

Abstract
The feasibility of using difference spectroscopy, i.e. subtraction of two correlation spectra at different mixing times, for substantially enhanced resolution in crowded two-dimensional 13C-13C chemical shift correlation spectra is presented. With the analyses of 13C-13C spin diffusion in simple spin systems, difference spectroscopy is proposed to partially separate the spin diffusion resonances of relatively short intra-residue distances from the longer inter-residue distances, leading to a better identification of the inter-residue resonances. Here solid-state magic-angle-spinning (MAS) NMR spectra of the full length M2 protein embedded in synthetic lipid bilayers have been used to illustrate the resolution enhancement in the difference spectra. The integral membrane M2 protein of Influenza A virus assembles as a tetrameric bundle to form a protonconducting channel that is activated by low pH and is essential for the viral lifecycle. Based on known amino acid resonance assignments from amino acid specific labeled samples of truncated M2 sequences or from time-consuming 3D experiments of uniformly labeled samples, some inter-residue resonances of the full length M2 protein can be identified in the difference spectra of uniformly 13C labeled protein that are consistent with the high resolution structure of the M2 (22–62) protein (Sharma et al. 2010).
Keywords: difference spectroscopy, solid-state MAS NMR, membrane protein, inter-residue correlation, 13C-13C chemical shift correlation
Introduction
In the past decade, solid-state magic-angle-spinning (MAS) NMR has rapidly emerged as a powerful technique for studying insoluble proteins, such as membrane proteins. Spectral resolution in these proteins is a major problem because of the uniform low dielectric environment in the transmembrane (TM) domain and the hydrophobic amino acid content. Resonance assignment is an essential step towards structural determination of uniformly 15N and 13C labeled proteins, routinely requiring multi-dimensional solid-state MAS NMR techniques, such as three-dimensional (3D) NCOCX and NCACX (Pauli et al. 2001). In such 3D experiments, the N-C heteronuclear correlations (NCO and NCA) are keys toward sequential assignments due to the fact that each backbone nitrogen covalently bonds with the carbonyl (C′) carbon of residue i and the Ca of residue i+1 providing a through-bond linkage between two sequential residues. While this approach does not eliminate the need for 3D experiments it does provide enhanced resolution spectra for the observation of long range couplings and for some resonance assignments in experiments on samples that may be quite sensitive to long experimental times if they are performed at elevated temperatures to achieve a native-like environment. Typically the heteronuclear polarization transfer efficiency between 13C and 15N is around 30–40%. More critically, the amide 15N chemical shift resonances are considered to be more sensitive to the heterogeneity and/or dynamics (Saito et al. 2010; Wylie et al. 2006) of the sample being investigated, so that the spectral resolution in the 15N dimension may be worse than that in the 13C dimension in solid-state MAS spectroscopy of membrane proteins Here, we demonstrate the feasibility of using difference spectroscopy to identify some inter-residue correlations for resonance assignments in the crowded 2D 13C-13C chemical shift correlation spectra of uniformly 13C, 15N-labeled full length M2 protein.
The Influenza A virus M2 protein assembles as a tetrameric bundle to form a protonconducting channel that is activated by low pH and is essential for the viral lifecycle (Sugrue and Hay 1991; Sakaguchi et al. 1997). The full length M2 is a 97-residue protein with a 22-residue N-terminal and a 51-residue C-terminal segment connected by a single TM helix of 24 residues. Its amphipathic helix (residues 47–62) (Schnell and Chou 2008; Tian et al. 2003; Nguyen et al. 2008) is essential for membrane trafficking, localization and viral budding (Ma et al. 2009; Rossman et al. 2010), while the α-helical TM tetramer (residues 25–46) is responsible for the proton conductance that triggers the release of viral RNA into the host cells. This tetrameric TM domain is an important drug target (Hu et al. 2007; Nishimura et al. 2002; Lamb et al. 1985; Grambas et al. 1992) and therefore this domain and constructs including both the TM and amphipathic helix (conductance domain, residues 22–62) have been the subject to a number of high-resolution structural studies in the past decade using both magic angle spinning (MAS) (Cady et al. 2010; Cady et al. 2009) and oriented sample (Du et al. 2012; Hu et al. 2007; Sharma et al. 2010; Wang et al. 2013; Wang et al. 2001) solid-state NMR as well as solution NMR (Schnell and Chou 2008; Pielak et al. 2009; Pielak et al. 2011; Pielak and Chou 2010) and x-ray crystallography (Stouffer et al. 2008; Acharya et al. 2010). In recognition of the importance of environments that support the native structure and function of membrane proteins (Cross et al. 2011; Duerr et al. 2012), solid-state NMR (ssNMR) is increasingly being recognized as an important tool. Additional sparse distance restraints from MAS NMR have also been obtained from samples in lipid environments to validate the tetrameric structure of the TM pore within the conductance domain (Can et al. 2012; Andreas et al. 2010). The observation of resonance pairs for His37 and Trp41 sites as well as others in the TM domain from specific (Hu et al. 2007; Andreas et al. 2010; Andreas et al. 2012) and uniformly (Can et al. 2012; Miao et al. 2012) labeled samples suggests that the structure is a dimer of dimer conformation.
Structural restraints obtained from 13C-13C chemical shift correlation experiments are important building blocks for structural determination. With the development of dipolar recoupling techniques (Detken et al. 2001; Takegoshi et al. 2001; Weingarth et al. 2009) in MAS NMR, spin diffusion is one of the most useful tools for obtaining carbon-carbon distance restraints for protein structural elucidation. In such experiments, a short mixing time (typically a few milliseconds) can generate cross peaks between directly bonded carbons to establish the intra-residue connectivity. However, a much longer mixing time is needed to obtain the correlations between those sterically close carbons from different residues. Since spin diffusion is based on through-space dipolar couplings, both intra- and inter-residue resonances coexist in the same spectrum, with the former giving rise to much stronger correlation signals than the latter, making it extremely difficult to identify those inter-residue resonances from the crowded spectrum. Understanding spin diffusion requires the knowledge of complete coupling networks of the system (Bloembergen et al. 1959; Vanderhart and Garroway 1979; Suter and Ernst 1985; Wei and Ramamoorthy 2001; Xu et al. 2008). Here, we analyze the spin diffusion behavior in simplified coupling systems and introduce difference spectroscopy, i.e. subtraction of two 13C-13C chemical shift correlation spectra at different mixing times, for partial cancellation of the influence of spin diffusion resulting from short-distance spin pairs leading to an aid for the identification of long-range inter-residue resonances, which is essential for structural determinations. Since the structure for the TM domain in the full length M2 protein is virtually the same as in the truncated conductance domain sequence (Miao et al. 2012), spectra of uniformly 13C, 15N-labeled full length M2 protein (M2FL), whose resonance assignments are known from the conductance domain (residues 22–62) (Can et al. 2012), will be used to demonstrate the feasibility of identifying the inter-residue correlation peaks via difference 13C-13C chemical shift correlation spectra.
Materials and methods
M2 Protein Sequence as expressed:
| His Tag: | MHHH-21 HHHSSGVDLG-11 TENLYFQSNA-1 |
| N-terminal Domain: | MSLLTEVETP10 IRNEWGCRSN20 DS |
| TM Helix: | SDPLVVAA30 SIIGILHLIL40 WILDRL |
| Amphipathic Helix: | FFKS50 IYRFFEHGLK60 RG |
| C-terminal Domain: | PSTEGVPE70 SMREEYRKEQ80 QSAVDADDSH90 FVSIELE97 |
Protein expression
Uniformly 13C-labeled glucose and 15N-labeled NH4Cl were purchased from Cambridge Isotope Laboratories, Inc. M2 protein having the amino acid sequence described above was expressed in E. Coli BL21DE3-RPcodon plus strain (Weiner et al. 1994) (Stratagene) using the pTBSG vector. 2 × 50ml of bacterial overnight culture (in LB with 0.1 mg/ml ampicillin) were transferred to 2 × 1L fresh media (LB with 0.1 mg/ml ampicillin) and grown at 37°C in a shaker incubator until the bacteria entered logarithmic growth stage (OD600 ~ 0.8). Cells were spun down by centrifugation at 6000× g for 10 minutes. The cell pellet was transferred into 2 × 1L minimal media (M9 with 4g/L 13C-glucose and 1g/L 15NH4Cl) and grown for 1 hour at 27°C before adding 0.4 mM IPTG to induce protein expression which was carried out for 18 hours at 27°C. Finally, the cells were spun down at 6000× g for 15 minutes and the cell pellet was stored at −80°C. (Sambrook et al. 1989)
Protein purification
Complete cell lysis and solubilization was performed by suspending the cells in 20mM Tris, 500mM NaCl, 3% empigen, 0.1% TritonX-100, 0.8M urea, 0.25mg/mL lysozyme, 0.2mg/mL phenylmethylsulfonyl fluoride, and 1 μl/50mL benzonase nuclease (Novagen), pH 8.0 at 4 °C overnight with agitation. The cell lysate was cleared by centrifugation at 12,000g for 20 minutes and filtered with a 0.2 μm filter before loading onto a 20mL HIS-FastFlow (GE Healthcare) column equilibrated in 20mM Tris, 500mM NaCl, 2.5% empigen, 20mM imidazole, pH 8.0 using an AKTA-FPLC system (GE Healthcare). The column was subsequently washed in three steps with the following buffers: (1) 20mM Tris, 500mM NaCl, 2.5% empigen, 20mM imidazole, pH 8.0, (2) 20mM Tris, 500mM NaCl, 0.7% empigen, 60mM imidazole, pH 8.0, (3) 20mM Tris, 0.5% octyl-β-D-glucopyranoside (OG), pH 8.0. Finally, the protein was eluted with 20mM Tris, 2% OG, 500mM imidazole, pH 8.0. The typical yield is ~40mg/L and the His-tag is not removed.
Protein reconstitution in DOPC/DOPE liposomes
Proteoliposomes containing DOPC/DOPE/M2 in a molar ratio of 4:1:150 were prepared using a detergent dialysis protocol (Basu and Basu 2002). For reconstitution, DOPC and DOPE lipids (Avanti Polar Lipids) were mixed in a round bottom flask in chloroform and the solvent was removed first by flowing N2 gas over the solution and then under high vacuum overnight. The lipid film was suspended in deionized water and sonicated until the solution became clear. The liposome solution was then made 20mM Tris, 2% OG, pH8.0. Purified M2 protein in 2% OG was mixed with this lipid/detergent preparation to a final lipid concentration of 5mM, which was incubated on a shaker bed at room temperature for one hour. The protein-lipid-detergent mixture was then transferred to 3.5 kDa cutoff dialysis bag and dialyzed against 20mM Tris buffer, pH 6.6 at room temperature. Detergent was removed through several rounds of buffer change over 2 days. The final dialysis steps were against 5mM Tris, pH 6.6 containing 2–4g/L of Amberlite to avoid high salt concentration in the liposome pellet and to remove any remaining detergent. The liposome suspension harvested after dialysis was centrifuged at 12,000× g for 20 minutes to remove any precipitate and then sonicated in a bath sonicator for 15 min to reduce the vesicle size and to achieve a more homogeneous liposome population. Finally, the liposome solution was ultracentrifuged at 400,000× g for 6 hours at 5°C producing a wet pellet. The proteoliposome pellet containing approximately 10mg of M2 protein was packed into a 3.2 mm thin-wall MAS rotor (36 μL sample volume).
NMR Spectroscopy
MAS NMR spectra were acquired on a Bruker Avance 600.1 MHz NMR spectrometer using an NHMFL 3.2 mm Low-E triple-resonance biosolids MAS probe (Gor’kov et al. 2007; McNeill et al. 2009). The sample spinning rate was controlled by a Bruker pneumatic MAS unit at 10 kHz±3Hz. The 13C magnetization was enhanced by cross polarization (CP) with a contact time of 1 ms, during which a 1H spin-lock field of 50 kHz was used and the 13C B1 field was ramped from 38 to 56 kHz (Peersen et al. 1994). The 13C 90° pulse length was 3.2 μs. A SPINAL64 decoupling sequence (Fung et al. 2000) with an 1H B1, field of 78 kHz was used during the t1 and t2 dimensions. TPPI was used for quadrature detection in the t1 dimension (Bodenhausen et al. 1980). A DARR irradiation (Takegoshi et al. 2001) was used during 13C-13C mixing time with a 1H B1, irradiation field of 10 kHz. The acquisition times for t1 and t2 dimensions were 6.5 and 10.3 ms, respectively. The data were zero-filled to a 2048 × 2048 matrix before Fourier transform and were processed with a Gaussian window function (LB=−50 Hz and GB=0.15) in both dimensions. The 13C chemical shifts were referenced to the carbonyl carbon resonance of glycine at 178.4 ppm relative to TMS.
Results and Discussions
13C-13C chemical shift correlation spectra
Fig. 1 shows the 13C-13C DARR spectra of the 13C,15N uniformly labeled full length M2 protein in DOPC/DOPE lipid bilayers at pH 6.6 using different mixing times (10, 50, and 200ms). For the 10 ms mixing time (Fig. 1A), the cross peaks mostly result from intra-residue resonances. These resonances are almost entirely from the TM and amphipathic helices (residues 23–62, i.e. the conductance domain). The residues for the terminal helices appear to be much broader. In fact, the resonance frequencies in Fig. 1A are so similar to those obtained from the DARR spectra of the M2 conductance domain sequences (Andreas et al. 2010; Can et al. 2012) that the resonance assignments can be transferred from the conductance domain to these spectra of the full length protein. Therefore, we have adopted their assignments, which are primarily restricted to the TM helix. It is clear that the multiple Leu and Ile resonances are not resolved due to the very small dispersion that is typical in TM helices. While frequencies with an error bar of less than a ppm can be assigned to each of these residues it is not possible to identify the inter-residue cross peaks with a sequence specific Leu or Ile based on these uniformly 13C labeled spectra alone. Knowing that the MAS spectral frequencies of the full length and conductance domain samples are the same suggests that the conductance domain structure can be used to help identify the sequence specific cross peak resonances in these DARR spectra.
Fig. 1.
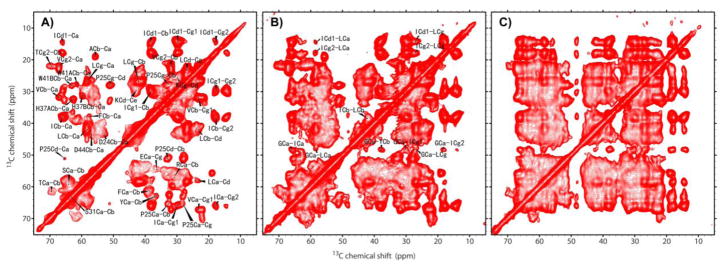
13C-13C chemical shift correlation spectra of uniformly 13C, 15N-labeled full length M2 in DOPC/DOPE liposomes at pH 6.6 using mixing times of (A) 10ms, (B) 50 ms, and (C) 200ms. The assignments were adopted from JACS (Andreas et al. 2010; Can et al. 2012).
At longer mixing times (e.g. 50 and 200 ms), with more inter-residue cross peaks appearing the spectral space in these 2D spectra become highly congested. Another contributing factor to this congestion is the broad resonances, which we anticipate are from the terminal regions. Since the inter-residue and inter-helical cross peaks are typically at longer distances and therefore require longer mixing times to build up, it becomes extremely difficult to identify the resonances. For instance, a small intensity for the inter-residue resonance ICd1-LCa at (58.1, 14.2) ppm can be observed in Fig. 1B owing to the lack of any intra-residue resonances in this region. Only in these regions can inter-residue resonances be easily identified, such as those residues that are in close proximity to the Gly residue (Ca at 48.2 ppm). While in the crowded regions (e.g. the areas between Ca-Ca, Ca-Cb, and Cb-Cb) where the inter- and intra-residue cross peaks are severely overlapped, as shown in Fig. 1B and 1C, it is extremely difficult, if not impossible, to extract the inter-residue cross peaks.
Spin diffusion transfer in simple spin systems
In order to understand the spin diffusion transfer behavior, we consider a simple three-spin system (Ca-Cb-Cg) where the spin diffusion between Ca <-> Cb, Cb <-> Cg, and Ca <-> Cg are characterized by the time constants τsdab, τsdbg, and τsdag, respectively. The proton-driven DARR/PARIS 13C-13C spin diffusion (Takegoshi et al. 2001; Weingarth et al. 2009) is a very complicated process. It highly depends on 13C-13C distances (proportional to 1/r6, where r refers to their inter-nuclear distance) but also is affected by other factors such as proton coupling network, dynamics, proton-proton dipolar interactions and dipolar recoupling efficiencies, etc (Ernst et al. 1989; Khitrin et al. 2011). For simplicity, we use this simplest model and assume that the spin diffusion time is 5 ms for the directly bonded carbons and 130 ms for the two non-bonded carbons based on their relative distances, i.e. τsdab = τsdbg = 5ms and τsdag = 130 ms. Fig. 2A shows the spin diffusion transfer from Ca to Cb (solid blue line) and then to Cg (dashed blue line) as a function of mixing time. Clearly, for the directly bonded Ca-Cb, the Cb magnetization builds up very fast and reaches a plateau at ~20 ms. While the indirect transfer from Ca to Cg (via Cb) is also fast and reaches a plateau at ~50 ms mixing time, even though the distance from Ca to Cg is relatively long owing to the presence of their bridging carbon, Cb. However, in the absence of the bridging carbon Cb, this two non-bonded 13Ca->13Cg transfer is rather slow, as indicated by the green solid line in Fig. 2A, because of their relatively long distance. Generally, this implies that relatively short mixing times allows for establishing the connectivity within each residue (each residue can be considered as a sub spin system) for uniformly 13C labeled samples, while for limited labeling by means of 1,3- and 2-glycerol samples a much longer mixing time is needed even for the connectivity within the same residues.
Fig. 2.

Magnetization buildup in simple spin systems. (A) a three-spin system. (B) a three-spin system with a remotely coupled carbon.
If there are carbons that are in close proximity to this three-spin system, those carbons will also receive the magnetization transfer from Ca through spin diffusion. Again for simplicity, we use Cx to represent a carbon from a different sub spin system in the close proximity, which could be one of the carbons in a nearby residue. Fig. 2B shows the spin diffusion transfer from Ca to Cb (solid blue line) and to Cg (dashed blue line), and then to Cx (solid green line) as a function of mixing time, assuming that the spin diffusion time between Cg and Cx is 300 ms. Apparently, the Cb and Cg magnetization buildup is similar to that in Fig. 2A. However, their magnetization will not remain at the plateau in the presence of Cx. Instead, their magnetization decreases as Cx magnetization slowly builds up, but only reaches 15% intensity at a 100ms mixing time compared to Cb or Cg magnetization. Clearly, the spin diffusion between relatively distant carbons, such as between residues, is inefficient and can only be observed with a long mixing time. However, it is severely obscured by the spin diffusion within residues, making it difficult to identify inter-residue resonances that are not within van der Waals contact.
Difference spectroscopy
It can be rationalized from Fig. 2 that the spin diffusion within a residue is generally very efficient while typically very ineffective between residues. In order to identify the weak interresidue spin diffusion, we introduce difference spectroscopy to reduce the complexity in the spectra by suppressing the spin diffusion resulting from the shorter distance carbon pairs. Fig. 3 shows the difference spectrum between two DARR spectra with 50 and 20 ms mixing times. It is noted that the spectra need to be normalized prior to the difference spectroscopy in order to minimize the T1 relaxation effect during the mixing time, although such an effect is small because the mixing time is usually shorter than the relaxation time. There are two ways to do the normalization: 1) Normalizing the total signal intensity of the first t1 increment in the 2D experiments; 2) Normalizing the specific signal intensity from the processed 2D spectra based on the fact the signal intensity for each resonance should be conserved at different mixing times in the absence of relaxation. In other words, the sum of all signals (the diagonal peak and its cross peaks) along either the t1 or t2 dimension resulting from a specific resonance should remain the same at different mixing times in the absence of relaxation. The latter processing would minimize the influence of the lipid signals, as opposed to the former one. Here, we used the IleCd1 resonance peaks, far away from the lipids resonances, for normalization before performing the difference spectroscopy.
Fig. 3.

Difference spectra of two 13C-13C chemical shift correlation spectra between 50 and 20 ms mixing times of the uniformly 13C,15N-labeled full length M2 in DOPC/DOPE liposomes at pH 6.6.
The resolution in Fig. 3 appears to be greatly enhanced as compared to Fig. 1B. There are three scenarios in the difference spectrum: null, positive (red), and negative (green). Compared to Fig. 1A, all of the one-bond resonances in Fig. 3 have either disappeared or become negative, except those for ACa-Cb that remain positive. These can be understood as follows: One-bond 13C-13C correlation quickly builds up to reach a plateau at ~20 ms and remains unchanged at a longer mixing time, provided that little magnetization transfer to other remote carbons occurs at 50 ms mixing time. In this case, the signals are canceled in the difference spectrum. However, if there are other carbons in close range and the magnetization transferring to those carbons becomes noticeable at this mixing time, these signals at the longer mixing time (e.g. 50ms) become smaller than at the shorter mixing time (e.g. 20ms), resulting in negative peaks in the difference spectrum. For ACa-Cb, there exists the internal rotation in the methyl group resulting in a relatively slow buildup of their correlations, so they cannot reach equilibrium within a 20 ms mixing time and therefore remain as positive signal intensities in the difference spectrum. Among the rest of the positive peaks, many of them are inter-residue correlations with some additional two-bond correlations (ICg2-Cg1, ICd1-Cb, LCd-Cb, ICg2-Ca, ICg1-Ca, VCg-Ca, LCg-Ca, P25Ca-Cg) and three-bond correlations (ICd1-Ca, LCd-Ca). For the diagonal resonances, they always become negative in the difference spectra as long as they participate in spin diffusion.
Therefore, the negative peaks in the difference spectrum indicate that those carbons have spin diffusion transfer to near-by carbons of other residues. It can be noticed from Fig. 3 that the negative peaks are mainly from H, P, L, V, and I residues and many intra-residue cross peaks for P, L, V, and I can be identified from the negative peaks in the difference spectrum, especially in the 20–40 ppm region (e.g. P25Cg-Cb, VCb-Cg2, VCb-Cg1, LCd-Cg) without interference from the diagonal peaks as compared to the spectra in Fig. 1. This also allows the revealing of some inter-residue correlations (e.g. H37BCb-LCg) that are close to the diagonal.
With the known amino acid assignments and the improved resolution in Fig. 3, some sequence specific inter-residue resonances (either intra- or inter-helical) can now be identified as consistent with the structure 2L0J. For instance, the ACb-Ca correlation is now resolved into two peaks: A29 and A30, in the difference spectrum and their correlations with other residues can be traced out (A30Cb-L26Cg, I35Cg1-A30Ca (inter-helical), V28Cb-A29Ca, I32Cb-A29Ca). While the V27 and V28 peaks as well as many L residues in the DARR spectra are severely overlapped (c.f. Fig. 1B), the improved resolution in the difference spectrum in Fig. 3 permits us to unambiguously trace the L26-V27 correlations, e.g. V27Cg2-L26Cb, V27Cb-L26Ca, L26Ca-V27Ca, as indicated by the grey solid lines. While in the Ca-Ca region, many inter-residue resonances are well resolved in the difference spectrum compared to the highly congested DARR spectra (Fig. 1B, C).
Various schemes have been proposed to simplify the crowded 13C-13C chemical shift correlation spectra of membrane proteins either through labeling strategies (specific labeling, reverse labeling, glucose labeling, or glycerol labeling) (Castellani et al. 2002; Higman et al. 2009; Habenstein et al. 2013) or via spectroscopic methods (Traaseth and Veglia 2011). The difference spectroscopy proposed here simplifies the spectra by suppressing the intra-residue resonances allowing for a better identification of the inter-residue resonances. However, a complete suppression of the intra-residue resonances in a difference spectrum is impossible, since the spin diffusion process and distances varies for the residues in a protein sequence. Thus, there is a need to collect several difference spectra using different mixing times in order to gain more structural information. Difference spectroscopy has been widely used in both solution (Ernst et al. 1989) and solid-state NMR (Gopinath et al. 2010), where the most common applications are to select two quadrature components in the indirect dimension through editing (sum and difference) two data sets for sensitivity enhancement. In our case, we simply subtract two 13C-13C chemical shift correlation spectra at different mixing times.
It is interesting to note from Fig. 3 that both of the His37 resonances, i.e. H37ACa-Cb at (62.6, 32.4) ppm and H37BCa-Cb at (59.7, 31.2) ppm, show negative intensities, indicating that some carbons from neighboring residues are in their close ranges. Fig. 4 shows their intensities as a function of mixing time. Obviously, the intensity for H37BCa-Cb is noticeably more intense than H37ACa-Cb at 10 ms mixing time but decays faster with the increase of mixing time. This supports the suggestion of two conformations/charged states of H37 in the H37 tetrad with H37B carrying more charge under this condition since more protons in its close proximity would strengthen the coupling network giving rise to more effective spin diffusion.
Fig. 4.

Plot of His37 Ca-Cb cross peak intensities versus the mixing time in the DARR 13C-13C chemical shift correlation spectra. An exponential decay was used to fit these data, yielding a decay rate of 36.5 and 43.7 ms for H37ACa-Cb and H37BCa-Cb, respectively.
Conclusion
Difference spectroscopy has been proposed to enhance spectral resolution to discriminate intra-and inter-residue correlations in the long mixing time 13C-13C chemical shift correlation spectra in solid-state MAS NMR of membrane proteins. The idea is to partially suppress the spin diffusion contributions from relatively short distance intra-residue carbons, leading to a better identification of long distance inter-residue resonances. This enhanced resolution allows us to obtain long-range distance restraints in crowded 2D 13C-13C correlation spectra based on the known amino acids assignments but without the involvement of the third 15N dimension. By using the uniformly 13C,15N –labeled full length M2 protein in DOPC/DOPE lipid bilayers at pH6.6 as an example, we have identified many of the inter-residue resonances for the conductance domain of the full length M2 protein, which agree very well with those previously assigned resonances from truncated M2 sequences obtained by using specific labelings or by time-consuming three-dimensional NCOCX/NCACX experiments. Thus, the difference spectroscopy offers an opportunity to make sequential 13C assignments without involving nitrogen dimension, which could be very useful and complementary to other multidimensional methods for structural determination of membrane proteins, particularly when the 15N resolution is limited.
Acknowledgments
This work was supported by NIH Grant AI23007. All NMR experiments were carried out at the National High Magnetic Field Lab (NHMFL) supported by the NSF Cooperative agreement N. DMR-0654118 and the State of Florida.
References
- Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci USA. 2010;107 (34):15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A. J Am Chem Soc. 2012;134 (17):7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas LB, Eddy MT, Pielak RM, Chou J, Griffin RG. Magic angle spinning NMR investigation of influenza A M2 (18–60): support for an allosteric mechanism of inhibition. J Am Chem Soc. 2010;132 (32):10958–10960. doi: 10.1021/ja101537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SC, Basu M. Liposome methods and protocols. Methods Mol Biol. 2002:199. [Google Scholar]
- Bloembergen N, Shapiro S, Pershan PS, Artman JO. Cross-relaxation in spin systems. Phys Rev. 1959;114 (2):445–459. [Google Scholar]
- Bodenhausen G, Void RL, Vold RR. Multiple quantum spin-echo spectroscopy. J Magn Reson. 1980;37 (1):93–106. [Google Scholar]
- Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol. 2009;385 (4):1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463 (7281):689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can TV, Sharma M, Hung I, Gor’kov PL, Brey WW, Cross TA. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza A M2 protein functional insights. J Am Chem Soc. 2012;134 (22):9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420 (6911):98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- Cross TA, Sharma M, Yi M, Zhou H-X. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36 (2):117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detken A, Hardy EH, Ernst M, Kainosho M, Kawakami T, Aimoto S, Meier BH. Methods for sequential resonance assignment in solid, uniformly C-13, N-15 labelled peptides: Quantification and application to antamanide. J Biomol NMR. 2001;20 (3):203–221. doi: 10.1023/a:1011212100630. [DOI] [PubMed] [Google Scholar]
- Du J, Cross TA, Zhou H-X. Recent progress in structure-based anti-influenza drug design. Drug Discov Today. 2012;17 (19–20):1111–1120. doi: 10.1016/j.drudis.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr UHN, Gildenberg M, Ramamoorthy A. The Magic of Bicelles Lights Up Membrane Protein Structure. Chem Rev. 2012;112 (11):6054–6074. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst RR, Bodenhausen G, Wokaun A. Principles of nuclear magnetic resonance in one and two dimensions. Clarendon Press; Oxford: 1989. [Google Scholar]
- Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142 (1):97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Gopinath T, Verardi R, Traaseth NJ, Veglia G. Sensitivity enhancement of separated local field experiments: application to membrane proteins. J Phys Chem B. 2010;114 (15):5089–5095. doi: 10.1021/jp909778a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gor’kov PL, Chekmenev EY, Li C, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J Magn Reson. 2007;185 (1):77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Grambas S, Bennett MS, Hay AJ. Influence of amantadine resistance mutations on the pH regulatory function of the M2-protein of influenza-A viruses. Virology. 1992;191 (2):541–549. doi: 10.1016/0042-6822(92)90229-i. [DOI] [PubMed] [Google Scholar]
- Habenstein B, Loquet A, Giller K, Becker S, Lange A. Structural characterization of supramolecular assemblies by 13C spin dilution and 2D solid-state NMR. J Biomol NMR. 2013;55:1–9. doi: 10.1007/s10858-012-9691-9. [DOI] [PubMed] [Google Scholar]
- Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, van Rossum B-J, Oschkinat H. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively C-13-labelled proteins. J Biomol NMR. 2009;44 (4):245–260. doi: 10.1007/s10858-009-9338-7. [DOI] [PubMed] [Google Scholar]
- Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys J. 2007;92 (12):4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khitrin AK, Xu JD, Ramamoorthy A. Cross-correlations between low-gamma nuclei in solids via a common dipolar bath. J Magn Reson. 2011;212 (1):95–101. doi: 10.1016/j.jmr.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA, Zebedee SL, Richardson CD. Influenza virus-M2 protein is an integral membrane-protein expressed on the infected-cell surface. Cell. 1985;40 (3):627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- Ma C, Polishchuk AL, Ohigashi Y, Stouffer AL, Schoen A, Magavern E, Jing X, Lear JD, Freire E, Lamb RA, DeGrado WF, Pinto LH. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci USA. 2009;106 (30):12283–12288. doi: 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill SA, Gor’kov PL, Shetty K, Brey WW, Long JR. A low-E magic angle spinning probe for biological solid state NMR at 750 MHz. J Magn Reson. 2009;197 (2):135–144. doi: 10.1016/j.jmr.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA. M2 proton channel structural validation from full-length protein samples in snthetic bilayers and E. coli membranes. Angew Chem. 2012;51 (33):8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PA, Soto CS, Polishchuk A, Caputo GA, Tatko CD, Ma C, Ohigashi Y, Pinto LH, DeGrado WF, Howard KP. pH-induced conformational change of the influenza M2 protein C-terminal domain. Biochemistry. 2008;47 (38):9934–9936. doi: 10.1021/bi801315m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Kim SG, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41 (44):13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain C-13 and N-15 signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 tesla. Chembiochem. 2001;2 (4):272–281. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Peersen OB, Wu XL, Smith SO. Enhancement of CP-MAS signals by variable-amplitude cross-polarization - compensation for inhomogeneous B-1 fields. J Magn Reson A. 1994;106 (1):127–131. [Google Scholar]
- Pielak RM, Chou JJ. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem Biophys Res Commun. 2010;401 (1):58–63. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak RM, Oxenoid K, Chou JJ. Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. Structure. 2011;19 (11):1655–1663. doi: 10.1016/j.str.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA. 2009;106 (18):7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT- independent membrane scission. Cell. 2010;142 (6):902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Ando I, Ramamoorthy A. Chemical shift tensor - The heart of NMR: Insights into biological aspects of proteins. Prog In NMR. 2010;57 (2):181–228. doi: 10.1016/j.pnmrs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Tu QA, Pinto LH, Lamb RA. The active oligomeric state of the minimalistic influenza virus M-2 ion channel is a tetramer. Proc Natl Acad Sci USA. 1997;94 (10):5000–5005. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. 1, 2, and 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York, USA: 1989. [Google Scholar]
- Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451 (7178):591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou H-X, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330 (6003):509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451 (7178):596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza-A viruses - evidence that it forms a tetrameric channel. Virology. 1991;180 (2):617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter D, Ernst RR. Spin diffusion in resolved solid-state NMR-spectra. Phys Rev B. 1985;32 (9):5608–5627. doi: 10.1103/physrevb.32.5608. [DOI] [PubMed] [Google Scholar]
- Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344 (5–6):631–637. [Google Scholar]
- Tian CL, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Prot Sci. 2003;12 (11):2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Veglia G. Frequency-selective heteronuclear dephasing and selective carbonyl labeling to deconvolute crowded spectra of membrane proteins by magic angle spinning NMR. J Magn Reson. 2011;211 (1):18–24. doi: 10.1016/j.jmr.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderhart DL, Garroway AN. C-13 NMR rotating frame relaxation in a solid with strongly coupled protons - polyethylene. J Chem Phys. 1979;71 (7):2773–2787. [Google Scholar]
- Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, Degrado WF. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci USA. 2013;110 (4):1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Prot Sci. 2001;10 (11):2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YF, Ramamoorthy A. 2D 15N-15N isotropic chemical shift correlation established by 1H-1H dipolar coherence transfer in biological solids. Chem Phys Lett. 2001;342 (3–4):312–316. [Google Scholar]
- Weiner MP, Anderson C, Jerpseth B, Wells S, Johnson-Browne B, Vaillancourt P. Studier pET system vectors and hosts. Strategies Mo Bio. 1994;7 (2):41–43. [Google Scholar]
- Weingarth M, Demco DE, Bodenhausen G, Tekely P. Improved magnetization transfer in solid-state NMR with fast magic angle spinning. Chem Phys Lett. 2009;469 (4–6):342–348. [Google Scholar]
- Wylie BJ, Franks WT, Rienstra CM. Determinations of N-15 chemical shift anisotropy magnitudes in a uniformly N-15, C-13-labeled microcrystalline protein by three-dimensional magic-angle spinning nuclear magnetic resonance spectroscopy. J Phys Chem B. 2006;110 (22):10926–10936. doi: 10.1021/jp060507h. [DOI] [PubMed] [Google Scholar]
- Xu J, Struppe J, Ramamoorthy A. Two-dimensional homonuclear chemical shift correlation established by the cross-relaxation driven spin diffusion in solids. J Chem Phys. 2008;128 (5):052308. doi: 10.1063/1.2826323. [DOI] [PubMed] [Google Scholar]