides (Integrated DNA Technologies) as described by the manufacturer. All secA mutants were verified by DNA sequence analysis (DNA sequence facility, University of Pennsylvania). The plasmids were transformed into BL21.19 cells and checked for secA complementation by comparing their plating efficiency at 42°C and 30°C as described previously 40. SecA proteins were overproduced and purified as previously described 33. Protein concentration was determined using the Bradford assay (BioRad) with bovine serum albumin as the standard.
Dye Labeling
Purified preparations of monocysteine SecA protein were divided into equal portions for labeling with either the donor or the acceptor dye. Each monocysteine mutant was labeled with the selected dye at a protein:dye ratio of 1:20 for 4 hours at room temperature in a 25 mM Tris-HCl (pH 7.5), 25 mM KCl, 1 mM EDTA (TKE) buffer. Free dye was removed using a dye removal column (Pierce) and the protein was stored at −80°C. The signal peptides were labeled and purified as previously described 33. The lyophilized signal peptide was dissolved in dimethyl sulfoxide to a final concentration of 3 mM and stored at −80°C. Peptide concentration was confirmed by amino acid analysis at the Keck Biotechnology Resource Laboratory (Yale University), and the degree of labeling was calculated as described by the dye manufacturer 41.
SecA Mutant ATPase Activity
Labeled and unlabeled SecA monocysteine mutant ATPase activities were determined by the Malachite green method 42 utilizing the modifications described by Mitchell and Oliver 43. ATPase activity was calculated using the following formulas: endogenous ATPase activity = ATPase activity in the presence of SecA – ATPase activity in its absence; membrane ATPase activity = ATPase activity in the presence of SecA and inverted membrane vesicles – endogenous ATPase activity; translocation ATPase activity = ATPase activity in the presence of SecA, inverted membrane vesicles, and preprotein – membrane ATPase activity.
Analysis of SecA monomer-dimer population
The SecA monomer-dimer population was assessed by SEC and static light scattering at the Yale KECK facility as described previously 11. In brief, static light scattering data were collected using a Superose 6 10/30 HR size exclusion chromatography column (GE Healthcare) connected to a high-performance liquid chromatography system (Alliance 2965;Waters Corp.) equipped with an autosampler. The eluate from the size exclusion chromatography column was monitored by using a photodiode array UV/Vis detector (996 PDA; Waters Corp.), a differential refractometer (OPTI-Lab or OPTI-rEx; Wyatt Corp.), and a static, multiangle laser light scattering detector (DAWN-EOS; Wyatt Corp.). The system was equilibrated with the indicated buffer at a flow rate of 0.3 ml/min. The weight average molecular mass (MW) for SecA protein was determined for the indicated concentration ranges. Two software packages were used for data collection and analysis; the Millennium software (Waters Corp.) controlled the high-performance liquid chromatograph operation and data collection from the multiwavelength UV/Vis detector, while the ASTRA software (Wyatt Corp.) collected data from the refractive index detector and the light scattering detector and recorded the UV trace at 280 nm sent from the 996 PDA detector.
Fluorescence measurements
Fluorescence anisotropy and steady-state fluorescence spectra were recorded on a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon) with a programmable water bath (Neslab RTE model 7, Thermo Scientific). The samples were placed in a quartz cuvette (Starna Cell, Inc.) with a 3 mm path length. The spectral bandwidths of the excitation and emission slits were set at 4 nm and 6 nm, respectively, for the IAE-IAN dye pair and 1 nm for both slits for the Alexa Fluor dye pairs. Spectra were collected with an integration time of 0.5 s/data point and a resolution of 1 nm/data point. Final values result from at least 3 separate experiments of at least 3 scans for each sample.
Anisotropy experiments were conducted with samples containing 1 μM IAN-labeled alkaline phosphatase signal peptide in TKE buffer. SecA was added to the samples over a concentration range from 0 to 30 μM and samples were incubated for 30 min before data collection. Samples were excited at 465 nm and measured at 550 nm for SP22 and excited at 473 nm and measured at 536 nm for SP41. Data were fit based on a 1:1 binding interaction (peptide/SecA monomer) 33 with the following equation using Origin v. 6.0:
| (1) |
where SP is the total concentration of signal peptide, P is the total concentration of SecA protein, Kd is the equilibrium dissociation constant, y is the measured anisotropy of the signal peptide at each concentration of SecA, Ao is the anisotropy of the signal peptide in the absence of SecA and Ai is the anisotropy under saturating binding conditions.
For FRET measurements, SecA labeled with the donor dye (SecA-donor) (2 μM) and SecA labeled with the acceptor dye (SecA-acceptor) (2 μM) in TKE were incubated together for a total SecA concentration of 4 μM for 30 min at room temperature. Since the t1/2 for SecA monomer-dimer subunit exchange is ∼5 sec 7, 30 min was a sufficient amount of time to form equilibrated SecA dimers containing both donor and acceptor. Signal peptide-induced changes in the SecA dimer were investigated in the presence of 15 μM signal peptide to ensure at least 62.5% SecA-bound signal peptide for the weakest binding mutant. Higher concentrations of peptide could not be used because of aggregation. The polarizers were set at 0° for excitation and 55° for emission and samples were scanned at a rate of 1 nm/s and resolution of 1 nm/data point at 20°C. The IAE-IAN dye pair samples were excited at 336 nm and measured from 346 nm to 660 nm. The AF488-AF568 dye pair samples were excited at 492 nm and measured from 502 nm to 750 nm. The AF568-647 dye pair samples were excited at 568 nm and measured from 578 nm to 750 nm.
FRET Calculation
All spectra were corrected for background and buffer contributions. Donor or acceptor only spectra were collected in the presence of the unlabeled counterpart to correct for any changes in fluorescence intensity as a consequence of binding. Three samples were prepared for each mutant and dye pair examined by mixing equal parts of the same mutant in the following manner: (A) SecA-donor with unlabeled SecA (donor only), (B) SecA-acceptor with unlabeled SecA (acceptor only), and (C) SecA-donor with SecA-acceptor (FRET). A population distribution of 1:2:1 was expected for all samples where, e.g. in the FRET sample (Sample C above), the distribution would be 1:2:1 of donor-donor: donor-acceptor: acceptor-acceptor. Thus, the concentration distribution of SecA-donor in the donor only and FRET samples is expected to be equivalent, and similarly for SecA-acceptor in the acceptor only and FRET samples. Spectra of donor only and acceptor only samples were used to correct the FRET sample scans for any changes in fluorescence intensity that did not result from energy transfer, specifically for peptide binding experiments. The FRET efficiency, E, was calculated from the quenching of the donor fluorescence intensity in the FRET sample relative to the donor only sample using the following equation44:
| (2) |
and FDA, FD, and FA are the fluorescence intensities of the FRET, donor only, and acceptor only samples respectively, and fA represents the efficiency of acceptor labeling. In this case FA represents the fluorescence intensity of the acceptor only sample excited at the donor excitation wavelength. This term is included to remove any contribution of the acceptor fluorescence intensity to the donor fluorescence intensity in the FRET sample and F′DA represents this corrected donor FRET intensity, which was used for the efficiency calculations. If needed, the efficiency of donor labeling was also taken into account. These corrections followed the methodologies as outlined by Clegg 45. Although the transfer efficiency was calculated from the decrease in donor emission, the observation of FRET was also confirmed by the appearance of enhanced acceptor emission (Figure 2). The R0, J(λ), QD values were calculated as previously reported 33, 44.
Figure 2.
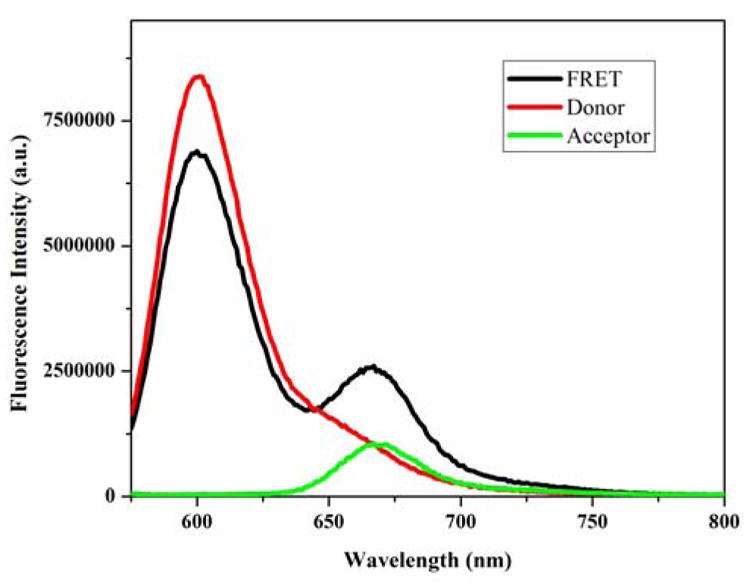
SecA donor quenching and acceptor enhancement. Fluorescence spectra of AF568-labeled SecA-506C with unlabeled SecA-506C (red), AF568-labeled SecA-506C with AF647-labeled SecA-506C (black), and AF647-labeled SecA-506C with unlabeled SecA-506C (green). Observation of FRET is detected in the doubly-labeled sample as both a decrease in donor fluorescence and increase in acceptor fluorescence.
The quantum yield of the donors in the absence of acceptor was measured relative to known quantum yield standards as previously described 44, 46 . The quantum yield of IAE-labeled SecA was measured relative to quinine sulfate (Φ = 0.56), the quantum yield of AF488-labeled SecA was measured relative to fluorescein (Φ = 0.925) 47, and the quantum yield of AF568-labeled SecA was measured relative to cresyl violet (Φ = 0.54)48.
Maximum and minimum values for κ2 were calculated from the steady-state anisotropy values of donor only or acceptor only samples, using the formalism of Dale et al. 49 as modified by Ivanov and co-workers 50. From the κ2 values, the ΔRDA value was determined. When presenting the data, the larger error was used and reported. The and values with associated Rmin and Rmax for each labeled residue are given in the Supporting information, Table 3. In general the percent error arising from our estimation of κ2 was approximately -16% to +25% for all of the dye-labeled proteins.
IV. Results and Discussion
Selection and Biochemical Characterization of SecA Mutants
We analyzed the available SecA crystal structures and chose nine spatially separated SecA mutants with unique cysteine substitutions for dye labeling that would potentially allow us to experimentally distinguish among the existing SecA dimers utilizing FRET. The mutants chosen were part of a collection of 63 monocysteine SecA mutants that have been shown to be functional in vivo by genetic complementation analysis 40. To assess the integrity of the purified SecA mutant proteins, ATPase activities were measured. SecA has three different ATPase activities: a low basal (endogenous) ATPase activity, which is weakly stimulated upon binding to SecYEG in inverted membrane vesicles (membrane ATPase activity), or which is greatly stimulated when bound to both SecYEG and an export-competent preprotein (translocation ATPase activity) 51. We found that all of the unlabeled and dye-labeled SecA mutant proteins utilized here exhibited SecA ATPase activities consistent with functional protein (i.e. none were inactive for ATPase activity), although such activities were affected by the cysteine substitution or dye attachment in certain cases (Supporting Information: Table 1 and Figure 1). Since previous studies have shown that SecA ATPase activity can be substantially affected by mutations in the NBF1 and NBF2 motor domains as well as the PPXD, HSD, and HWD regions that allosterically regulate the SecA DEAD motor while still resulting in functional mutant protein in vivo and in vitro (e.g. see for example Kourtz et al.52 and Sianidis et al.53) such perturbation of SecA ATPase activity was neither unanticipated nor disqualifying. However in the FRET study below, we strove to create a redundant dataset that is not dependent on the results obtained from any one mutant or dye pair combination. In addition, we have explicitly explored the labeling effects on SecA dimer stability of two representative mutants, and these results are given below.
Energy Transfer Efficiencies and Distances
We determined the energy transfer efficiencies of our various dye-labeled SecA mutant donor-acceptor pairs based on donor fluorescence quenching in the presence of the acceptor molecule according to the method described by Lakowicz 44 (Figure 2). R0 values or the distances at which energy transfer is 50% efficient were calculated for all donor-acceptor pairs from spectral data obtained for each labeled SecA mutant. Since the calculated distances have a steep sixth-order dependence on the measured efficiencies, we considered the 0.1 to 0.9 efficiency range to yield the most reliable distance measurements 44. The IAE-IAN pair had an average R0 value of 34 Å, yielding measurable distances from 21 to 49 Å. This value is in good agreement with previously reported R0 values for this pair 33. The average R0 values calculated for the AF488-AF568 and AF568-AF647 dye pairs were 57 and 76 Å, respectively, yielding measurable distance ranges from 35 Å to 82 Å and from 53 Å to 110 Å, respectively. Similar to the IAE-IAN pair, these values are in line with expected R0 values for these dyes, indicating that the local environment of the dyes did not noticeably affect their spectral properties (Supporting Information: Table 2).
There are different sources of error associated with the generation and analysis of FRET data. In particular, the mobility of the dyes and their associated linkers can influence the measurement. To ensure that the SecA-attached dyes were relatively mobile, we measured the fluorescence anisotropy of our dye-labeled SecA mutants (Supporting Information: Table 3), and we discarded from our study any mutant that exhibited an anisotropy value of 0.24 or higher, indicative of a relatively fixed orientation. For the remaining dye pairs, we used the steady state anisotropy values to estimate maximum and minimum values of the orientation factor, κ2, from which we could estimate errors for the calculated R values 49, 50 (Supporting Information: Table 3).
Verification of SecA dimer population
Our FRET samples contained a total SecA concentration of 4 μM to ensure that the majority of SecA would be dimeric (>98% dimer based on the previously reported monomer-dimer association constant 5, 54). We also performed SEC and static light scattering measurements under our buffer conditions and SecA concentration range to verify the dimeric state. The measured molecular weight of the cysteine-less SecA mutant, SecA-0C, remained relatively constant at 200 kD from 0.1 to 3.5 μM, suggestive of a near homogenous dimer population (Supporting information: Figure 2). The FRET signal obtained with the dye-labeled SecA-340C mutant was examined over a concentration range from 0.1 to 10 μM protein (Figure 3A). This mutant was chosen because of its strong transfer efficiency observed with the AF488–AF568 dye pair and consistent distance measurements (Table 1). Similar to the results from SEC and light scattering, the data suggest that there is no change in monomer-dimer equilibrium over the 0.1-10 μM SecA protein concentration range. The relatively constant and high FRET efficiency observed correlates well with a predicted dimer population of 93-99% based on a monomer-dimer dissociation constant of 1 nM.
Figure 3.
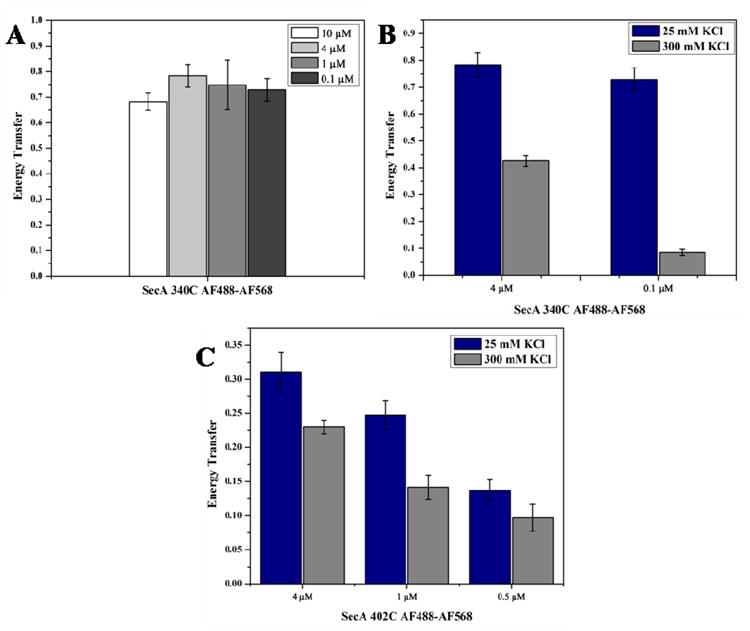
Effect of protein concentration and salt on SecA dimer state as assessed by energy transfer efficiency. (A) Effect of protein concentration on energy transfer efficiency of AF488-AF568-labeled SecA-340C. (B) Effect of salt and protein concentration on energy transfer efficiency of AF488-AF568-labeled SecA-340C. (C) Effect of salt and protein concentration on energy transfer efficiency of AF488-AF568-labeled SecA-402C.
Table 1. FRET distances compared with distances predicted from X-ray Crystal Structures.
| FRET pair | SecA Domain | EFRETa | Distance (Å)b | Distance predicted from Crystal Structure (Å) | ||||
|---|---|---|---|---|---|---|---|---|
| 1M6Nc | 2IBMd | 2IPCe | 2FSFf | 1NL3g | ||||
| 59IAE-59IAN 59AF568-59AF647 |
NBF1 | 0.38 ± 0.09 ∼1 |
33 ± 10 ≤ 44 (0.5 R0) |
20 | 91 | 83 | 93 | 106 |
| 402IAE-402IAN 402AF488-402AF568 |
NBF1 | 0.19 ± 0.10 0.31 ± 0.03 |
43 ± 25 54 ± 14 |
16 | 43 | 38 | 68 | 55 |
| 59IAE-402IAN | NBF1-NBF1 | 0.40 ± 0.16 | 33 ± 15 | 30 | 66 | 61 | 81 | 81 |
| 340IAE-340IAN 340AF488-340AF568 |
PPXD | 0.12 ± 0.08 0.78 ± 0.08 |
≥32 (1.4 R0) 45 ± 11 |
50 | 50 | 30 | 21 | 69 |
| 427IAE-427IAN 427AF488-427AF568 427AF568-427AF647 |
NBF2 | 0.10 ±.09 0.19 ± 0.02 0.25 ± 0.02 |
≥48 (1.4 R0) 65 ± 17 68 ± 17 |
71 | 35 | 19 | 64 | 44 |
| 458AF488-458AF568 | NBF2 | 0.43 ± 0.05 | 62 ± 15 | 63 | 11 | 49 | 27 | 36 |
| 470IAE-470IAN 470AF488-470AF568 470AF568-470AF647 |
NBF2 | 0.08 ±0.01 0.24 ± 0.02 0.35 ± 0.03 |
≥49 (1.4 R0) 72 ± 16 87 ± 22 |
101 | 13 | 62 | 50 | 55 |
| 506AF488-506AF568 506AF568-506AF647 |
NBF2 | 0.30 ±0.03 0.70 ± 0.07 |
71 ± 16 67 ± 17 |
63 | 31 | 48 | 24 | 53 |
| 696IAE-696IAN 696AF488-696AF548 696AF568-696AF647 |
HWD | 0.08 ± 0.02 0.23 ± 0.02 0.59 ± 0.06 |
≥53 (1.4 R0) 76 ± 18 73 ± 18 |
89 | 78 | 25 | 128 | 120 |
| 734AF568-734AF647 | HWD | 0.43 ± 0.04 | 85 ± 21 | 91 | 94 | 51 | 140 | 127 |
FRET efficiency (EFRET) was measured by the quenching of the donor fluorescence intensity as described.
The donor-acceptor distance (R) was calculated as described.
Coordinates for the antiparallel dimeric B. subtilis SecA crystal structure (PDB ID: 1M6N) were generously provided by John Hunt.
Zimmer et al. B. subtilis structure (PDB ID: 2IBM).
Vassylyev et al. T. thermophilus structure (PDB ID: 2IPC).
Papanikolau et al. E. coli structure (PDB ID: 2FSF). This structure lacked resolution in the PPXD, predicted distance measurements were obtained by modeling the PPXD based on the B. subtilis structure. Coordinates generously provided by Anastasios Economou.
Sharma et al. M. tuberculosis structure (PDB ID: 1NL3).
To further probe whether SecA oligomeric state can be measured by FRET, we measured transfer efficiencies at 25 mM KCl and compared them with those obtained at 300 mM KCl (Figure 3): the latter condition shifts the SecA monomer-dimer equilibrium towards the monomer 5, 27, 54. Using SEC-static light scattering, we previously measured the wildtype SecA monomer-dimer association constant to be 2.2 μM at 300 mM KCl—1000-fold higher than that observed at 25 mM KCl 11. The sensitivity of FRET measurements to the dimer population is shown by efficiency measurements performed at two SecA concentrations in 300 mM KCl (Figure 3). At a total protein concentration of 4 μM SecA-340C in 300 mM KCl, 59% of SecA should be a dimer, predicting a theoretical energy transfer efficiency of 0.47, which is in good agreement with our measured value of 0.43 (Figure 3B). At 0.1 μM SecA-340C in 300 mM KCl, only 7.7% of SecA should be a dimer, giving rise to a theoretical energy transfer efficiency of 0.061, which again agrees well with our measured value of 0.085. The consistency between our experimental results and expected dimer populations demonstrates that FRET measurements can detect SecA dimer state.
We have exploited this sensitivity to oligomeric state to examine the effect of the dyes at a potential SecA dimer interface and specifically studied the behavior of the SecA-402C mutant that is predicted to have the dye located at the interface of the 1M6N dimer (Figure 1A). When the behavior was analyzed in this fashion, the changes in transfer efficiency with increased salt and reduced protein concentration were not as dramatic as for SecA-340C (Figure 3C). At 4 μM SecA-402C concentration, the interprotomer transfer efficiency changed only from 0.31 to 0.23 when going from 25 mM to 300 mM KCl, respectively. Importantly, at 0.5 μM SecA, the change in energy transfer efficiency was even less pronounced: 0.15 and 0.1 at 25 mM and 300 mM KCl, respectively. The relatively low transfer efficiencies observed at this reduced protein concentration suggest that this mutant is strongly biased towards the monomer even under the low salt condition. We attribute this alteration in the labeled SecA-402C monomer-dimer equilibrium to the potential interaction of the dyes at an interfacial location, which is predicted to perturb protomer association in the Hunt (1M6N) dimer (vide infra) (Figure 1).
Determination of Protomer Orientation
The dominant solution state SecA dimer configuration was determined by comparing experimentally-measured FRET distances with those predicted from the five existing SecA X-ray structures, the B. subtilis antiparallel dimer (1M6N) 21, the B. subtilis orthogonal dimer (2IBM) 25, the T. Thermophilius parallel dimer (2IPC) 24 the E. coli antiparallel dimer (2FSF) 22, and the M. tuberculosis antiparallel dimer (1NL3)23. Ten unique interprotomer distances were measured utilizing nine cysteine residues that were widely distributed throughout SecA and are contained within domains critical for nucleotide binding (NBF1, NBF2), signal peptide and preprotein binding (PPXD), or an essential but unknown SecA function(s) (HWD) (Table 1). Since the predicted interprotomer distances varied widely among the possible dimers, three different dye pairs, IAEDANS-IANBD (IAE-IAN), Alexa Fluor 488-Alexa Fluor 568 (AF488-AF568), and Alexa Fluor 568-Alexa Fluor 647 (AF568-AF647), were utilized to obtain measurements over the full range of predicted distances (16 Å to 140 Å). We also employed multiple dye pairs at certain locations to confirm distances calculated from transfer efficiencies measured at the limit of the workable range, which allowed us to more effectively rule out specific dimer structures.
Given logical sources of uncertainty in the experimental distances such as the flexibility of the dye tethers or the measurement of predicted distances utilizing Cα positions rather than the actual fluorophore, a tight correspondence between experimentally measured and predicted distances was not expected. Thus, we readily consider predicted distances within one standard deviation of the experimental distances to be in good agreement. Other sources of variation in our measurements arise from the inevitable structural differences between SecA in crystalline form versus solution state as well as the existence of conformational heterogeneity in the latter case. For example, an NMR study showed that SecA exists in both ‘open’ and ‘closed’ states that differ in the extent of PPXD-HWD separation 34, whereas by crystallography, these are distinct states with their own crystalline form 21, 26. Below, we discuss our FRET data organized by the domain locations of the attached dyes used for the interprotomer measurements.
NBF1 measurements
Three interprotomer distances were determined for residues in NBF1 utilizing three different dye pairs appropriate for measuring the predicted distances (Table 1). The FRET efficiency measured from residue 59 of one protomer to residue 59 of the other protomer, labeled with the IAE-IAN dye pair was 0.38, which yields a distance of 33 Å. A FRET efficiency of ∼1 was obtained with the AF568-AF647 dye pair at this position, which is too high for an accurate distance calculation; however, based on our experimentally determined R0 values for the AF568-AF647 dye pair, we can estimate that the distance is shorter than 44 Å. These data are most consistent with the distance predicted from the 1M6N structure (20 Å); however, the 1M6N predicted distance lies just outside the error range (3 Å). The FRET distances do not correspond well with the distances predicted from the four other crystal structures, which are all between 83-106 Å. These longer distances are well within the range of the AF568-AF647 dye pair (R0=76 Å), and if such orientations were present at a significant population density, they should have been detected.
Interprotomer distances for residue 402, also located in NBF1, were determined with both the IAE-IAN and AF488-AF568 dye pairs, and they resulted in weak (0.19) and moderate (0.31) FRET efficiencies, respectively, corresponding to distances of 43 Å and 54 Å (Table 1). The difference in these values probably results from the relatively low transfer efficiency and correspondingly high error in the IAE-IAN measurement, although interfering dye-protein interactions may also be a contributing factor in either case (see below). The two measured distances are within one standard deviation of those predicted from the 2FSF (68 Å), 2IBM (43 Å) and 1NL3 (55 Å) structures. Although the IAE-IAN measured distance (43 Å) is in good agreement with that predicted from the 2IPC structure (38 Å), this predicted distance is slightly outside of the range of the AF488-AF568 measured distance (54 Å), which was measured at higher efficiency and should be more accurate. Both FRET distances are too long for the distance predicted from the 1M6N structure (16 Å). However, our analysis of the dimeric state of the dye-labeled SecA-402C mutant (shown above in Figure 3C) suggested the existence of a significant population of SecA monomers under our experimental conditions, which would compromise this measurement. In particular, the significant monomer population would lead to an apparent longer distance since the FRET measurement would result from a weighted average of the true dimer distance and an infinitely long distance for the monomer population.
To gain more information about the relative orientation of this domain, we also determined an interprotomer distance between residue 59 and 402 of adjoining protomers using the IAE-IAN dye pair (Table 1). A moderate (0.40) FRET efficiency was obtained in this case, and the calculated distance (33 Å) corresponds closely to that predicted from the 1M6N structure (30 Å). The distances predicted from the other crystal structures are all significantly longer (>60 Å) and are outside the error range of the measured value.
PPXD measurements
The interprotomer distance for residue 340 was determined with the IAE-IAN and AF488-AF568 dye pairs, which yielded similar distances of ≥ 32 Å and 45 Å, respectively (Table 1). These distances are in relatively good agreement with that of 50 Å predicted from the 1M6N and 2IBM structures. Furthermore, the IAE-IAN dye pair with a specific R0 value of 22 Å for residue 340 should have readily detected the distances of 30 Å and 21 Å predicted from the 2IPC and 2FSF structures, respectively, if those protomer orientations constituted a large portion of our sample. Similarly, the 69 Å distance predicted from the 1NL3 structure is well within the distance range of the AF488-AF568 dye pair, but is outside the range of the FRET measurement.
NBF2 measurements
Four interprotomer distances were determined with one or more dye pairs for each set of mutants in this domain (Table 1). For residue 427 the interprotomer FRET efficiency obtained with the IAE-IAN dye pair was 0.10, which is too low for an accurate distance calculation, but suggests a distance longer than 48 Å. The AF488-AF568 and AF568-AF647 dye pairs at this position yielded weak FRET efficiencies of 0.19 and 0.25, respectively, corresponding to distances of 65 Å and 68 Å. Thus, all three FRET measurements are consistent with each other and indicate an interprotomer distance that is relatively long and in the 65-68 Å range. These experimental values are in good agreement with the predicted distances from the 1M6N (71 Å) and 2FSF (64 Å) structures, but their range does not include the shorter distances predicted from the 2IBM (35 Å), 2IPC (19 Å) and 1NL3 (44 Å) structures.
The other NBF2 residues yielded similarly long interprotomer distances (Table 1). For example, residue 458 labeled with the AF488-AF568 dye pair yielded a moderate (0.43) FRET efficiency corresponding to a distance of 62 Å. This is consistent with the distances predicted from the 1M6N (63 Å) and 2IPC (49 Å) structures, but it is significantly longer than those predicted from the 2IBM (11 Å), 2FSF (27 Å) and 1NL3 (36 Å) structures. The interprotomer distance measured between residues at position 470 labeled with the AF488-AF568 dye pair yielded a relatively weak (0.24) FRET efficiency corresponding to a distance of 72 Å, while the AF568-AF647 dye pair gave a more moderate (0.35) FRET efficiency and a distance of 87 Å. Similar to residue 427, the transfer efficiency measured with the IAE-IAN dye pair was too low to calculate an accurate distance but suggests that the distance is longer than 49 Å. Thus collectively, these dye pairs indicate the 470 interprotomer distance is approximately 80 Å and do not provide evidence for significant populations of either the 2IBM (13 Å) or 2FSF (50 Å) structures. The range of the AF488-AF568 FRET measured distance does include the predicted distances from the 2IPC (62 Å) and 1NL3 (55 Å) structures, but not the 1M6N (101 Å) structure. The higher efficiency AF568-AF647 measurement (0.35), which is considered more accurate, is not in good agreement with either the 2IPC or the 1NL3 structure; although the 2IPC distance lies just outside the experimental range (3 Å). For this dye pair, only the 1M6N predicted distance (101 Å) is within the range of the measurement (87 Å).
Additional probing of interprotomer distances in NBF2 was accomplished utilizing residue 506 labeled with AF488-AF568 or AF568-AF647, which yielded FRET efficiencies of 0.30 and 0.70, corresponding to interprotomer distances of 71 and 67 Å, respectively (Table 1). The two different dye pairs are in excellent agreement with each other, indicative of well-determined distances. This distance corresponds best to that predicted from the 1M6N structure (63 Å) and also agrees with 1NL3 (53 Å), but does not correspond to the shorter distances of 31 Å (2IBM), 48 Å (2IPC), and 24 Å (2FSF) predicted from the other structures.
HWD measurements
Two interprotomer distances were determined in the HWD using residues at positions 696 and 734. For residue 696, the two Alexa Fluor dye pairs yielded efficiencies from which distances could be calculated; whereas, the efficiency of the IAE-IAN pair was very low but indicates the distance is at least 53 Å or longer (Table 1). Although the AF488-AF568 dye pair at this site yielded a relatively weak (0.23) FRET efficiency, the calculated distance of 76 Å is in very good agreement with the more accurate distance of 73 Å measured with the higher FRET efficiency (0.59) of the AF568-AF647 dye pair. These distances correspond most closely with that predicted from the 2IBM structure (78 Å), although the range also includes the distance predicted from the 1M6N structure (89 Å). The experimental distances are not in good agreement with the distances predicted from the 2IPC (25 Å), 1NL3 (120 Å) and 2FSF (128 Å) structures.
Only the AF568-AF647 dye pair was used at residue 734, given the relatively long predicted distances in this case (Table 1). The moderate (0.43) FRET efficiency measured corresponds to a distance of 85 Å. Similar to residue 696, the measured distances correspond best to the 1M6N (91 Å) and 2IBM (94 Å) structures but not to those predicted from the 2IPC (51 Å), 1NL3 (127 Å) and 2FSF (140 Å) structures. If any of these latter three dimer structures were present to any great extent in our sample, much stronger or weaker FRET efficiency, respectively, would have been observed.
Correlation of distances with dimer structure
Collectively, the FRET measured distances are suggestive of a dimer structure in which the PPXD and NBF1 regions of one protomer are relatively proximal (≤ 50 Å) to the same region in the other protomer. In contrast, the other domains examined (NBF2 and HWD) appear to be situated further apart (≥ 60 Å). Since many of our individual FRET measurements were compatible with more than a single protomer orientation, to refine our structure determination we examined the degree of correspondence between all of the measured and predicted distances to evaluate which dimer structure is most consistent with all of the data. A total of 10 interprotomer distances were measured in four out of the five well-structured SecA domains, and accounting for the different dye pairs that gave usable data (i.e. based on reasonable FRET efficiencies), a total of 15 measurements were considered in this analysis. The correspondence between the experimental and predicted distances was evaluated through linear correlation plots (Figure 4). In these plots, if the FRET-determined distances were in exact agreement with those predicted by a given crystal structure, then the points should fall along the solid line shown. Points above the line indicate experimental distances that are longer than the predicted distance and points below the line indicate experimental distances that are shorter than the predicted distances.
Figure 4.

Correlation between FRET distances and predicted distances from dimer crystal structures. FRET distances are plotted versus those predicted from the indicated SecA X-ray crystal structures. Distances and errors are given in Table 1. The linear solid line depicts the perfect case of a 1:1 correspondence when the FRET distances exactly match the crystal structure distances. The degree of correspondence and goodness of fit are evaluated through parameters, R2 and χ2; all points are evaluated except where indicated. (A) Hunt et al. B. subtilis structure (PDB ID: 1M6N) 21. Distances measured between 402IAE-402IAN and 402AF488-402AF568 are shown with open squares, (B) Zimmer et al. B. subtilis structure (PDB ID: 2IBM) 25, (C) Vassylyev et al. T. thermophilus structure (PDB ID: 2IPC) 24, (D) Papanikolau et al. E. coli structure (PDB ID: 2FSF) 22, and (E) Sharma et al. M. tuberculosis structure (PDB ID: 1NL3) 23.
Quantitatively, the correspondence between the FRET and crystal structure distances was evaluated by the correlation coefficient (R2) and the χ2 parameter. In the case of the R2 parameter a value close to one is indicative of a good fit, while a low χ2 parameter indicates good agreement between observed and predicted values. By these measures only the 1M6N crystal structure distance yielded a reasonable fit to the experimental data with values of 0.78 for R2 and 4.28 for χ2 (Figure 4A). All the other crystal structures exhibited R2 values less than 0.1 and χ2 values greater than 15 (Figure 4B-E). These statistical parameters are consistent with the fact that for these other structures less than half of the predicted distances matched the experimental distances. Thus, our data quantitatively and visually yield the greatest linear correlation with those predicted from the 1M6N dimer structure. The statistical parameters are reflective of the fact that 12 of the FRET-measured distances were consistent with prediction (Figure 4A). In addition, at least 2 of these measured distances uniquely correspond to the 1M6N orientation: 59IAE-59IAN and 59IAE-402IAN (Table 1). This evaluation, then, strongly points to the identification of this protomer orientation as the dominant one in solution.
The distance measured at residue 470 with the AF488-AF568 dye pair was one of the ones that did not correspond with the 1M6N structure, but when measured with the greater transfer efficiency of the AF568-AF647 dye pair, the experimental range included the 1M6N predicted distance and none of the other structures (Table 1). Our data for residue 402 measured with either the IAE-IAN or AF488-AF568 dye pairs were also not consistent with the 1M6N structure (shown as open squares in Figure 4A). However, our analysis of monomer-dimer association (Figure 3C) suggested that this labeled mutant had a significant monomer population present that would lead to an apparent longer distance. An examination of all five of the SecA crystal structures indicates that the 1M6N structure is the only one where residue 402 is close enough to the dimer interface to potentially perturb dimer stability upon dye attachment at this site (blue residues in Figure 1). In fact, exclusion of these points from the fit significantly improves both the R2 (0.87) and χ2 (1.96) values for the 1M6N structure (Figure 4A).
Examination of the other dimer structures reveals that dye labeling at our selected sites could similarly perturb the dimer interface and result in apparent longer FRET distances; however, this concern is not supported by the data. For example, labeling of residue 340 in both the 2FSF and 2IPC structures (yellow residues: Figure 1) has the potential to disrupt these dimer interfaces. As the energy transfer efficiency of the SecA-340C mutant labeled with AF488-AF568 remains relatively constant from 4.0 to 0.1 μM SecA, labeling at this site does not appear to alter the monomer-dimer equilibrium (Figure 3). Dye attachment at residue 458 or 470 (brown and purple residues: Figure 1) might similarly disrupt the dimer interface in the 2IBM structure and have led to longer observed distances; however, FRET measurements performed with other NBF2 residues that are not proximal to the interface (Figure 1) do not agree with the 2IBM structure. Furthermore, the distances determined between NBF2 domains taken as a whole do not suggest an interfacial location, since they are all at least 60 Å or longer (Table 1). Although other labeled residues besides residue 402 may perturb the SecA dimer interface, the overall FRET data do not support these possibilities.
The prevalence of the 1M6N antiparallel protomer orientation in solution is further confirmed by previous results in which removal of N-terminal residues 2-11 of SecA destabilizes the dimer interface and shifts the equilibrium toward the monomer 10. In fact, this monomer-biased SecA mutant is poorly functional both in vivo and in vitro 8, although it can be reactivated by over expression that favors dimer formation 11. Only in the 1M6N and 2IPC structures do these N-terminal residues contribute to the interprotomer interaction 21, 24. However, our FRET measurements and linear correlation data do not support the presence of a substantial concentration of protomers arranged in a parallel fashion similar to the 2IPC structure (Table 1; Figure 4C).
The 1M6N and 2IBM dimers with the largest amounts of solvent accessible surface area buried at the interface (∼5400 Å2) 21, 25 are predicted to be the most energetically stable, followed in rank order by the 2FSF (3300 Å2), 2IPC (3200 Å2), and 1NL3 (1300 Å2) dimers 22-24. Thus, a consideration of all the FRET data, dimer stability and energetics of the five dimer structures leads us to conclude that, in solution, the two SecA protomers are primarily arranged in an antiparallel dimer similar to the 1M6N structure as proposed originally by Hunt et al. 21. In any one measurement we cannot exclude the existence of at most 10-15% population of an alternative dimer in solution, nor can our results speak to transient fluctuations in SecA protomer association that could be captured by disulfide crosslinking experiments given standard reaction time frames 13, 24, 25. Nevertheless, our FRET results in their totality convincingly point to a protomer arrangement analogous to the 1M6N structure, which gives us a working model for SecA dimer in solution. The actual SecA dimer is, of course, expected to differ in a number of ways from this model based on its conformational flexibility and dynamics in solution and in the presence of ligands, all of which merit further investigation.
Conformational changes in SecA upon signal peptide binding
Using our experimentally determined knowledge of SecA dimer structure, we elected to explore the effects of signal peptide binding on SecA conformation and dimer stability. For this purpose we utilized four dye pairs located in PPXD, NBF2, and HWD that were chosen because of their potential sensitivity to conformational changes associated with signal peptide binding, their robust energy transfer efficiencies, and their overall coverage of the protein. FRET measurements were performed with alkaline phosphatase signal peptides. The peptides, SP2 and SP22, consist of the natural 21 amino acid alkaline phophatase signal peptide from E. coli with cysteine residues inserted at positions 2 and 22, respectively, while SP41 is similar to SP2 but also contains 19 amino acid residues of the early mature region of E. coli alkaline phosphatase. This latter peptide allowed us to probe whether incorporation of this mature region would alter the conformation of signal peptide-bound SecA.
To ensure that FRET measurements were being performed under conditions where SecA was fully bound by signal peptide, binding affinities were determined using fluorescence anisotropy where wildtype or mutant SecA was titrated into solutions containing 1 μM of IAN-labeled SP2, SP22 or SP41. These peptides were labeled at cysteine residues located at positions 2, 22, and 2, respectively. Wildtype SecA displayed a higher affinity for SP22 than SP2 or SP41 (Figure 5), consistent with our previous finding that IAN labeling of SP2 at residue 2 reduces its binding activity nearly 10 fold (1.2 μM vs 10.7 μM for unlabeled vs IAN labeled SP2, respectively 33). The lower anisotropy value is attributed to the mobility of the N-terminus as detected in the NMR structure of bound peptide (34). The binding affinity of the SecA mutants for SP22 was found to be comparable to that of wildtype SecA (1.4 ± 0.2 μM) within error (Figure 6A, Table 2). The exception was the SecA-696C mutant, which exhibited a modestly lower binding affinity (2.5 μM) possibly because of the placement of the mutation in a region that interacts with the peptide. Binding affinities to SP41 for all four mutants were modestly weaker (approx. Kd = 6 μM) relative to SP22 and were comparable to that of wildtype SecA (Kd = 6.2 ± 0.8 μM) (Figure 6B, Table 2). This latter result was not surprising since SP41 like SP2 is labeled with IAN at residue 2, which we have previously shown reduces SecA signal peptide binding affinity 33. Based on these results, we performed our FRET study at 15 μM SP22 or SP41; at this concentration of peptide, 81% or 65% of SecA should be bound to SP22 or SP41, respectively.
Figure 5.
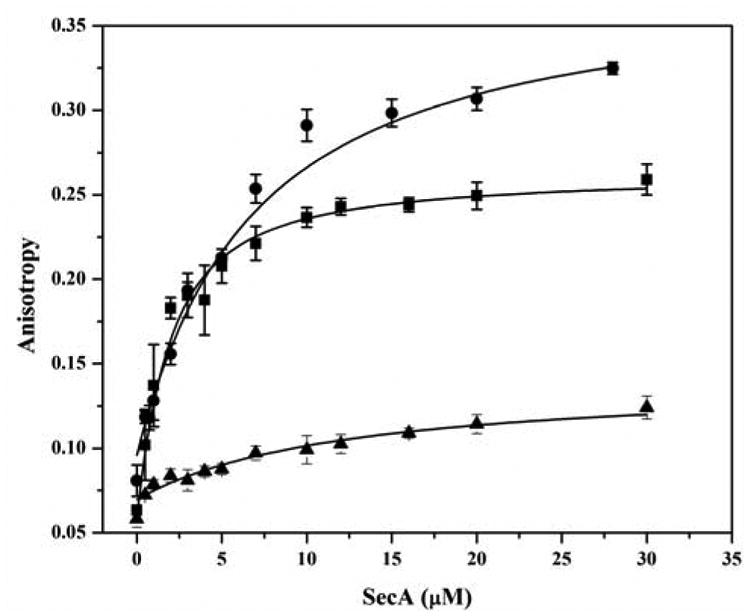
Measurement of signal peptide-binding affinity to SecA protein. Binding of IAN-labeled SP22 ■, SP41 ●, and SP2 ▲ signal peptides to wildtype SecA was determined by fluorescence anisotropy as described in “Experimental Procedures”.
Figure 6.
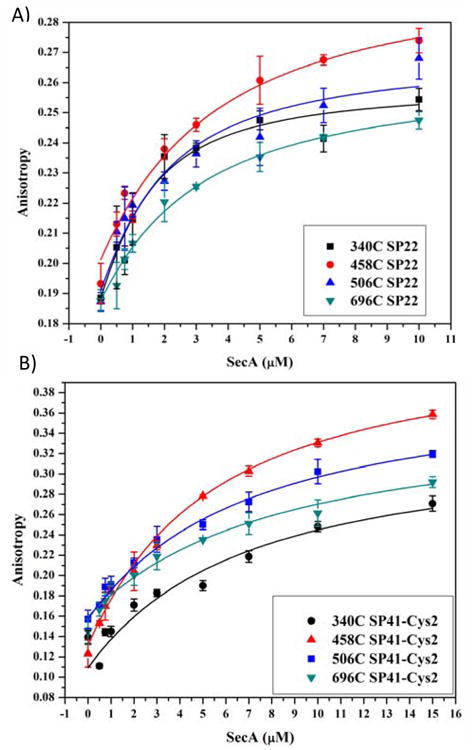
Measurement of signal peptide-binding affinity to SecA mutant proteins. Binding of IAN-labeled SP22 (A) or SP41 (B) to the indicated SecA mutant was determined by fluorescence anisotropy as described in “Experimental Procedures”.
Table 2. Signal Peptide Kd values and Energy Transfer efficiencies measured with signal peptides bound.
| FRET pair | With SP22a | With SP41b | ||
|---|---|---|---|---|
| EFRETc | Kd (μM) | EFRETa | Kd (μM) | |
| 340AF488-340AF568 | 0.64 ± 0.06 | 1.1 ± 0.4 | 0.23 ± 0.02 | 6.2 ± 4.3 |
| 458AF488-458AF568 | 0.45 ± 0.05 | 3.0 ± 1.7 | 0.45 ± 0.05 | 4.8 ± 0.6 |
| 506AF568-506AF647 | 0.67 ± 0.07 | 1.5 ± 0.7 | 0.69 ± 0.07 | 6.3 ± 1.0 |
| 696AF568-696AF647 | 0.42 ± 0.04 | 2.5 ± 0.2 | 0.41 ± 0.04 | 7.5 ± 1.3 |
A 22 residue peptide labeled with IAN at residue 22.
A 41 residue peptide labeled with IAN at position 2.
FRET efficiency (EFRET) was measured as described in Table 1.
A final consideration in setting up our SecA signal peptide binding study was the effect of signal peptide binding on SecA monomer-dimer status. Recently it was reported that signal peptide binding leads to a ∼10-fold increase in the SecA dimer dissociation constant 54. We note, however, that under the salt conditions of our study (25 mM KCl), the SecA monomer-dimer dissociation constant is extremely low (∼1 nM). Thus even with a 10-fold increase in this value, 96% of SecA is predicted to be a dimer under our conditions (4 μM SecA).
We found that the binding of the two signal peptides to SecA produced distinctly different results on SecA global conformation depending on the domain investigated and signal peptide utilized. For example, the binding of either SP22 or SP41 to the dye labeled SecA-458C or SecA506C mutant did not lead to any significant changes in energy transfer efficiency (Table 1 and Table 2). This suggests that the interprotomer distance between NBF2 regions did not change significantly upon peptide binding and that SecA remains as a dimer in its peptide-bound state. A considerably different result was obtained with the SecA-340C mutant, where the energy transfer efficiency decreased moderately upon binding SP22 (from 0.78 to 0.64) and dramatically upon binding SP41 (0.23) (Table 2). Assuming transfer efficiencies are a weighted average of free and bound forms, we determined the efficiency of the peptide-bound population based on the dissociation constants measured above and estimate that the distance between interprotomer PPXD residues increased by 6 Å upon binding SP22 and by over 34 Å upon binding SP41. Finally, the SecA-696C mutant also exhibited a moderate decrease in its initial energy transfer efficiency (0.59), but the magnitude of the change was similar for SP22 (0.42) and SP41 (0.41) (Table 1 and Table 2), corresponding to an increased interprotomer distance between HWD residues of 11 Å and 15 Å, respectively. Thus, similar to our findings for PPXD, binding of the signal or extended peptide resulted in a more open conformation of the SecA dimer between interprotomer HWD regions as well. In this case, however, a quantitatively similar conformational change was observed with the two peptides, suggesting that signal peptide binding solely triggers the change with no involvement of the early mature region.
Our results are supportive of a model in which signal peptide binding leads to an active, open conformational state of SecA that is dimeric with considerable motion of PPXD and HWD while NBF2 remains fairly rigid. This view is consistent with previous studies demonstrating the formation of an open conformation of SecA upon signal peptide or phospholipid binding 27, 29, 55. A more ‘open’ state of SecA with PPXD rotated away from HWD has been captured in a B. subtilis SecA monomer crystal and was proposed to be important for preprotein interaction 26. However, our result is novel in that it suggests that the SP41 extended peptide induced a much larger opening, thereby potentially accommodating additional residues into a binding site for the mature region of the preprotein. Indeed, further displacement of PPXD from that found in its ‘open’ state to one adjacent to NBF2 was observed in the T. maritima SecA-SecYEG crystal structure 17. A recent study that utilized disulfide crosslinking to map the polypeptide pathway through SecA-bound SecYEG protein demonstrated that the PPXD and NBF2 interaction constitutes a ‘clamp’ for preprotein capture by SecA and subsequent delivery to the protein-conducting channel 56. Our results here should allow an assessment of an earlier stage of peptide capture by SecA in solution utilizing some of the tools developed in this study. These results are also consistent with recent electron microscopy studies of SecA interacting with SecB and proOmpA, where an asymmetric interaction of the SecA dimer was observed after the SecA-SecB complex bound proOmpA. The electron microscopy images were most consistent with one protomer of the SecA dimer adopting an open conformation in the ternary complex 57. Finally, while the matter of whether SecA exists as a monomer or dimer when bound to SecYEG has remained a controversial one, we note that recent fluorescence burst experiments detected a significant population of SecA dimer after binding to SecYEG 20. This suggests that additional characterization of the SecA dimer state at SecYEG is warranted.
Modeling of the ‘open’ dimer
We modeled two ‘open’ SecA dimers utilizing the preferred 1M6N dimer interface and compared them to the original ‘closed’ B. subtilis SecA dimer published by Hunt et al. (Figure 7A) 21. For this purpose either the ‘open’ B. subtilis SecA monomer X-ray structure 26 or the lsqb;open’ E. coli SecA monomer NMR structure 34 was utilized to build the corresponding dimer employing the 1M6N interface, resulting in the two ‘open’ dimers shown (Figure 7B,C, respectively). Although the B. subtilis and E. coli SecA proteins are highly homologous and contain ∼50% sequence identity overall 58, minor structural differences are observed between the two proteins that are, in part, attributed to small insertions and deletions contained along the differential length of the two proteins (841 vs 901 amino acid residues, respectively). This difference makes a strict comparison between these homologs somewhat difficult. However, the modestly longer interprotomer distances that were consistently observed for the E. coli‘open’ dimer compared to its B. subtilis counterpart are suggestive of a more open structure overall (Figures 7B,C). This view is also supported by the greater observed sensitivity to a variety of proteases with different cleavage specificities (trypsin, V8 protease, and proteinase K) for the former protein compared to the latter one 59 (D. Oliver, unpublished results). Of interest, when compared to the ‘closed’ state structure, both ‘open’ state structures displayed greater PPXD separation as predicted by our signal peptide-binding data for the SecA-340C mutant (Figure 7A vs B,C). By contrast, the correlation between the ‘open’ structures and FRET results from the SecA-696C mutant in the HWD was more equivocal, since only the E. coli SecA ‘open’ structure showed a slightly longer distance in this case, and it was unclear whether this marginal difference simply related to the global differences between the B. subtilis and E. coli proteins. Strictly comparing the B. subtilis ‘closed’ and ‘open’ dimers revealed that only the PPXD distance changed dramatically, suggesting that this latter ‘open’ dimer structure has not captured the signal peptide-induced change in the HWD. Our findings are in good agreement with an opening between the PPXD and HWD regions upon binding of the signal peptide as observed by NMR 34.
Figure 7.

Comparison of the ‘closed’ and ‘open’ states of the SecA antiparallel dimer. (A) The ‘closed’ B. subtilis SecA anti-parallel dimer (PDB ID: 1M6N) is compared to ‘open’ dimers that were modeled utilizing this preferred interface and either the (B) ‘open’ B. subtilis SecA monomer (PDB ID: 1TF5) or (C) ‘open’ E. coli SecA monomer (PDB ID: 2VDA). (D) The SecYEG-bound SecA monomer (PDB ID: 3DIN) colored according to domain: NBF1-yellow, NBF2-dark blue, PPXD-orange, HSD-green, and HWD-cyan. The color coordination for the residues, in E. coli SecA coordinates, is as follows: 340-yellow, 458-brown, 506-red, and 696-green. Predicted distances between identical residues of adjacent protomers are indicated.
The interaction of SecA with SecYEG as determined by x-ray crystallography 17 depicts a major conformational change where the PPXD moves closer to the NBF2 domain and further away from the HWD (Figure 7D). Thus, our study suggests that the peptide-bound SecA dimer adopts an activated ‘open’ state for SecYEG binding. Given that binding of SP41 induced a significantly larger conformational change than SP22, we propose that in solution SecA dimers primarily exist in a compact form, and that binding of signal peptides initiates formation of a partially ‘open’ state; however, interaction with portions of the mature preprotein are required to reach the fully ‘open’ form of SecA, in which the PPXD swings open farther away from the HWD forming the PPXD-NBF2 ‘clamp’ for preprotein capture. This view is consistent with a recently proposed model for Sec translocation, which requires activated dimeric SecA to bind to SecYEG 20.
In summary, we have utilized a FRET approach to determine the protomer orientation of the E. coli SecA dimer in solution. Our measurements are most consistent with distances determined from the B. subtilis 1M6N antiparallel dimer 21 and suggest that this is the dominant solution state interface. The FRET measurements further suggest that SecA retains its dimer structure upon interaction with signal peptide, but that the PPXD and HWD experience large conformational changes, as detected by increased interprotomer distances between these domains. Based on a modeled ‘open’ dimer with an anti-parallel orientation, we speculate that binding of an extended signal peptide, containing a portion of the early mature region of the preprotein, creates a SecA state that is activated for SecYEG binding. This study sets the stage for future work to explore the oligomeric state of SecA in the presence of its other ligands and deepens understanding of the mechanism of SecA action.
Supplementary Material
Acknowledgments
We are grateful to John Hunt and Anastassios Economou for providing dimer coordinates for the B. subtilis anti-parallel SecA and E. coli anti-parallel SecA dimer structures, respectively. We also thank Rich Olson for assistance in creating the ‘open’ dimer structures, and Lorry Grady, Sanchaita Das, and Dylan Maxwell Reilly for intellectual discussions during the course of the study as well as feedback on this paper.
Footnotes
This work was supported by grants from the National Institutes of Health awarded to D.O. (GM42033) and the National Science Foundation awarded to I.M. (MCB-0843656).
Abbreviations: AF, Alexa Fluor; FRET, Förster resonance energy transfer; HPLC, high-performance liquid chromatography; HSD, Helical scaffold domain of SecA; HWD, helical wing domain of SecA; IAEDANS, 5-((((2-iodoacetyl)amino)-ethyl)amino)naphthalene-1-sulfonic acid; IAE, IAEDANS; IANBD, N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole; IAN, IANBD; NBF1, nucleotide-binding fold-1 domain of SecA; NBF2, nucleotide-binding fold-2 domain of SecA; PPXD, preprotein-crosslinking domain of SecA; SEC, size exclusion chromatography; SP2, 22 residue long E. coli alkaline phosphatase signal peptide with cysteine at position 2; SP22, 22 residue long E. coli alkaline phosphatase signal peptide with cysteine at position 22; SP41, 41 residue long extended E. coli alkaline phosphatase signal peptide with cysteine at position 2; TKE, 25 mM Tris-HCl, pH 7.5, 25 mM KCl, 1 mM EDTA
Supporting Information Available. ATPase activities of labeled and unlabeled monocysteine SecA mutants (Table 1). ATPase activities of monocysteine SecA mutants (Figure 1). Assessment of SecA oligomeric state. (Figure 2) Donor quantum yields and FRET pair R0 values (Table 2). Evaluation of κ2 Distributions and Effect on Distances using Steady State Anisotropy Values (Table 3). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.du Plessis D, Nouwen N, Driessen A. The Sec translocase. Biochim Biophys Acta. 2011;1808:851–865. doi: 10.1016/j.bbamem.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Rusch S, Kendall D. Oligomeric states of SecA and SecYEG core components of the bacterial Sec translocon. Biochim Biophys Acta. 2007;1768:5–12. doi: 10.1016/j.bbamem.2006.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akita M, Shinkai A, Matsuyama S, Mizushima S. SecA, an essential component of the secretory machinery of Escherichia coli, exists as homodimer. Biochem Biophys Res Commun. 1991;174:211–216. doi: 10.1016/0006-291x(91)90507-4. [DOI] [PubMed] [Google Scholar]
- 4.Cabelli RJ, Dolan KM, Qian L, Oliver DB. Characterization of membrane-associated and soluble states of SecA protein from wild-type and secA51(Ts) mutant strains of Escherichia coli. J Biol Chem. 1991;266:24420–24427. [PubMed] [Google Scholar]
- 5.Woodbury RL, Hardy S, Randall L. Complex behavior in solution of homodimeric SecA. Protein Science. 2002;11:875–882. doi: 10.1110/ps.4090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sardis M, Economou A. SecA: a tale of two protomers. Mol Microbiol. 2010;76:1070–1081. doi: 10.1111/j.1365-2958.2010.07176.x. [DOI] [PubMed] [Google Scholar]
- 7.Or E, Navon A, Rapoport T. Dissociation of the dimeric SecA ATPase during protein translocation across the bacterial membrane. EMBO J. 2002;21:4470–4479. doi: 10.1093/emboj/cdf471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jilaveanu LB, Zito CR, Oliver D. Dimeric SecA is essential for protein translocation. Proc Natl Acad Sci U S A. 2005;102:7511–7516. doi: 10.1073/pnas.0502774102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duong F. Binding, activation, and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J. 2003;22:4375–4384. doi: 10.1093/emboj/cdg418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Or E, Boyd D, Gon S, Beckwith J, Rapoport TA. The bacterial ATPase SecA functions as a monomer in protein translocation. J Biol Chem. 2005;280:9097–9105. doi: 10.1074/jbc.M413947200. [DOI] [PubMed] [Google Scholar]
- 11.Das S, Stivison E, Folta-Stogniew E, Oliver D. Reexamination of the role of the amino terminus of SecA in promoting its dimerization and functional state. J Bacteriol. 2008;190:7302–7307. doi: 10.1128/JB.00593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Na B, Yang H, Tai PC. Additional in vivo and in vitro evidence for SecA funtioning as dimers in the membrane: dissociation into monomers is not essential for protein translocation in Escherichia coli. J Bacteriol. 2008;190:1413–1418. doi: 10.1128/JB.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jilaveanu LB, Oliver D. SecA dimer cross-linked at its subunit interface is functional for protein translocation. J Bacteriol. 2006;188:335–338. doi: 10.1128/JB.188.1.335-338.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Or E, Rapoport TA. Cross-linked SecA dimers are not functional in protein translocation. FEBS Letters. 2007;581:2616–2620. doi: 10.1016/j.febslet.2007.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Keyzer J, van der Sluis E, Spelbrink R, Nijstad N, de Kruijff B, Nouwen N, van der Does C, Driessen A. Covalently dimerized SecA is functional in protein translocation. J Biol Chem. 2005;280:35255–35260. doi: 10.1074/jbc.M506157200. [DOI] [PubMed] [Google Scholar]
- 16.Tziatzios C, Schubert D, Lotz M, Gundogan D, Betz H, Schagger H, Hasse W, Duong F, Collinson I. The bacterial protein-translocation complex: SecYEG dimers associate with one or two SecA molecules. J Mol Biol. 2004;340:513–524. doi: 10.1016/j.jmb.2004.04.076. [DOI] [PubMed] [Google Scholar]
- 17.Zimmer J, N Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musial-Siwek M, Rusch S, Kendall D. Probing the affinity of SecA for signal peptides in different environments. Biochemistry. 2005;44:13987–13996. doi: 10.1021/bi050882k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benach J, Chou YT, Fak JJ, Itkin A, Nicolae DD, Smith PC, Wittrock G, Floyd DL, Golsaz CM, Gierasch LM, Hunt JF. Phospholipid-induced monomerization and signal-peptide-induced oligomerization of SecA. J Biol Chem. 2003;278:3628–3638. doi: 10.1074/jbc.M205992200. [DOI] [PubMed] [Google Scholar]
- 20.Kusters I, van den Bogaart G, Kedrov A, Krasnikov V, Fulyani F, Poolman B, Driessen A. Quaternary structure of SecA in solution and bound to SecYEG probed at the single molecule level. Structure. 2011;19:430–439. doi: 10.1016/j.str.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 22.Papanikolau Y, Papadovasilaki M, Ravelli R, McCarthy A, Cusack S, Economou A, Petratos K. Structure of dimeric SecA, the Escherichia coli preprotein translocase motor. J Mol Biol. 2007;366:1545–1557. doi: 10.1016/j.jmb.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 23.Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, Jacobs WR, Sacchettini JC. Crystal structure of Mycobacterium tuberculosis SecA, a preprotein tranlsocating ATPase. Proc Natl Acad Sci USA. 2003;100:2243–2248. doi: 10.1073/pnas.0538077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassylyev D, Mori H, Nassylyeva M, Tsukazaki T, Kimura Y, Tahirov T, Ito K. Crystal structure of the translocation ATPase SecA from Thermus thermophilus reveals a parallel, head-to-head dimer. J Mol Biol. 2006;364:248–258. doi: 10.1016/j.jmb.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 25.Zimmer J, Li W, Rapoport TA. A novel dimer interface and conformational changes revealed by an X-ray structure of B. subtilis SecA. J Mol Biol. 2006;364:259–265. doi: 10.1016/j.jmb.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 26.Osborne AR, Clemons WM, Rapoport TA. A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci U S A. 2004;101:10937–10942. doi: 10.1073/pnas.0401742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding H, Hunt JF, Mukerji I, Oliver D. B. subtilis SecA ATPase exists as an antiparallel dimer in solution. Biochemistry. 2003;42:8729–8738. doi: 10.1021/bi0342057. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Pan X, Tang Y, Quan S, Tai PC, Sui SF. Full-length Escherichia coli SecA dimerizes in a closed conformation in solution as determined by cryo-electron microsopy. J Biol Chem. 2008;283:28783–28787. doi: 10.1074/jbc.C800160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding H, Mukerji I, Oliver D. Lipid and signal peptide-induced conformational changes within the C-domain of Escherichia coli SecA protein. Biochemistry. 2001;40:1835–1843. doi: 10.1021/bi002058w. [DOI] [PubMed] [Google Scholar]
- 30.Natale P, den Blaauwen T, van der Does C, Driessen A. Conformationa state of the SecYEG-bound SecA probed by single tryptophan fluorescence spectroscopy. Biochemistry. 2005;44:6424–6432. doi: 10.1021/bi047488r. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt M, Ding H, Ramamurthy V, Mukerji I, Oliver D. Nucleotide binding activity of SecA homodimer is conformationally regulated by temperature and altered by prlD and azi mutations. J Biol Chem. 2000;275:15440–15448. doi: 10.1074/jbc.M000605200. [DOI] [PubMed] [Google Scholar]
- 32.Ding H, Mukerji I, Oliver D. Nucleotide and phospholipid-dependent control of PPXD and C-domain association for SecA ATPase. Biochemistry. 2003;42:13468–13475. doi: 10.1021/bi035099b. [DOI] [PubMed] [Google Scholar]
- 33.Auclair S, Moses J, Musial-Siwek M, Kendall D, Oliver D, Mukerji I. Mapping of the signal peptide-binding domain of Escherichia coli SecA using Forster resonance energy transfer. Biochemistry. 2010;49:782–792. doi: 10.1021/bi901446r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gelis I, Bonvin A, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos C. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baud C, Karamanou S, Sianidis G, Vrontou E, Politou AS, Economou A. Allosteric communication between signal peptides and the SecA protein DEAD motor ATPase domain. J Biol Chem. 2002;277:13724–13731. doi: 10.1074/jbc.M200047200. [DOI] [PubMed] [Google Scholar]
- 36.Musial-Siwek M, Rusch S, Kendall D. Selective photoaffinity labeling identifies the signal peptide binding domain on SecA. J Mol Biol. 2007;365:637–648. doi: 10.1016/j.jmb.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Miller A, Kendall DA. Signal peptide determinants of SecA binding and stimulation of ATPase activity. J Biol Chem. 2000;275:10154–10159. doi: 10.1074/jbc.275.14.10154. [DOI] [PubMed] [Google Scholar]
- 38.Studier WF, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 39.Ramamurthy V, Oliver D. Topology of the integral-membrane form of Escherichia coli SecA protein. J Biol Chem. 1997;272:23239–23246. doi: 10.1074/jbc.272.37.23239. [DOI] [PubMed] [Google Scholar]
- 40.Jilaveanu LB, Oliver D. In vivo membrane topology of Escherichia coli SecA ATPase reveals extensive periplasmic exposure of multiple functionally important domains clustering on one face of SecA. J Biol Chem. 2007;282:4661–4668. doi: 10.1074/jbc.M610828200. [DOI] [PubMed] [Google Scholar]
- 41.Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. Sixth. Molecular Probes, Inc.; Eugene, OR: 1996. [Google Scholar]
- 42.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell C, Oliver DB. Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol Microbiol. 1993;10:483–497. doi: 10.1111/j.1365-2958.1993.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 44.Lakowicz JR. Principles of Fluorescence Spectroscopy. second. Plenum Publishers; New York: 1999. Energy transfer; pp. 367–394. [Google Scholar]
- 45.Clegg RM. Fluorescence Resonance Energy Transfer and Nucleic Acids. Meth Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 46.Demas JN, Crosby GA. The measurement of Photoluminescence quantum yields. A review. J Phys Chem. 1971;75:991–1024. [Google Scholar]
- 47.Magde D, Wong R, Seybold PG. Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: improved absolute standards for quantum yields. Photochem Photobiol. 2002;75:327–334. doi: 10.1562/0031-8655(2002)075<0327:fqyatr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 48.Magde D, Brannon JH, Cremers TL, Olmsted J. Absolute luminescence yield of cresyl violet. A standard for the red. J Phys Chem. 1979;83:693–699. [Google Scholar]
- 49.Dale RE, Eisinger J, Blumberg WE. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ivanov V, Li M, Mizuuchi K. Impact of emission anisotropy on fluorescence spectroscopy and FRET distance measurements. Biophys J. 2009;97:922–929. doi: 10.1016/j.bpj.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lill R, Cunningham K, Brundage LA, Ito K, Oliver D, Wickner W. SecA protein hydrolyzes ATP and is an essential component of the protein translocation ATPase of Escherichia coli. EMBO J. 1989;8:961–966. doi: 10.1002/j.1460-2075.1989.tb03458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kourtz L, Oliver D. Tyr-326 plays a critical rile in controlling SecA-preprotein interaction. Mol Microbiol. 2000;37:1342–1356. doi: 10.1046/j.1365-2958.2000.02078.x. [DOI] [PubMed] [Google Scholar]
- 53.Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A. Cross-talk between catalytic and regulatory elements in a DEAD motor domain is essential for SecA function. EMBO J. 2001;20:961–970. doi: 10.1093/emboj/20.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wowor AJ, Dongmei Y, Kendall DA, Cole JL. Energetics of SecA dimerization. J Mol Biol. 2011;408:87–98. doi: 10.1016/j.jmb.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JY, Kim M, Ahn T. Effects of signal peptide and adenylate on the oligomerization and membrane binding of soluble SecA. J Biochem Mol Biol. 2006;39:319–328. doi: 10.5483/bmbrep.2006.39.3.319. [DOI] [PubMed] [Google Scholar]
- 56.Bauer BW, Rapoport TA. Mapping polypeptide interactions of the SecA ATPase during translocation. Proc Natl Acad Sci U S A. 2009;106:20800–20805. doi: 10.1073/pnas.0910550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Y, Pan X, Chen Y, Tai PC, Sui SF. Dimeric SecA couples the preprotein translocation in an asymmetric manner. PLOS One. 2011;6:e16498. doi: 10.1371/journal.pone.0016498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sadaie Y, Takamatsu H, Nakamura K, Yamane K. Sequencing reveals similiarity of the wild-typediv+ gene of Bacillus subtilis to the Escherichia coli secA gene. Gene. 1991;98:101–105. doi: 10.1016/0378-1119(91)90110-w. [DOI] [PubMed] [Google Scholar]
- 59.Cabelli RJ. Biochemical characterization of the role of SecA protein in protein export in Escherichia coli. State University of New York; Stony Brook, NY: 1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.