

Abstract
Objective
Phenobarbital is the first-line treatment for neonatal seizures. Many neonates with hypoxic ischemic encephalopathy (HIE) are treated with therapeutic hypothermia (TH) and about 40% have clinical seizures. Little is known about pharmacokinetics of phenobarbital in infants with HIE who undergo TH. The objective of this study was to determine the effect of TH on phenobarbital pharmacokinetics, taking into account maturational changes.
Setting
Level 3 neonatal intensive care unit.
Patients
Infants with HIE and suspected seizures, all treated with phenobarbital. Some of these infant also received treatment with TH.
Interventions
None.
Design
A retrospective cohort study of 39 infants with HIE treated with phenobarbital (20 were treated with TH and 19 were not).
Measurements and main results
Data were collected on phenobarbital plasma concentrations on 39 subjects with HIE with or without TH. Using nonlinear mixed-effects modeling, population pharmacokinetics of phenobarbital were developed with a total of 164 plasma concentrations. A one-compartment model best described the pharmacokinetics. The clearance (CL) of phenobarbital was linearly related to body weight and matured with increasing age with a maturation half-life of 22.1 days. TH did not influence the pharmacokinetic parameters of phenobarbital.
Conclusions
TH does not influence the CL of phenobarbital after accounting for weight and age. Standard phenobarbital dosing is appropriate for initial treatment of seizures in neonates with HIE treated with TH.
Keywords: phenobarbital, pharmacokinetics, neonates, seizures, therapeutic hypothermia, neonatal encephalopathy
INTRODUCTION
Clinical seizures occur in up to 40% of neonates with hypoxic-ischemic encephalopathy (HIE) [1, 2]. These seizures have been associated with increased mortality and significant morbidity, including cerebral palsy, epilepsy, and long-term functional deficits in memory, behavior, and cognition [3]. Phenobarbital is the most common initial treatment for neonatal seizures [4, 5], although it has only modest efficacy [6]. Phenobarbital has been shown to induce inappropriate apoptosis in the neonatal rodent brain [7], raising concerns regarding the long term effects of this medication on the developing human brain.
Many neonates with HIE are treated with therapeutic hypothermia (TH) as a neuroprotection strategy. There is clinical and experimental evidence that moderate cooling after acute perinatal HIE can improve medium-term neurological recovery [1, 2, 8]. In neonates treated with TH, seizures are a predictor of adverse outcome [3]. Since most infants with HIE and seizures are treated with phenobarbital, there is a pressing need to understand the factors which affect the variability of phenobarbital pharmacokinetics (PK) and the impact of TH on the disposition of phenobarbital in this population.
Hypothermia is associated with physiologic changes that affect many organ systems, including the liver and the kidney. Thus, there is the potential for altered PK and pharmacodynamics of various drugs used during TH [9]. TH alters P450 enzyme activity in animal models [10] and there is evidence of altered enzyme activity in humans as well. For example, morphine plasma concentrations are significantly higher in newborn human infants who undergo TH than in normothermic controls[11]. These findings suggest that the TH may affect the capability of liver to metabolize drugs. Therefore, it is plausible that phenobarbital, which is metabolized by the cytochrome P450 system in the liver would be affected by TH [12-14]. However, little is known about impact of TH on phenobarbital disposition in neonates with HIE. The purpose of this study was to identify and quantify the effects of TH and patients’ maturational changes on phenobarbital PK in neonates with HIE, using a population PK model.
METHODS
Case identification and data collection
This study was approved by the Institutional Review Board at the University of Michigan. The Vermont-Oxford Database for our neonatal intensive care unit (NICU) and pediatric neurology consultation logs from January 2002 through June 2007 were reviewed for patient identification. 215 neonates of ≥36 weeks gestation who were treated for seizures with phenobarbital were identified. Controls were newborn infants ≥36 weeks gestation who were diagnosed with HIE and were treated with phenobarbital for seizures but did not undergo therapeutic hypothermia. Cases were infants with HIE complicated by seizures, who were treated with therapeutic hypothermia according to standard, published protocols [1, 2].
To ensure maximal clinical applicability, data for the TH subjects were derived from both the cooling and rewarming periods (i.e. temperature <37°C). We used 37°C as the upper limit in the TH group’s included data, which is slightly higher than the 36.5°C designated in the published protocols, because of the differences in the procedures for rewarming. For selective head cooling, systemic temperature control during rewarming is not actively regulated by the cooling device. The head cooling protocol uses a radiant warmer and the infant’s own thermogenesis to rewarm. Therefore, there is potential for less-controlled rewarming and subjects often attain somewhat higher than the goal temperature. Conversely, the whole body hypothermia rewarming protocol utilizes active regulation by the servo-controlled cooling device throughout the rewarming period.
Target hypothermia temperatures are somewhat different in the selective head cooling and whole-body hypothermia protocols (rectal temperature of 34.5°C ±0.5 °C versus esophageal temperature 33.5°C ±0.5 °C, respectively). However, published data suggest that the effect on multiorgan system dysfunction is similar for both methods of cooling [15]. Since end-organ function is most relevant to phenobarbital metabolism, it is rational to include both types of TH patients in a single category.
From these patients’ medical records, all phenobarbital plasma concentrations and doses (including time and date) were recorded. The temperature documented in the medical records nearest to the time plasma concentrations were measured was recorded. Demographic information, including gender, birth weight, gestational age, Apgar scores, liver function tests, and clinical diagnoses were also recorded. Liver function tests were often measured repeatedly in these infants; results nearest to the time of seizure diagnosis are reported. Descriptive statistics were used to compare the demographic and population parameters between the two groups.
Population pharmacokinetics
The population PK parameters for phenobarbital were derived from routine clinical data using nonlinear mixed-effects modeling (NONMEM® version 7.2, ICON Development Solutions, Ellicott City, MD, USA). The first-order conditional estimation (FOCE) method with interaction was used throughout the model-building procedure. Two different basic structural models, a one- and two-compartmental linear PK model with IV infusion, were fit to phenobarbital concentration-time data. An exponential variance model was used to describe the variability of PK parameters across individuals in the form:
in which Pi is the estimated parameter value for the individual subject i; θj is the typical population value of PK parameter j; ηji is the inter-individual random effect for individual i and parameter j. Models were explored with various inter-individual random effect covariance structures. Additive, proportional, and combined (additive and proportional) residual error models were considered during the model-building process.
After the PK structural model was selected, covariate models were built using a 3-step approach. First, the impact of weight (body size) on all pharmacokinetic parameters was investigated using an allometric model:
in which TVP is the typical value of a PK parameter, θTVP is an estimated parameter describing the typical PK parameter value for an individual with weight equal to the reference weight, WTref ; WTi is the body weight of individual subject i; and θa is an allometric power parameter. During the model-building process, the median weight of the studied population (3.47kg) was used as WTi and standard 70kg was used as WTref; θa was either estimated or fixed at a value of 0.75 for clearance and a value of 1 for >volumes on the basis of physiologic consideration of size impact on metabolic process[16, 17].
After the effects of weight were accounted for in the model, a model for other biological covariates (i.e., gestational age, Apgar scores, and liver function tests) was built using a stepwise selection approach. The biological covariates that resulted in the greatest statistically significant decrease in the values of objective function were added to the model, and the entire procedure was repeated stepwise until all significant covariates were included in the model (forward selection). Then the biological covariates from the full model were removed one by one and the covariates whose removal resulted in statistically significant increases in objective function were retained in the model (backward elimination). Several covariate models, including linear model and saturation model with baseline and continuous post-natal age (PNAc), were used to explore the age-related changes in PK parameters[16].
After the effect of the biological covariates was incorporated in the model, the effect of TH on PK parameters was tested to yield the final model. This sequential approach was taken to distinguish any changes in PK parameters related to weight and other biological covariates from changes resulting from TH.
Comparison of alternative structural models and construction of the covariate model were based on the typical goodness-of-fit diagnostic plots and likelihood ratio tests. When comparing alternative nested models, the differences in the values of the objective function are approximately in a chi-square distributed with n degrees of freedom (n is the difference in the number of parameters between the models). To discriminate two nested models and select significant covariates, a difference in an objective function of greater than 6.64 (1 degree of freedom), which corresponds to a significant level of p<0.01, was used. Akaike criterion (AIC) was used to select non-nested models.
A bootstrap resampling technique was used to evaluate the stability of the final model and estimate the confidence interval of parameters. This model evaluation technique consists of repeatedly fitting the model to bootstrap replicates of the data set and parameters estimates for each of the replicate data sets were obtained. The results from 1000 successful runs were obtained, and the mean and 2.5th and 97.5th percentiles (denoting the 95% confidence interval) for the population parameters were determined and compared with the estimates of the original data.
RESULTS
Each of the 39 identified neonates with HIE received phenobarbital as treatment for their seizures. Twenty received TH (6 whole body hypothermia[2] and 14 selective head cooling[1]) and were included as cases. The remaining 19 subjects with HIE, who did not receive TH, were included as normothermic controls.
There was no difference in gender distribution, gestational age, birth weight, 5-minute Apgar scores, or aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels between the TH subjects and the normothermic controls (Table 1). As expected, the mean body temperature recorded at the time phenobarbital concentration was measured was lower in the TH group, compared to controls (p<0.0001;Figure 1).
Table 1.
Demographic data
|
Hypothermia (n=20) N(%) or Mean±S.D. |
Normothermia (n=19) N(%) or Mean±S.D. |
p-value | |
|---|---|---|---|
| Gender | |||
| Male | 14 (70%) | 10 (53%) | 0.27a |
| Female | 6 (30%) | 9 (47%) | |
| Gestational Age | 39.5 weeks±1.7 | 38.7±1.8 | 0.16b |
| Birth Weight | 3493g±578 | 3450g±670 | 0.83b |
|
5-minute Apgar
|
3; 2 | 4; 2 | 0.60d |
| Score c | |||
| ALT | 83.9±93.4 | 105.4±113.6 | 0.53b |
| AST | 276.8±283.4 | 272.5±367.1 | 0.97b |
| Temperature e | 34.95°C±1.75 | 36.80°C±0.49 | <0.0001b |
Chi-Square Test
Two-tailed two sample t-test
Median; interquartile range
Wilcoxon two-sample test
Temperature recorded at the time nearest to phenobarbital concentration measurement
S.D. = standard deviation
ALT= alanine aminotransferase
AST= aspartate aminotransferase
Figure 1.
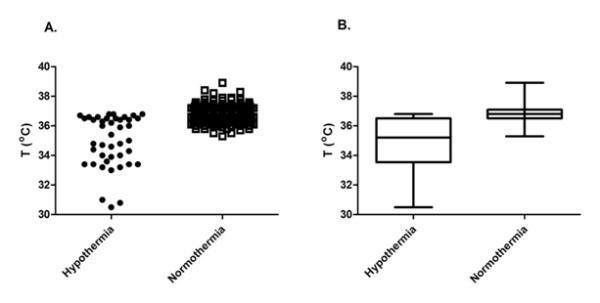
Temperatures recorded nearest to the phenobarbital plasma concentration measurements are presented for the hypothermia and normothermia (control) groups. Panel A demonstrates the range of temperatures at which the phenobarbital concentrations were sampled. In the hypothermia group, there are three groups of several data points: there is a cluster less than 32°C, suggesting over-cooling that may have occurred during the initial phases of hypothermia (for example during transport); there are many data points between 33°C and 35°C, implying the interval during hypothermia in our NICU; finally, there are data points above 36°C, which represent the rewarming period. Panel B displays boxplots of the same temperature data. The length of the boxes represents the interquartile range, while the horizontal line in the box is the median value and the whiskers indicate the minimum and maximum data points.
Pharmacokinetic and Statistical Analysis
A total of 164 phenobarbital concentration data from 39 subjects with HIE were included in the analysis. Both one- and two-compartment models were tested initially as base models, but the two-compartment model did not provide a better fit for the data based on change of objective function and diagnostic plots. The removal of inter-individual variability terms of volume of distribution (Vc) did not result in a statistically significant increase in the objective function (p>0.01). Therefore, only the inter-individual variability term of clearance (CL) was retained.
Among several residual error models tested, the combined proportional and additive error model best described the data. Hence, the best structural model for phenobarbital in the studied population is one-compartment linear PK model with an exponential error model for inter-individual variability of CL and combined proportional and additive error model for the residual variability. This final structural model was then used to build the final model with covariates.
To assess the effect of body weight on the CL of phenobarbital, several different covariate models were explored, as outlined in the methods section. However, the ability of these models to describe the relationships between PK parameters and body weight were very similar and could not differentiate from each other based on likelihood ratio test and AIC model selection criteria. In order to develop a parsimonious model, the effects of body weight and CL was described in the model with WTref of 70Kg and allometric scaling coefficients (to account for body size) were fixed at 0.75 for CL. The allometric scaling coefficient was fixed at 1.00 for Vc to describe the effect of body weight on Vc.
Once the effects of body weight on PK parameters were accounted for in the model, a stepwise approach was used to examine the effects of other biological covariates on the PK parameters of phenobarbital. PNAc was the only biological covariate that had a significant effect on the CL of phenobarbital in neonates with HIE. The relationship between the PNAc and CL was best described by the sigmoidal saturation model with PNAc and slope factor fixed to one. TH treatment did not have significant effects on CL and Vc of phenobarbital after the effects of body weight and PNA were included in the model. Therefore, the final model was as follows:
Final parameter estimates, inter-individual variability, and residual variability are represented in Table 2, with the respective standard errors of the point estimates. The typical CL and Vc were 0.672 L/hr and 64.9L, respectively for an adult weighting 70 kg. With allometric scaling, this would translate to a CL of 0.0241 L/hr (0.0069 L/kg/hr) and Vc of 3.22L (0.92 L/kg) for neonates with median body weight of 3.47 kg and median PNAc of 13.1 days in the studied population. PNAc50, a maturation half-time, was 22.1 days, indicating that the phenobarbital clearance was expected to reach 50% and 90% of the adult level at 22.1 days (3.16 weeks) and 198 days (28.4 weeks), respectively (Figure 2). The inter-individual variance was 0.175 for CL.
Table 2.
Final PPK Model and Population Parameter Estimates 509 from original data and bootstrap parameter estimates obtained from bootstrap procedure
| Original Data Set | 1000 Bootstrap Replicates | |||
|---|---|---|---|---|
| Estimate | SE | Mean | 95% CI | |
| Structural Model CL (L/hr) Vc (L) |
0.672 64.9 |
0.177 2.05 |
0.665 64.7 |
0.388, 0.958 58.3, 72.2 |
| Inter-individual Variability ω2CL ω2Vc |
0.175 - |
0.0616 - |
0.180 - |
0.0175, 0.469 - |
| Covariate Model WT Effect on CL PNAc50 on CL WT Effect on Vc |
0.75 (Fixed) 22.1 1.0 (Fixed) |
- 8.50 - |
- 21.6 - |
- 8.61, 36.5 - |
| Residual Variability Proportional error σprop, Additive error |
0.197 6.12 |
0.0194 0.830 |
0.187 5.60 |
0.101, 0.258 1.19, 7.61 |
SE = standard error of estimates
CL = clearance
Vc = volume of distribution
WT = body weight
ω2CL = interindividual variance of CL
ω2Vc = interindividual variance of Vc
PNAc50 = the postnatal age value at which clearance reaches half its maximal value.
CI = confidence interval.
Figure 2.
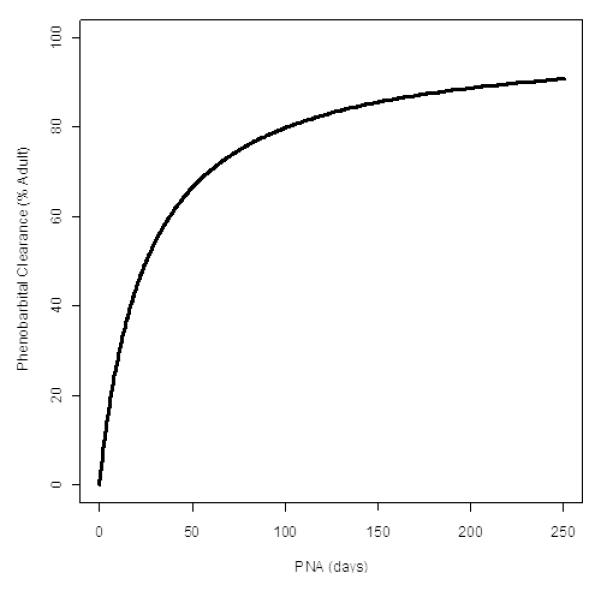
Maturational changes in phenobarbital clearance with postnatal age (PNA) are presented for a reference subject with median body weight of 3.47 kg, based on data obtained from the studied population.
The ratio between the inter-subject variance from the model with covariates to the model without covariates gives an indication of how important covariate information is in explaining the population variability of the PK parameters. For example, the ratio of 0.368 achieved for clearance in this study indicates that (1 – 0.368)*100 = 63.2% of the overall variability in clearance is predictable from covariate information. Weight and PNAc explained 14.6% and 44.8% of the inter-individual variability of the phenobarbital clearance, respectively. Weight alone explained 66.7% of the inter-individual variability of the distribution volume for phenobarbital. After adjusting for body weight, PNAc remained a significant effect, with clearance increasing with age. The dependency of CL on PNA in the weight-adjusted covariate model is accounted for in the model. Observed versus individual and standard model diagnostic plots suggest that the model can describe the observed plasma phenobarbital concentrations in hypothermia and normothermia subjects and reveal no major systematic bias in prediction of plasma phenobarbital concentrations (Figures 3 and 4).
Figure 3.
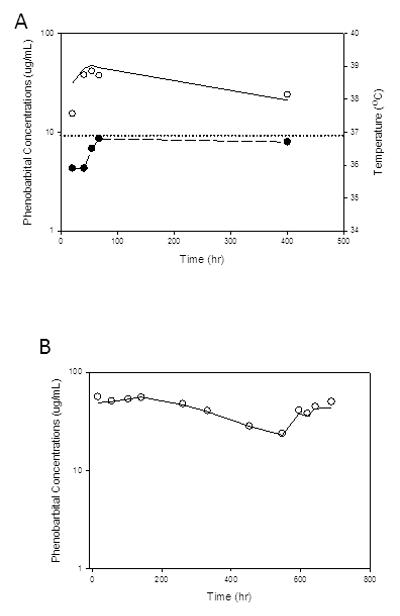
Individual observed phenobarbital concentration-time profiles and model prediction from A) hypothermia and B) normothermia subjects. Open circle – observed phenobarbital concentrations; solid line – model predicted phenobarbital concentrations; solid circle with dashed line – body core temperature of hypothermia subject; dotted line – normal core temperature.
Figure 4.
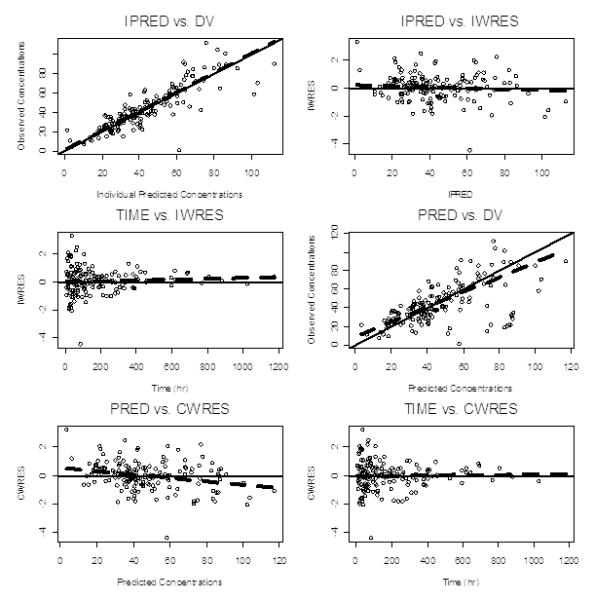
Standard Model Diagnostic Plot. IPERD – individual predicted concentrations; DV – observed concentrations; IWRES – individual weighted residuals; PRED – population predicted concentrations; CWRES – conditional weighted residuals. Standard model diagnostic plots reveal no significant systematic bias in the prediction of plasma phenobarbital concentrations.
From the original data set, 1000 replicate data sets were generated and used for the evaluation of the stability of the final model and the result is shown in Table 2. The mean population parameters obtained from the bootstrap procedure were similar to the parameter estimates of the original data set, again indicating that the developed PK model was stable. The 95% confidence intervals for the PK parameters except inter-individual variance of CL indicated good precision. The relatively large 95% confidence interval for the inter-individual variance of CL from the bootstrap procedure compared to the standard error of estimates of CL obtained from the original data set may be due to presence of an outlier whose CL deviated significantly from the population reference value. Therefore, excluding this subject in some of the replicated data may result in relatively smaller inter-individual variances, as shown in the Table 2.
DISCUSSION
This is the first study evaluating the effect of therapeutic hypothermia on population pharmacokinetics of phenobarbital in newborn infants with HIE. To our knowledge, this is also the first report to describe the age-dependent maturation of the phenobarbital metabolism in this patient population. We demonstrate that therapeutic hypothermia has no effect on the clearance and volume of distribution of phenobarbital in neonates with HIE. This is a reassuring finding for clinicians treating such patients.
Other studies have examined the effect of TH on the pharmacokinetics of morphine and gentamicin in newborn infants [11, 18], but none have employed the population PK approach. These previous studies employed traditional pharmacokinetic methods, which require a homogeneous group of study subjects to decrease inter-individual variation. Such traditional pharmacokinetic studies also require dense plasma concentration sampling (6-10 samples during dosing interval).
In contrast, population PK methods are based on statistical models that allow interpolation of data and provide a better estimation of between-subject and residual variability in drug absorption, distribution, metabolism, and excretion. This technique also allows for a sparse sampling strategy, a property that allows for use of routine clinical data to evaluate drug disposition. This is an obvious advantage in the study of newborn infants, for whom dense sampling raises concerns about the volume of blood required for the study.
Morphine levels were found to be significantly higher in cooled newborn human infants (n=10) than in normothermic controls with HIE (n=6) who were treated with the same morphine infusion regimen[11]. In that study, subjects treated with TH were much more likely to achieve potentially toxic morphine levels (>300ng/mL) than normothermic controls (13/42 blood samples versus 2/28 samples, p<0.025) and the mean serum morphine concentration at 72 hours of life was nearly 60% higher among the hypothermia group than the controls (373ng/mL vs. 222ng/mL, p=0.02), although the morphine clearance was not different between the two groups. In contrast, there was no significant difference in the metabolism of gentamicin in newborn infants or piglets treated with TH compared to those who were not [18, 19].
It might be expected that hypothermia would have a different effect on the metabolism of morphine, a hepatically metabolized medication, than gentamicin, a renally cleared drug. However, we did not detect any significant effect of therapeutic hypothermic on the clearance of phenobarbital. Morphine is largely metabolized by uridine 5′-diphosphate UGT2B7 to morphine-3-glucuronide and morphine-6-glucuronide [20]. Phenobarbital is eliminated both by hepatic metabolism and renal excretion and the main metabolic pathway is aromatic hydroxylation to form p-hydroxyphenobarbital by P450 CYP2C isoenzymes [12]. Therefore, it is possible that the therapeutic hypothermia may have different effects on the different metabolizing enzymes in the liver that mediate drug clearance.
The most important biological co-variates for predicting phenobarbital clearance in our patient population were body size (weight) and postnatal age. Each of these factors is related to the stage of development of newborn infants. The final model suggested that clearance of phenobarbital increases proportionately with increasing body weight. Furthermore, the model indicated that the phenobarbital clearance gradually reaches the adult level with increasing postnatal age and the older newborn is expected to have a higher rate of clearance and hence lower elimination half-life than a younger newborn of equal body weight. This finding is consistent with prior studies of phenobarbital, which showed that the half-life of normothermic newborns decreases with increasing age [21-23].
The mean population CL among our study subjects with median weight of 3.47 kg and PNAc of 13.1 days was 7.6 mL/kg/hr and the elimination half-life was 84.9 hours. The reference range for phenobarbital half-life in neonates reported in the literature is wide (45-500 hours)[24]. Grasela & Donn [25] examined phenobarbital population PK in a cohort of premature and term newborns treated with phenobarbital for various indications. Their results were: phenobarbital elimination half-life 141 hours, and CL 4.7 mL/kg/hr. Although the results from the Grasela & Donn study are somewhat different than ours, many of their study subjects were born prematurely and most were administered phenobarbital for prevention of intraventricular hemorrhage. Therefore, these findings suggest that different disease states, post-conceptional ages, and body size may have significant effects on the phenobarbital clearance.
In a prospective, traditional pharmacokinetics study, Filippi et al [26] assessed phenobarbital plasma concentrations among 19 infants treated with whole body hypothermia for HIE. Their analysis focused on plasma concentrations resulting from different phenobarbital dosing regimens, rather than on the effect of hypothermia on pharmacokinetics, and all of their subjects were co-treated with topiramate. After loading phenobarbital doses of up to 35mg/kg, the first seven infants received 2.5mg/kg maintenance dosing every 12 hours, while the remaining 12 subjects were treated with 1.5mg/kg maintenance dosing every 12 hours. There were no normothermic controls and liver function tests were not reported. Plasma phenobarbital levels were drawn 27 times during the 72 hours of therapeutic hypothermia, with four levels drawn immediately before maintenance doses. Using these very different methods from the present report, the authors report a phenobarbital half-life of 173.9 ±62.5 hours; CL 6.38 ±2.7 mL/kg/hr; and volume of distribution 1.56 ±1.0 L/kg. In our study a typical neonate with HIE had a phenobarbital elimination half-life of 84.9 hours, CL of 7.6 mL/kg/hr, and volume of distribution 0.92 L/kg. However, since the methods of the two studies are so different, it is difficult to make direct comparisons.
Due to the retrospective nature of our data and the large inter-individual and intra-individual variations, simulation of appropriate dosage regimens may not be accurate. Based on our results, however, it seems appropriate to initiate treatment of phenobarbital at the currently recommended doses and then individualize the dose based on subsequent plasma concentration measurements to achieve a desired effect.
Because our results were derived from clinically recorded data, we were unable to specifically evaluate the relationship between phenobarbital PK parameters and exact body temperatures, or to correlate them with the timing of phenobarbital doses relative to either age or hours of cooling. Rather than study PPK at specific body temperatures, we grouped the subjects into hypothermia and normothermia categories. Since the half-life of phenobarbital is very long, the effect of TH on its metabolism might not be seen immediately during the cooling phase. Rather, it is possible that the effect could be cumulative over time and might not be seen until rewarming, or even later in the treatment course. Therefore, for this initial study, we sought to include data derived from both the cooling and rewarming phases. Future prospective studies, which would likely need to be designed as nested protocols within large multicenter trials, could benefit from larger sample sizes, such that they would have sufficient data to comment upon the pharmacokinetics at particular temperature ranges.
Our study has some other limitations. The population PK approach is designed to allow for use of routine clinical data to determine drug disposition, so our retrospective design did not significantly detract from the analysis. Because we employed routine clinical data, we were not able to analyze free phenobarbital levels, but only total phenobarbital plasma concentrations. Some prior studies of phenobarbital in newborns with seizures utilized free levels, because of concern about variable protein binding in critically ill newborns[6]. However, the free plasma phenobarbital level is tightly correlated to the total serum concentration [27, 28]. Finally, our study did not include premature infants, so the findings cannot be directly applied to that population.
Like Grasela & Donn [25] and Yukawa, et al, [29] we demonstrated that population PK is a feasible approach for neonates, even in a retrospective study design. . Nevertheless, a prospective study with a large sample size may reduce some of the variability observed in this retrospective study. However, because TH is now the standard of care for HIE, it is unlikely that there will be further opportunities for prospective studies of phenobarbital pharmacokinetics that would include a normothermic HIE control group.
CONCLUSIONS
Based on our results, it appears that neonates with HIE complicated by seizures who undergo TH as a neuroprotection strategy can be treated initially with standard doses of phenobarbital, with further treatment guided by monitoring of treatment response and routine measurement of serum concentrations.
ACKNOWLEDGEMENT
The authors thank Ms. Amanda Barks for her assistance in preparing this manuscript.
FINANCIAL DISCLOSURE: Funding for this research was provided in part by the Clinical and Translational Science Award (UL1RR024986; RAS) and the University of Michigan’s Undergraduate Research Opportunity Program (CHD).
Footnotes
COMPETING INTERESTS: The authors affirm that they have no significant conflict of interest relevant to this manuscript.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663–670. doi: 10.1016/S0140-6736(05)17946-X. [DOI] [PubMed] [Google Scholar]
- 2.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, et al. Whole body hypothermia for neonates with hypoxic-ischemic encephalopathy. New England Journal of Medicine. 2005;353:1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- 3.Wyatt JS, Gluckman PD, Liu PY, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, et al. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119:912–921. doi: 10.1542/peds.2006-2839. [DOI] [PubMed] [Google Scholar]
- 4.Bartha AI, Shen J, Katz KH, Mischel RE, Yap KR, Ivacko JA, Andrews EM, Ferriero DM, Ment LR, FS. S. Neonatal Seizures: Multicenter Variability in Current Treatment Practices. Pediatric Neurology. 2007;37:85–90. doi: 10.1016/j.pediatrneurol.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Blume HK, Garrison MM, Christakis DA. Neonatal seizures: treatment and treatment variability in 31 United States pediatric hospitals. Journal of Child Neurology. 2009;24(2):148–154. doi: 10.1177/0883073808321056. [DOI] [PubMed] [Google Scholar]
- 6.Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, Paneth N, Minnigh B, Alvin J. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. New England Journal of Medicine. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- 7.Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, Dzietko M, Pesditschek S, Mai I, Dikranian K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99(23):15089–15094. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. New England Journal of Medicine. 2009;361:1349–1358. doi: 10.1056/NEJMoa0900854. [DOI] [PubMed] [Google Scholar]
- 9.Zanelli S, Buck M, Fairchild K. Physiologic and pharmacologic considerations for hypothermia therapy in neonates. Journal of Perinatology. 2010:1–10. doi: 10.1038/jp.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tortorici MA, Kochanek PM, Bies RR, Poloyac SM. Therapeutic hypothermia-induced pharmacokinetic alterations in CYP2E1 chlorzoxazone-mediated metabolism in a cardiac arrest rat model. Critical Care Medicine. 2006;34:785–791. doi: 10.1097/01.ccm.0000201899.52739.4f. [DOI] [PubMed] [Google Scholar]
- 11.Roka A, Melinda KT, Vasarhelyi B, Machay T, Azzopardi D, Szabo M. Elevated morphine concentrations in neonates treated with morphine and prolonged hypothermia for hypoxic ischemic encephalopathy. Pediatrics. 2008;121(4):e844–e849. doi: 10.1542/peds.2007-1987. [DOI] [PubMed] [Google Scholar]
- 12.Hvidberg EF, Dam M. Clinical pharmacokinetics of anticonvulsants. Clinical Pharmacokinetics. 1976;1(3):161–188. doi: 10.2165/00003088-197601030-00001. [DOI] [PubMed] [Google Scholar]
- 13.Mann MW, Pons G. Various pharmacogenetic aspects of antiepileptic drug therapy: a review. CNS Drugs. 2007;21(2):143–164. doi: 10.2165/00023210-200721020-00005. [DOI] [PubMed] [Google Scholar]
- 14.Kwan P. Antiepileptic drug research in Asia: where do we go from here? Neurol J Southeast Asia. 2003;8:81–85. [Google Scholar]
- 15.Sarkar S, Barks JD, Bhagat I, Donn SM. Effects of therapeutic hypothermia on multiorgan dysfunction in asphyxiated newborns: whole-body cooling versus selective head cooling. Journal of Perinatology. 2009;29(8):558–563. doi: 10.1038/jp.2009.37. [DOI] [PubMed] [Google Scholar]
- 16.Anderson BJ, McKee AD, Holford NH. Size, myths, and the clinical pharmacokinetics of analgesia in pediatric patients. Clinical Pharmacokinetics. 1997;33:313–327. doi: 10.2165/00003088-199733050-00001. [DOI] [PubMed] [Google Scholar]
- 17.West GB, Brown JH, Enquist BJ. Vancomycin pharmacokinetics in preterm neonates and the prediction of adult clearance. British Journal if Clinical Pharmacology. 1999;63:75–84. doi: 10.1111/j.1365-2125.2006.02725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Borooah M, Stone J, Chakkarapani E, Thoresen M. Serum gentamicin concentrations in encephalopathic infants are not affected by therapeutic hypothermia. Pediatrics. 2009;124:310–315. doi: 10.1542/peds.2008-2942. [DOI] [PubMed] [Google Scholar]
- 19.Satas S, Hoem NO, Melby K, Porter H, Lindgren CG, Whitelaw A, Thoresen M. Influence of mild hypothermia after hypoxia-ischemia on the pharmacokinetics of gentamicin in newborn pigs. Biology of the neonate. 2000;77(1):50–57. doi: 10.1159/000014195. [DOI] [PubMed] [Google Scholar]
- 20.Stone AN, Mackenzie PI, Galetin A, Houston JB, Miners JO. Isoform selectivity and kinetics of morphine and 3- and 6-glucuronidation by human udp-glucuronosyltransferases: evidence for atypical glucuronidation kinetics by UGT2B7. Drug Metab Dispos. 2003;31(9):1086–1089. doi: 10.1124/dmd.31.9.1086. [DOI] [PubMed] [Google Scholar]
- 21.Alonso GAC, Ortega VL, Santos BD, SMJ. G, Santos BJ, Monzon CL, Dominguez-Gil HA. Dosage programming of phenobarbital in neonatal seizures. Journal of Clinical Pharmacy and Therapeutics. 1993;18(4):267–270. doi: 10.1111/j.1365-2710.1993.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 22.Heimann G, Gladtke E. Pharmacokinetics of phenobarbital in childhood. European Journal of Clinical Pharmacology. 1977;12(4):305–310. doi: 10.1007/BF00607431. [DOI] [PubMed] [Google Scholar]
- 23.Pitlick W, Painter M, Pippenger C. Phenobarbital pharmacokinetics in neonates. Clin Pharmacol Ther. 1978;23(3):346–350. doi: 10.1002/cpt1978233346. [DOI] [PubMed] [Google Scholar]
- 24.Takemoto CK, Hodding JH, Kraus DM, editors. The Pediatric Dosage Handbook. 16 edition American Pharmacist Association; Hudson, OH: 2010-2011. [Google Scholar]
- 25.Grasela TH, Donn SM. Neonatal population pharmacokinetics of phenobarbital derived from routine clinical data. Dev Pharmacol Ther. 1985;8:374–383. doi: 10.1159/000457062. [DOI] [PubMed] [Google Scholar]
- 26.Filippi L, la Marca G, Cavallaro G, Fiorini P, Favelli F, Malvagia S, Donzelli G, Guerrini R. Phenobarbital for neonatal seizures in hypoxic ischemic encephalopathy: a pharmacokinetic study during whole body hypothermia. Epilepsia. 2011;52(4):794–801. doi: 10.1111/j.1528-1167.2011.02978.x. [DOI] [PubMed] [Google Scholar]
- 27.Tokugawa K, Fujito H, Kurokawa T. Correlation between the saliva and free serum concentration of phenobarbital in epileptic children. European Journal of Pediatrics. 1986;145:401–402. doi: 10.1007/BF00439247. [DOI] [PubMed] [Google Scholar]
- 28.Nishihara K, Uchino K, Saitoh Y, Honda Y, Nakagawa F, Tamura Z. Estimation of plasma unbound phenobarbital concentration by using mixed saliva. Epilepsia. 1979;20:37–45. doi: 10.1111/j.1528-1157.1979.tb04774.x. [DOI] [PubMed] [Google Scholar]
- 29.Yukawa E, Suematsu F, Yukawa M, Minemoto M. Population pharmacokinetic investigation of phenobarbital by mixed effect modelling using routine clinical pharmacokinetic data in Japanese neonates and infants. Journal of Clinical Pharmacy and Therapeutics. 2005;30:159–163. doi: 10.1111/j.1365-2710.2005.00619.x. [DOI] [PubMed] [Google Scholar]