

Abstract
The variable lymphocyte receptors of lamprey and hagfish are comprised of leucine-rich repeat modules, instead of the immunoglobulin-like domain building blocks of antibodies and T-cell receptors in jawed vertebrates. Both types of vertebrate rearranging antigen receptors are similarly diverse, with repertoires that can potentially exceed 1014 unique receptors. In order to characterize antigen binding properties of the VLRs, we developed a high-throughput yeast surface display platform for isolation of monoclonal VLRs. We have isolated VLRs that specifically bind hen egg lysozyme, β-galactosidase, cholera toxin subunit B, R-phycoerythrin, and the blood group trisaccharides A and B, with binding affinities in the mid-nanomolar to mid-picomolar range. VLRs may thus be excellent single-chain alternatives to Ig-based antibodies for biotechnology applications.
Keywords: Lamprey, variable lymphocyte receptors, recombinant antibodies, yeast surface display
1. Introduction
The variable lymphocyte receptors (VLRs) of jawless fish, such as lamprey and hagfish, are the only known rearranging antigen receptors that are built from leucine-rich repeats (LRR), instead of the immunoglobulin (Ig) superfamily domains that are building blocks of the B- and T-cell receptors of jawed vertebrates from shark to man (1-3). Members of the LRR-containing protein superfamily serve as cardinal microbial recognition molecules in the innate immune systems of plants and animals, for instance the LRR-containing plant Disease Resistance genes, Toll and Toll-like receptors and the cytoplasmic nucleotide-binding site (NBS)-LRR proteins (4). These innate microbial recognition molecules have diverged to serve specific functions over very long evolutionary periods, like nearly all other genes in plant and animal genomes. In sharp contrast, in vertebrate lymphocytes the rearranging antigen receptors are combinatorially assembled from hundreds of gene fragments, resulting in repertoires that can potentially exceed 1014 unique receptors (5,6).
Experimental data indicates that Ig-based antibodies can specifically bind virtually all types of antigens with high affinity. Little is known, however, about the antigen-binding properties of VLRs. Antigen recognition by VLRs from immunized lamprey have been shown for spore coats of anthrax (Bacillus anthracis) and their BclA glycoprotein component, for the human blood group trisaccharide antigens, and for hen egg lysozyme (HEL) (5,7). In order to isolate and characterize VLR binders of specific antigens, we developed a yeast surface display (YSD) platform for the VLRs. Thus far we have isolated clones that bind HEL, E. coli β-galactosidase, cholera toxin subunit B, R-phycoerythrin (RPE), and the blood group trisaccharides A and B, with binding affinities in the mid-nanomolar to mid-picomolar range, comparable to high affinity IgG antibodies with KDs in the low nanomolar range (8). These monoclonal VLRs were isolated from libraries originating from immunized lamprey, as well as from non-immunized animals, indicating that for most antigens there is no need to for immunization in order to isolate specific ligand-binding clones (7). VLRs may thus be excellent single-chain alternatives to Ig-based antibodies for biotechnology applications, since both of these antigen receptors were optimized over hundreds of millions of years of evolution (9).
The VLR diversity regions consist of sets of LRR modules, each with a highly variable sequence, as shown in Fig.1. At both ends of the diversity region there are capping modules, the N-terminal LRR (LRRNT) and the C-terminal LRR (LRRCT), which are stabilized by two sets of intramodular disulfide bonds (10,11). These disulfide bonds are essential for proper folding and stability of the VLR structure. Expression of recombinant VLRs, therefore, requires a eukaryotic host, such as the yeast Saccharomyces cerevisiae that possess an efficient oxidative protein folding machinery and secretory pathway, and is amenable to high-throughput screens (12,13). We also noted that for optimal antigen binding, the VLRs require free N-termini. We therefore developed a YSD vector based on C-terminal fusion of the VLRs to the yeast surface-anchored flocculation protein Flo1p, as shown in Fig. 2a,b.
Figure 1.
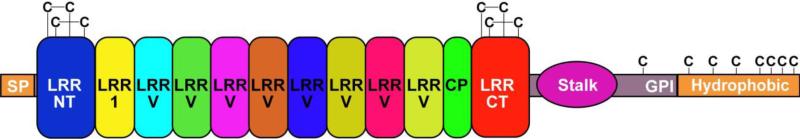
A stick model of a lamprey mature VLRB. The VLR comprises a set of highly diverse LRR modules capped by disulfide-bonded N-terminal LRR (LRRNT, 24-32 amino acids) and C-terminal LRR (LRRCT, 45-62 amino acids). The 25-residue LRR1 is followed by one to ten 24-residue LRRVs and then a 16-residue truncated LRR, the connecting peptide (CP). The invariant portions of VLRBs include an N-terminal secretory signal peptide (SP) and an 81-residue C terminus that contains a threonine/proline-rich stalk (33 amino acids) and a glycosyl phosphatidylinositol (GPI) membrane anchor motif, which tethers the VLR to the lymphocyte surface. Seven cysteines in the 22-residue hydrophobic C-terminal domain may participate in VLR oligomerization.
Figure 2.

(A) Yeast surface display of VLRs fused to the C-terminus of the Flo1p anchor. The hemagglutinin (HA)-tag serves for VLR detection via Alexa 488 labeled antibodies. Biotinylated ligands are detected via R-phycoerythrin conjugated to streptavidin (SA-PE). (B) The pYSD2 vector for VLR yeast surface display. The VLRs are expressed under the tightly regulated GAL1 promoter, fused between the authentic Flo1p leader and the yeast Flo1p C-terminus, includes the surface-anchoring domain. The vector replicates in bacteria and yeast (ColE1, CEN6/ARS4), selected by kanamycin/geneticin resistance. (C) VLRs are cloned directionally between two unique SfiI sites. Protein detection and purification tags, and the annealing sites for primers are indicated (YSD.F, YSD.R). D. The homologous recombination cassette consists of two 49-bp direct repeats, separated by a linker with a PmeI restriction site for plasmid linearization. HR.F, HR.R indicate primers for rolling-circle amplification across of the plasmid.
2. Materials
2.1. Construction of VLR YSD library
The pYSD2 vector is available upon request following MTA.
Primers for PCR amplification of VLRA and VLRB (see Note 1). The primers carry overhangs with 2 unique SfiI sites (underlined). VLRA.F 5-aaaaaaggccaccggggccAAAACGTGTGAAACGGTC; VLRA.R 5-aaaaaaggccccagaggccccCTCCACGAATGGGCACT; VLRB.F aaaaaaggccaccggggccGCATGTCCCTCGCAGTGT; VLRB.R aaaaaaggccccagaggccccTGGGCATTTCGAGGGGCT.
QIAquick PCR purification kit (QIAGEN).
TempliPhi 100 amplification kit (GE Healthcare).
Primers for the homologous recombination cassette: HR.F 5-AAACGGAATTAACCCTCCACT, HR.R 5-AAACCGGCGTAGAGGATGCA
dNTPs, 25 mM each (Roche).
Yeast inorganic pyrophosphatase, phi29 DNA polymerase, Bovine serum albumin (BSA) and PmeI restriction enzyme (all from New England Biolabs).
2.2. Yeast transformation
Yeast strain BJ5464 (ATCC 208288).
Bacto Peptone, Bacto Yeast Extract and Bacto Agar (BD Biosciences).
Salmon sperm carrier DNA, MB-grade (Roche).
YPD Plus (Zymo Research).
Geneticin (G-418 Sulfate, American Bioanalytical)
1 L YPD medium: 20 g Bacto Peptone and 10 g Bacto Yeast Extract. For YPD plates, add 18 g Bacto Agar. Add water to 950 mL and autoclave. Allow to cool to 55°C and add 50 mL of filter sterilized 40% glucose. When needed, add in YPD medium G-418 to 100 μg/mL, and for plates add G-418 to 300 μg/mL. Store at room temperature for up to 1 month or at 4°C for up to 6 months.
1 L YPD medium (pH 4.5): YPD including 10.4 g sodium citrate and 7.4 g citric acid monohydrate.
1 L 2x YPD medium: 40 g Bacto Peptone and 20 g Bacto Yeast Extract. Add water to 900 mL and autoclave. Allow to cool to 55°C and add 100 mL of filter sterilized 40% glucose.
Transformation buffer 1: 0.1 M lithium acetate (LiAc), 10 mM Tris-HCl, pH 7.5, 1 mM EDTA. Prepare fresh from stock of 1 M LiAc (10.2 g of lithium acetate dihydrate in 100 mL water. Autoclave and store at room temperature).
Transformation buffer 2: 40% PEG, 0.1 M LiAc, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA. Prepare fresh from stocks of 1 M LiAc and 50% PEG (50 g of PEG 3350 in water in 100 mL volume. Stir to dissolve at 70°C on a heating plate. Autoclave and store at room temperature securely capped to prevent evaporation. See Note 2).
2.3. Isolation of antigen specific monoclonal VLRs
1 L YPG medium: 20 g Bacto Peptone and 10 g Bacto Yeast Extract. Add water to 933 mL and autoclave. Allow to cool to 55°C and add 67 mL of filter sterilized 30% galactose.
Penicillin, 5,000 units per mL, and Streptomycin, 5,000 μg per mL (Invitrogen).
MidiMACS with LS column (Miltenyi). Can separate 1×109 labeled yeast cells from a total of 1×1010 cells.
MiniMACS with MS column (Miltenyi). Can separate 5×107 labeled yeast cells from a total of 5×108 cells.
MACS buffer: PBS with 0.5% BSA, 2 mM EDTA, 0.1% Tween 20. Filter sterilize and store at 4°C for up to 6 months.
FACS buffer: PBS with 0.5% BSA, 2 mM EDTA. Filter sterilize and store at 4°C for up to 6 months.
Anti-biotin microbeads (Miltenyi).
Rat anti-HA (high affinity clone 3F10, Roche) or rat anti-FLAG (Stratagene)
Streptavidin R-phycoerythrin (Invitrogen)
Alexa Fluor 488 labeled donkey anti-rat IgG (Invitrogen)
3. Methods
The pYSD2 vector consists the Flo1p leader peptide connected via a short linker to the cloning sites region, which consist of two unique SfiI sites for directional cloning, followed by a set of tags for protein detection and purification, and then the Flo1p stalk and C-terminal anchor, as shown in Fig. 2b,c. The shuttle vector can propagate both in E. coli and yeast, selected for kanamycin or geneticin resistance.
To characterize the natural VLR repertoire we developed a procedure for efficient library construction that circumvents recombination among the VLR inserts during yeast transformation. In S. cerevisiae, linear fragments of double stranded DNA can efficiently recombine based on homology regions spanning 30-50 bp, and even several bases of identity suffice to initiate recombination events. Thus, co-transformation of yeast with a gapped vector and molar excess of PCR amplicons of library inserts, which include overhangs homologous to the gap flanking regions in the vector, results in highly efficient recombination between the vector and amplicons, as well as between related amplicons, reshuffling the genes in the library (14). Library construction by means of gap-repair in vivo is about 100-fold more efficient than transformation with an equivalent aliquot of a plasmid library. However, this method produces low quality VLR libraries, perhaps due to the presence of multiple potential recombination sites in VLRs, which result in disrupted open reading frames.
To take advantage of the high efficiency of yeast transformation with linear DNA, we created a cassette for intra-plasmid homologous recombination in the pYSD2 vector, consisting of two 49-bp direct repeats separated by an 8-bp PmeI restriction enzyme site, as shown in Fig. 2d. After ligation of the inserts into the vector SfiI sites, the library is amplified via rolling-circle amplification. The amplified circular library is then linearized by PmeI digest and used to transform yeast. The vector is then re-circularized in vivo by homologous recombination between the two direct repeats in the pYSD2 plasmid.
3.1. Construction of VLR YSD library
Amplify the diversity regions of lamprey VLRs from lymphocyte cDNA or genomic DNA (see Notes 1, 3).
Digest 500 ng of the pYSD2 vector with SfiI restriction enzyme and gel purify the digested plasmid. Digest also 300 ng of the amplicon of VLR diversity regions with SfiI and column purify with QIAquick PCR. Set a ligation in 10 μL volume using 50 ng of the digested vector and 30 ng of the VLRA insert, or 25 ng of the VLRB insert (molar ratio of about 5:1 insert to vector). Ligate overnight at 16°C.
Use 2 μL of the ligated library for rolling-circle amplification in 10 μL reaction of TempliPhi. Incubate for 4 hours at 30°C.
Add 100 pmoles of each of the primers HR.F and HR.R, then in a PCR cycler heat for 2 min at 95°C and chill to 4°C. Increase the volume of the reaction to 400 μL (can be split into 2 tubes of 200 μL) adding dNTPs to 1 mM (16 μL of 25 mM stock), 8 μL BSA (10 mg/mL), 4 μL pyrophosphatase (100 units/mL), 40 μL of the 10X buffer and 8 μL of phi29 DNA polymerase (10 units/μL). Incubate 16 hours at 30°C, then add 100 pmoles of the primers HR.F and HR.R, and 6 μL of PmeI restriction enzyme (10 units/μL). Incubate at 30°C for 3 hours, then at 37°C for 1 hour. Finally, heat inactivate the enzymes at 65°C for 20 min. Purify the amplified DNA using 2 columns of QIAquick PCR. Reapply the flow through to increase the yield, and elute each sample using 100 μL of the kit elution buffer heated to 70°C. Typical yields are 10-15 μg of the linearized library ready for transformation.
3.2. Yeast transformation
Inoculate a yeast colony from a freshly streaked plate (see Note 4) into 20 mL YPD medium and grow overnight shaking at 250-300 RPM at 30°C (or longer at room temperature, 20-22°C).
Determine the culture cell density using a spectrophotometer. Dilute a sample 1:10 in water (10 μL culture in 90 μL water) and prepare a blank similarly (10 μL YPD medium in 90 μL water). For optimal results, only use a culture that have reached an OD546 between 2-4.
Dilute 30 OD units of the yeast culture into 200 mL of 2x YPD pre-warmed to 30°C (OD546 of 0.15).
Grow the cells at 30°C to an OD546 of 0.6 (3-5 hours; see Note 5).
Prepare Salmon sperm carrier DNA (10 mg/mL stock): thaw the DNA on ice just before transformation. Aliquot 100 μL DNA in a tube and boil for 5 min at 100°C. Immediately place the DNA tube in an ice/water bath for 5 min. Repeat the boiling and quenching once more and keep the DNA on ice.
Spin the 200 mL culture at 700 × g for 5 min.
Resuspend the pellet in 120 mL of sterile water by vortexing. Pipet up and down if necessary.
Spin again at 700 × g for 5 min.
Decant supernatant and resuspend the pellet in 4 mL of Transformation buffer 1.
Spin at 700 × g for 5 min.
Decant supernatant and spin briefly again to remove all residual fluid. Resuspend the pellet in 2.4 mL of Transformation buffer 1.
Set a 50 mL tube on ice and add 10-15 μg of the linearized library DNA.
Add 80 μL of the denatured carrier DNA.
Add the yeast cells from step 11 and vortex to mix.
Add 10 mL of Transformation buffer 2 and vortex for 1 min to thoroughly mix the components.
Incubate in a heat block at 30°C for 15 min, then 30 min at 30°C shaking at 100 RPM.
Remove the tube from the shaker and add 640 μL DMSO. Immediately mix by gently swirling the tube (at this stage the cells are becoming fragile).
Heat shock in a heat block at 42°C for 5 min, then 20 min in an incubator shaking gently at 50 RPM.
Pellet the cells at 700 × g for 5 minutes, decant supernatant and spin briefly again. Remove all residual fluid.
Resuspend the pellet in 10 mL YPD Plus by gentle pipetting up and down a 10 mL pipette (takes about 5 min reach a single-cell suspension).
Allow the cells to recover for 2 hours at 30°C shaking at 150-200 RPM.
Pellet the cells at 700g for 5 min.
Resuspend the cells in 10 mL of YPD (pH 4.5) supplemented with 100 μg/mL G-418 (citrate buffer at pH 4.5 inhibits growth of contaminating bacteria, which may be resistant to G-418).
Check the titer of the library by plating aliquots on YPD agar plates supplemented with 300 μg/mL G-418. Typical yields are 5-50 × 106 individual clones.
Transfer all the transformed cells to a 2 L baffled flask containing 400 mL YPD (pH 4.5) supplemented with 100 μg/ml G-418. Measure the OD546 at the start of culture.
Culture for 2 days at 30°C shaking at 250-300 RPM. After that, measure again the OD546 to calculate the actual growth of the library. Then, based on the original titer of the library, passage an aliquot representing at least 10-fold of the calculated library size, in a 2 L baffled flask containing 400 mL YPD (pH 4.5) supplemented with 100 μg/mL G-418. For strain BJ5464, 1 unit of OD546 represents 3×107 cells. Repeat the passage once more (during of the second and third passages, culture saturation should take less than a day).
The library can be stored for up to one month at 4°C. After that, passage an aliquot representing at least 10-fold of the library size.
For long term storage of the library prepare frozen aliquots. Culture an aliquot representing at least 10-fold of the library size in 100 mL YPD (pH 4.5) supplemented with 100 μg/mL G-418, at 30°C for 3 days (freezing cells in stationary phase enhances their survival).
Measure the OD546 of the culture to estimate cell number.
Spin the culture 10 min at 3,000 × g and decant supernatant.
Resuspend the pellet in YPD, 100 μg/mL G-418, at a final volume of 2.6 mL.
Prepare three 2 mL cryogenic tubes. To each tube add 150 μL of sterile glycerol and 850 μL of the cell suspension (each tube should contain about 109 cells).
Chill the cells gradually to −80°C. First, place the tubes in a Styrofoam box at −20°C. After 24 hours, transfer the box with the tubes to a −80°C freezer.
To initiate culture from a frozen aliquot, thaw the cells at room temperature, transfer into 100 mL YPD, 100 μg/mL G-418, and culture at 30°C. Passage the library 2-3 times in order to dilute the dead cells.
3.3. Isolation of antigen-specific monoclonal VLRs
In the morning, start a culture with an aliquot representing at least 10-fold of the library size. Inoculate the culture at an OD546 between 0.05-0.1 in YPD (pH 4.5), 100 μg/mL G-418. Incubate at 30°C with shaking at 250 RPM. The best results are obtained with cultures expanded to an OD546 between 1-3. If the culture grew beyond OD546 of 3, dilute with fresh medium to an OD546 of 0.5 and culture at 30°C for about 2 hours (doubling time is about 1.5 hours at 30°C) to reach OD546 of 1, and then proceed to induction of the library.
Inoculate the starter cells into pre-warmed YPG supplemented with 100 μg/mL G-418, at an OD546 of 0.05. Culture overnight at 30°C shaking at 250 RPM. The best results are obtained for induced cultures that reached an OD546 between 1-2. Library passages and induction can also be done at room temperature (20-22°C), with culture periods of the starter and induction extended to 16-24 hours (in YPD yeast grow nearly twice as fast as in YPG).
Spin 1×1010 induced cells in a centrifuge at 2,500g for 5 min and decant supernatant.
At this point cells are prepared for magnetic separation (see Note 6). Wash the cell pellet with 50 mL of MACS buffer at room temperature, vortex to resuspend.
Repeat for a total of 3 washes. Resuspend the cell pellet in 5 mL MACS buffer.
Add biotinylated antigen to a final concentration of 0.5-1 μM (up to 10 antigens may be used simultaneously). Rotate for 60 min at room temperature, followed by 10 min incubation on ice.
Pellet the cells in a refrigerated centrifuge at 4°C for 5 min at 2,500g and decant supernatant.
Wash the cell pellet with 50 mL of ice-cold MACS buffer. Repeat for a total of 3 washes.
Resuspend the cell pellet in 5 mL of ice-cold MACS buffer and add 100 μl anti-biotin Microbeads (up to 200 μl anti-biotin Microbeads may be used for maximal enrichment). Rotate the tube for 30 min at 4°C.
Pretreat an LS column, loaded onto the magnet, by flowing 3 mL of ice-cold MACS buffer.
Pellet the cells at 4°C for 5 min at 2,500g and decant supernatant.
Resuspend the cell pellet in 50 mL of ice-cold MACS buffer. Vortex to break any cell aggregates.
Immediately load 7 mL of the cell suspension onto the column on magnet. After the flow has stopped, briefly remove the column from magnet, in order to release captured unlabeled cells, and immediately place it back in the magnet. Add 1 mL of ice-cold MACS buffer and let flow through.
Repeat until all cells have been loaded.
Wash the column with 3 mL of ice-cold MACS buffer. Make sure the upper loading chamber is washed of all the cells.
To elute, remove the column from the magnet and place in a culture tube. Add 7 ml of YPD (pH 4.5) supplemented with 100 μg/mL G-418 and 1:100 dilution of Pen-Strep (to inhibit contaminating bacterial growth), and use the plunger to push the eluted cell suspension into the tube. Check the titer of eluted cells by plating aliquots.
Expand the eluted cell population and passage an aliquot representing at least 10-fold the size of the enriched cell population. Set a culture for induction of the enriched cell population, for further enrichment of antigen-binders via fluorescence-activated cell sorting (FACS, see Note 7).
Pellet the induced cells in a microfuge at full speed (16,000-21,000 × g) for 1 min and carefully aspirate the supernatant.
Wash the cells with 1 mL MACS buffer at room temperature. Vortex to resuspend the pellet.
Repeat for a total of 3 washes and resuspend the cells in MACS buffer (see Note 8).
Label the cells with 1:1000 dilution in MACS buffer of rat anti-HA (100 μg/mL stock) and with the biotinylated antigen at the desired concentration.
Rotate the cells at room temperature for 25 min, then incubate on ice for 5 min.
Pellet the cells at full speed for 30 sec at 4°C and wash with 1 mL ice-cold MACS buffer. Repeat for a total of 3 washes.
Label the yeast cells with a 1:200 dilution in ice-cold MACS buffer, of Alexa Fluor 488 labeled donkey anti-rat IgG, and of Streptavidin R-phycoerythrin (SA-PE).
Incubate the cells on ice for 15-20 min shielded from light. Mix the tubes once or twice during the incubation.
Spin the cells at full speed at 4°C and aspirate supernatant, then wash with 1 mL ice-cold FACS buffer for a total of three washes (FACS buffer used here, instead of the Tween-20 containing MACS buffer, to prevent distortion in fluid dynamics in the flow cytometer). Spin the cells for the last time, decant and keep the pellets on ice. Immediately prior to sorting, resuspend the cells in ice-cold FACS buffer at a concentration that is appropriate for your cytometer. Set a gate to sort and collect the population of double positive cells (see Note 9).
Successful enrichment of antigen-binding clones will result in at least 3-5 fold increase in the population of double positive cells after the second round of MACS, and after each subsequent round of FACS. An example of the process is shown in Fig. 3.
Figure 3.

Isolation of HEL binding VLRB clones from a YSD library of 1×107 clones. Each dot-plot represents 10,000 events. (A) The unsorted library is stained with 500 nM HEL, with no visible antigen binders. (B) After one round of enrichment with anti-biotin magnetic microbeads (MACS), the small population of double positive cells in the gate was sorted. (C) The output of the first fluorescence activating cell sorting (FACS) separation shows an enriched population of true binders, (D) which were further enriched during the subsequent sort. (E) Representative individual clones resulting from this screen.
Acknowledgments
The authors would like to thank Dr. K. Dane Wittrup and Ms. S. Annie Gai from Massachusetts Institute of Technology for introducing us to the fascinating world of yeast surface display. This work was supported by NSF grant MCB-0614672, NIH grant AI083892 and Intercenter Collaboration Grant, UMBI.
Footnotes
A set of primers devoid of overhangs may perform better for the first round of amplification of VLRs from cDNA or genomic DNA. Use these primers first, and then switch to the set listed in section 2.1.2: VLRANT.F 5-AAAACGTGTGAAACGGTCACAG; VLRACT.R 5-CTCCACGAATGGGCACTCATA; VLRBNT.F 5-GCATGTCCCTCGCAGTGTTC; VLRBCT.R 5-TGGGCATTTCGAGGGGCTAG.
It is essential to store the 50% PEG solution securely capped, to prevent evaporation, which over time will increase the PEG concentration and affect the efficiency of transformation.
PCR amplification of VLRAs yields substantial amounts of amplicons also from the non-assembled germline VLRA gene, which are invariant and include stop codons in all three frames. The germline amplicons are shorter than the amplicons of mature VLRAs, and in the intervening sequence include a unique AviII restriction site (TGCGCA). To eliminate the germline amplicons, amplify VLRA amplicons for the minimal number of PCR cycles required to see a product in an agarose gel, then digest the PCR product with AviII and gel purify the remaining longer band, which corresponds to the mature VLRA amplicons. These can be amplified again to obtain sufficient amount of DNA.
A colony from a freshly streaked plate yields the highest transformation efficiency.
Optimal transformation efficiencies are achieved only if the majority of cells have undergone 2 cell divisions. Adjust the inoculum in 2x YPD accurately to an OD546 of 0.15 and only proceed to the next step once the culture has reached an OD546 of 0.6. This may take 3-5 hours, or longer.
Using a flow cytometer (FACS), it is impractical in most cases to enrich antigen binding clones from a primary library of 1×106 clones, or larger, since the fraction of positive clones is usually below 0.01%. One or two rounds of enrichment with magnetic beads (MACS) should increase this fraction to a size that is practical for FACS. We describe here one round of MidiMACS enrichment. To use the MiniMACS, adjust the volumes proportionally.
Several cell samples are required every time the FACS is turned on, in order to set the parameters for the cytometer and for color compensation: 1) sample of unstained cells; 2) sample of VLR surface display level (rat anti-HA followed by Alexa Fluor 488 labeled donkey anti-rat IgG); 3) sample of antigen binding level (a known control clone can be used, stained with biotinylated antigen followed by SA-PE). For any new antigen it is recommended to stain uninduced cells with the biotinylated antigen followed by SA-PE, to detect non-specific biding on the surface of yeast. Since binders of the secondary reagents can also be enriched, it is important to frequently stain induced cell populations with Alexa Fluor 488-labled donkey anti-rat IgG, and separately with SA-PE, which should stain <0.01% of the cells. In case of high background staining, the secondary reagents may be replaced with alternatives.
Typical labeling volume is 1 mL for up to 1×109 cells, and 50-100 μL for 1-5×106 cells. It is recommended to maintain at least ten fold molar excess of antigen over the yeast-displayed receptors to prevent depletion of the antigen. Assuming 2.5-10×103 receptors per yeast cell, the receptor concentration for 106 cells in 100 μL is about 0.17 nM (104 × 106) and the lowest antigen concentration is therefore 1.7 nM. For lower antigen concentrations, increase the labeling volume proportionally. For the stage of labeling with secondary reagents use 0.5 mL volume for 1×109 cells, and 50-100 μl for 1-5×106 cells.
Yeast cells grow poorly in liquid culture at concentrations of less than 104 cells per mL. When sorting small numbers of cells, use plates instead of liquid culture to recover the cells.
References
- 1.Pancer Z, Amemiya CT, Ehrhardt GR, Ceitlin J, Gartland GL, Cooper MD. Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey. Nature. 2004;430:174–80. doi: 10.1038/nature02740. [DOI] [PubMed] [Google Scholar]
- 2.Pancer Z, Saha NR, Kasamatsu J, Suzuki T, Amemiya CT, Kasahara M, Cooper MD. Variable lymphocyte receptors in hagfish. Proc Nat Acad Sci USA. 2005;102:9224–9. doi: 10.1073/pnas.0503792102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Litman GW, Dishaw LJ, Cannon JP, Haire RN, Rast JP. Alternative mechanisms of immune receptor diversity. Curr Opin Immunol. 2007;19:526–34. doi: 10.1016/j.coi.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pancer Z, Cooper MD. The evolution of adaptive immunity. Annu Rev Immunol. 2006;24:497–518. doi: 10.1146/annurev.immunol.24.021605.090542. [DOI] [PubMed] [Google Scholar]
- 5.Alder MN, Rogozin IB, Iyer LM, Glazko GV, Cooper MD, Pancer Z. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science. 2005;310:1970–3. doi: 10.1126/science.1119420. [DOI] [PubMed] [Google Scholar]
- 6.Rogozin IB, Iyer LM, Liang L, Glazko GV, Liston VG, Pavlov YI, Aravind L, Pancer Z. Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat Immunol. 2007;8:647–56. doi: 10.1038/ni1463. [DOI] [PubMed] [Google Scholar]
- 7.Tasumi S, Velikovsky CA, Xu G, Gai SA, Wittrup KD, Flajnik MF, Mariuzza RA, Pancer Z. High-affinity lamprey VLRA and VLRB monoclonal antibodies. Proc Natl Acad Sci USA. 2009;106:12891–96. doi: 10.1073/pnas.0904443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marks JD, Bradbury A. Selection of human antibodies from phage display libraries. Methods Mol Biol. 2004;248:161–76. doi: 10.1385/1-59259-666-5:161. [DOI] [PubMed] [Google Scholar]
- 9.Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–68. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 10.Kim HM, Oh SC, Lim KJ, Kasamatsu J, Heo JY, Park BS, Lee H, Yoo OJ, Kasahara M, Lee JO. Structural diversity of the hagfish variable lymphocyte receptors. J Biol Chem. 2007;282:6726–32. doi: 10.1074/jbc.M608471200. [DOI] [PubMed] [Google Scholar]
- 11.Velikovsky CA, Deng L, Tasumi S, Iyer ML, Kerzic MC, Aravind L, Pancer Z, Mariuzza RA. Structure of a lamprey variable lymphocyte receptor in complex with a protein antigen. Nat Struc Mol Biol. 2009;16:725–30. doi: 10.1038/nsmb.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 13.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–473. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swers JS, Kellogg BA, Wittrup KD. Shuffled antibody libraries created by in vivo homologous recombination and yeast surface display. Nucleic Acids Res. 2004;32:e36. doi: 10.1093/nar/gnh030. [DOI] [PMC free article] [PubMed] [Google Scholar]