

Abstract
Protein kinase G (PKG) plays an important role in the regulation of vascular smooth cell contractility and is a critical mediator of nitric oxide signaling, which regulates cardiovascular homeostasis. PKG-I–knockout (Prkg1−/−) mice exhibit impaired nitric oxide/cGMP-dependent vasorelaxation and systemic hypertension. However, it remains unknown whether PKG-I deficiency induces pulmonary hypertension. In this study, we characterized the hypertensive pulmonary phenotypes in Prkg1−/− mice and delineated the underlying molecular basis. We observed a significant increase in right ventricular systolic pressure in Prkg1−/− mice in the absence of systemic hypertension and left-sided heart dysfunction. In addition, we observed marked muscularization of distal pulmonary vessels in Prkg1−/− mice. Microangiography revealed impaired integrity of the pulmonary vasculature in Prkg1−/− mice. Mechanistically, PKG-I–mediated phosphorylation of Rho A Ser188 was markedly decreased, and the resultant Rho A activation was significantly increased in Prkg1−/− lung tissues, which resulted in Rho kinase activation. The i.t. administration of fasudil, a Rho kinase inhibitor, reversed the hypertensive pulmonary phenotype in Prkg1−/− mice. Taken together, these data show that PKG-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation–mediated vasoconstriction and pulmonary vascular remodeling.
Pulmonary hypertension (PH), characterized by progressive increases in pulmonary vascular resistance and vascular remodeling, causes right-sided heart failure and mortality without treatment.1,2 Increased pulmonary vascular tone and severe structural remodeling of distal pulmonary vasculature are primary determinants of increased pulmonary vascular resistance. Because the molecular mechanisms responsible for pulmonary vascular remodeling and vasoconstriction remain elusive, there are limited options available for the prevention and treatment of progressive PH.3,4
Protein kinase G (PKG), a serine/threonine kinase, plays an important role in the regulation of vascular smooth cell contractility.5,6 Activation of soluble guanylyl cyclase by nitric oxide (NO) leads to increased synthesis of cGMP and activation of PKG. The PKG activation results in vasorelaxation via several mechanisms, including lowering of intracellular free Ca2+ levels and desensitization of the contractile apparatus to Ca2+.7,8 Two mammalian Prkg genes have been identified, encoding PKG-I and PKG-II.9 PKG-I is the prevalent PKG isoform expressed in the cardiovascular system, with a greater level in smooth muscle cells (SMCs) and a lower level in endothelial cells.10,11 The N-terminus (the first 90 to 100 residues) of PKG-I is encoded by two alternative exons generating two isoforms: PKG-Iα and PKG-Iβ.9 The enzyme is activated at submicromolar to micromolar concentrations of cGMP. The PKG-I is composed of three functional domains: an N-terminal domain, a regulatory domain, and a catalytic domain. The regulatory domain contains two cGMP-binding sites with high and low affinity. The catalytic domain contains the Mg-ATP– and peptide-binding pockets. Binding of cGMP to the regulatory domain releases the inhibition of the catalytic center by the N-terminal autoinhibitory/pseudosubstrate site and allows the phosphorylation of serine/threonine residues in target proteins.6,12 The PKG-I phosphorylates Rho A at Ser188, which inhibits its membrane association and, thereby, prevents activation of its downstream targets, such as Rho kinase.13,14 Rho kinase–mediated phosphorylation of human myosin phosphatase target subunit 1 at Thr696 (corresponding to Thr694 in mouse) inhibits smooth muscle myosin phosphatase activity and induces SMC contraction.15
PKG-I–knockout (Prkg1−/−) mice exhibit impaired NO/cGMP-dependent vasorelaxation and systemic hypertension in their early life.16–20 These mutant mice also exhibit a decreased life span (50% of these mice die before the age of 5 to 6 weeks),17 diminished ischemia-induced angiogenesis,21 and disturbed platelet adhesion and activation.22 Overexpression of PKG-I increases the sensitivity of cultured vascular SMCs to the anti-proliferative and pro-apoptotic effects of NO/cGMP,23 whereas down-regulation of PKG-I induces the change of the SMC phenotype from a contractile, differentiated to a synthetic, dedifferentiated state,24,25 an underlying feature of pulmonary vascular remodeling. Although PKG dysfunction has been indicated in the pathogenesis of PH, there is no genetic evidence demonstrating that PKG-I deficiency induces PH. Herein, we report that deletion of Prkg1 induces vascular remodeling and vasoconstriction and, thereby, PH in mice.
Materials and Methods
Mice
All mice were bred and maintained at the University of Illinois at Chicago facility, according to NIH guidelines. The generation of Prkg1−/− mice by homologous recombination has been described previously.17 Prkg1−/− mice were obtained from Dr. Xiaoping Du (University of Illinois College of Medicine). Littermates of Prkg1−/− and WT mice in C57BL/6 background were used. Approval for animal care and use in these experiments was granted by the Animal Care and Use Committee.
Primary Cultures of Mouse Pulmonary SMCs
Primary cultures of mouse lung SMCs from wild-type (WT) or Prkg1−/− mice were isolated as previously described.26 Briefly, a 21-gauge needle was inserted into the right ventricle to perfuse the pulmonary circulation with hot HBSS (50°C) and then with 2-μm irons in 0.5% agarose. The lung was then dissected, minced, and collected in a 15-mL tube. After incubation with collagenase A (1 mg/mL in HBSS; Roche Applied Sciences, Indianapolis, IN) at 37°C for 45 minutes with shaking, the rosetted cells were isolated and washed using a magnetic particle concentrator (Dynal MPC-15; Dynal, Oslo, Norway). The freshly dispersed SMCs were plated onto a 60-mm cell culture dish and incubated with Dulbecco's modified Eagle's medium containing 10% newborn calf serum, penicillin (100 U/mL), and streptomycin (100 μg/mL) in a humidified atmosphere of 5% CO2 in air at 37°C and grown to 90% to 95% confluence. These primary SMCs were used at passage 2 for all experiments.
Hemodynamic Measurements
Right ventricular systolic pressure (RVSP) was determined with a 1.4F pressure transducer catheter (Millar Instruments, Houston, TX) and AcqKnowledge software (Biopac Systems Inc., Goleta, CA), as previously described.26 Briefly, the 1.4F pressure transducer was inserted through the right external jugular vein of anesthetized mice (100 mg ketamine/5 mg xylazine/kg body weight, i.p.) and threaded into the right ventricle. RVSP was then recorded and analyzed with AcqKnowledge software. To inhibit Rho kinase, fasudil (60 mmol/L in 25 μL of saline) was administered into each mouse through tracheal instillation. RVSP was monitored continuously for 30 minutes after administration of fasudil. Carotid artery pressure was also determined with a 1.4F pressure transducer catheter.
Echocardiography
Transthoracic two-dimensional targeted M-mode echocardiography was performed with a 15-MHz linear array transducer attached to a commercially available Sequoia C256 system (Siemens, Mountain View, CA). Mice were anesthetized with methoxyflurane (Metofane; Dow Chemical Company, Hawthorn Woods, IL), intubated, and maintained on isoflurane anesthesia. Images of the left ventricle (LV) were taken from the parasternal short-axis view at the level of the papillary muscles. Interventricular septal and LV posterior wall thickness and LV internal dimensions at the end of diastole and systole were measured by the American Society of Echocardiography leading-edge method from at least three consecutive cardiac cycles on the M-mode tracings, with a paper speed of 200 mm/second. The percentage fractional shortening of the LV was calculated from digital images by subtracting the LV systolic internal dimension from the LV diastolic internal dimension and dividing by the LV diastolic internal dimension.
Fluorescent Microangiography
A 1% low melting point agarose (Sigma-Aldrich, St. Louis, MO) solution containing 0.2-μm yellow-green fluorescent microspheres was prewarmed and injected into the pulmonary artery through the left atrial appendage. The lungs were then fixed in 4% paraformaldehyde in PBS for 48 hours at 4°C, and sections (150 to 200 μm thick) were prepared for imaging. Confocal optical sectioning was used to produce a Z-stack of images spanning 150 μm of section depth. Projection images were generated from the Z-series to demonstrate vessel architecture throughout the entire thickness of the section. Fluorescence intensity was quantified in three separate sections per animal and three representative regions per section by a blinded observer. For this analysis, a single confocal optical section was taken at a predefined depth (10 μm) using fixed gain and power settings. The perfusion index was calculated as the area ratio of the image surpassing the threshold value. The perfusion index represents the proportion of the optical section filled by vascular perfusion.27
Immunofluorescent Staining
Lung tissues were fixed for 5 minutes by instillation of 10% PBS–buffered formalin through tracheal catheterization at a transpulmonary pressure of 15 cm H2O, and then overnight at 4°C with agitation.26 After paraffin processing, the tissues were cut into semithin sections (4 to 5 μm thick). For immunofluorescent staining, antigen retrieval was performed by incubating the slides in 10 mmol/L sodium citrate (pH 6.0) at 95°C for 10 minutes. After 1-hour incubation at room temperature in a blocking solution containing 2% bovine albumin serum (Sigma-Aldrich), 0.1% Triton X-100, and 2% normal goat serum, the sections were incubated for 2 hours at room temperature with anti–α smooth muscle actin (α-SMA) monoclonal antibody (1:400; Sigma-Aldrich). Nuclei were counterstained with DAPI. The anti–α-SMA–positive pulmonary arterial vessels per field (original magnification, ×200) were counted based on the diameter (<40 versus >40 μm). Twenty fields per section were randomly identified and counted.
Western Blot Analysis
Western blot analysis was performed using the following antibodies: anti-Rho A phosphorylated Ser188 (1:500; Santa Cruz, Santa Cruz, CA), anti-PKG-I (1:1000; Assay Designs, Inc. Ann Arbor, MI), anti-vasodilator-stimulated phosphoprotein (VASP; 1:1000; Axxora, San Diego, CA), and anti-VASP phosphorylated Ser239 (1:200; Axxora). Anti-β-actin (1:4000; BD Biosciences, San Jose, CA) was used as a loading control. Signals from immunoreactive bands were visualized by fluorography using an enhanced chemiluminescence (ECL) reagent (Thermo Fisher Scientific, Waltham, MA). The intensity of each band was quantified using NIH ImageJ software (Bethesda, MD).
RhoGTPase Activity Assay
The GTP-bound active forms of Rho A were determined by pull-down assay.28 Cells were washed with ice-cold PBS five times and lysed in a lysis buffer (50 mmol/L Tris, pH 7.4; 1% Triton X-100; 0.5% sodium deoxycholate; 0.1% SDS; 500 mmol/L NaCl; 10 mmol/L MgCl2; 10 μg/mL each of aprotinin and leupeptin; and 1 mmol/L phenylmethylsulfonyl fluoride). Similarly, lung tissues were homogenized with the same lysis buffer. After centrifugation at 18,000 × g at 4°C for 2 minutes, the extracts were incubated at 4°C for 60 minutes with glutathione-Sepharose beads coupled with glutathione-S-transferase–rhotekin fusion protein for determination of Rho A activity, following the manufacturer's instructions (Cytoskeleton Inc., Denver, CO). Bound Rho A proteins were quantified by using Western blot analysis, as previously described. Total Rho A levels were also determined by using direct Western blot analysis.
Statistical Analysis
Differences between groups were examined for statistical significance using the Student's t-test. P < 0.05 was considered significant.
Results
PKG-I Deficiency Induces PH in Prkg1−/− Mice
To determine the consequences of genetic deletion of Prkg1, we measured RVSP (to reflect the pulmonary artery systolic pressure) in Prkg1−/− mice. Prkg1−/− mice had significantly increased RVSP compared with WT mice (Figure 1, A–C). Genetic deletion of Prkg1 was confirmed using Western blot analysis (Figure 1D). Accordingly, decreased phosphorylation of VASP, a substrate of PKG-I, was observed in Prkg1−/− lungs compared with WT lungs. These data indicate that PKG-I deficiency induces PH in mice. Because structural remodeling of distal pulmonary arteries is the underlying feature of pulmonary vascular remodeling in the pathogenesis of PH, we stained lung sections with anti–α-SMA and quantified the number of muscularized distal pulmonary vessels. Prkg1−/− lungs exhibited a >10-fold increase in muscularized distal arteries (<40 μm in diameter) compared with WT, whereas a similar number of muscularized large vessels (>40 μm in diameter) was seen in Prkg1−/− and WT lungs (Figure 2, A and B).
Figure 1.
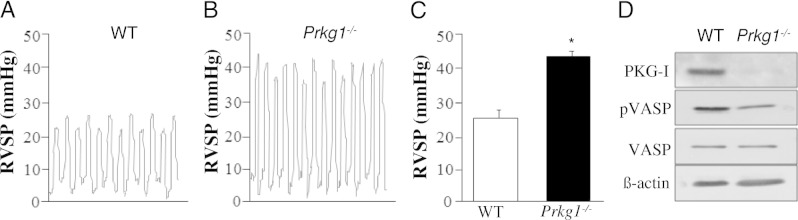
PKG-I deficiency induces PH in Prkg1−/− mice. Representative RVSP tracings in WT (A) or Prkg1−/− mice (B). C: Marked increase of RVSP in Prkg1−/− mice. RVSP was measured in age- and sex-matched 7- to 8-week-old mice. Data are expressed as mean ± SD. *P < 0.01 versus WT (n = 4 to 6). D: Western blot analysis of PKG-I expression and phosphorylation of VASP in lungs from 7-week-old mice. Lung lysate (30 μg per lane) was loaded and immunoblotted with antibodies against PKG-I, phosphorylated VASP (pVASP), VASP, and β-actin (loading control). Consistent with deletion of Prkg1, phosphorylation of VASP was significantly decreased in Prkg1−/− lungs compared with WT lungs.
Figure 2.

Increased muscularization of distal pulmonary arteries in Prkg1−/− mice. A: Representative micrographs of immunostaining of WT and Prkg1−/− lung sections from 7-week-old mice with anti–α-SMA (red). Arrows, muscularized distal pulmonary arterial vessels in Prkg1−/− lung sections. B: Quantification of muscularized pulmonary arterial vessels in mouse lung sections. α-SMA–positive vessels were counted in 20 fields (original magnification, ×200) of each lung section. Data are expressed as mean ± SD. *P < 0.001 versus WT vessels with a diameter <40 μm (n = 4 mice in each group). Prkg1−/− lungs exhibited a >10-fold increase in muscularized distal arteries (<40 μm in diameter) compared with WT.
To assess the integrity of the pulmonary vasculature, we performed fluorescent microangiography by perfusing the lungs with fluorescent microspheres (green) suspended in agarose.27 Normal filling of the microvasculature was observed in WT mouse lungs, whereas Prkg1−/− lungs showed a marked loss of microvascular perfusion and widespread pre-capillary occlusion (Figure 3, A and B). These data suggest that microvascular remodeling and pre-capillary occlusion cause impaired pulmonary vascular integrity in Prkg1−/− mice.
Figure 3.

Impaired pulmonary vascular perfusion in Prkg1−/− mice. A: Representative micrographs of fluorescent microangiography of lung sections perfused with fluorescent microspheres (green) suspended in agarose. Normal filling of the microvasculature was observed in WT lungs, whereas Prkg1−/− lungs showed a marked loss of microvascular perfusion and widespread pre-capillary occlusion. B: Quantification of pulmonary microvascular perfusion. Data are expressed as mean ± SD. *P < 0.01 versus WT (n = 3).
PH in Prkg1−/− Mice Is Independent of Left-Sided Heart Disease and Systemic Hypertension
To determine whether left-sided heart dysfunction is involved in the pathogenesis of PH in Prkg1−/− mice, echocardiography in anesthetized mice was performed to evaluate in vivo cardiac function of age-matched littermates. The Prkg1−/− mice, at the age of 45 days, exhibited a similar LV function as the WT mice (Figure 4A and Table 1). There was no evidence of LV hypertrophy and hypertension in Prkg1−/− mice compared with WT mice. A histological examination of the Prkg1−/− heart revealed similar sizes of ventricular chambers and thickness of the posterior wall and septum (Figure 4B). Measurement of carotid artery pressure revealed no systemic hypertension in Prkg1−/− mice at the age of 45 days (Figure 4C). These data suggest that PH in Prkg1−/− mice is not secondary to LV dysfunction and systemic hypertension.
Figure 4.
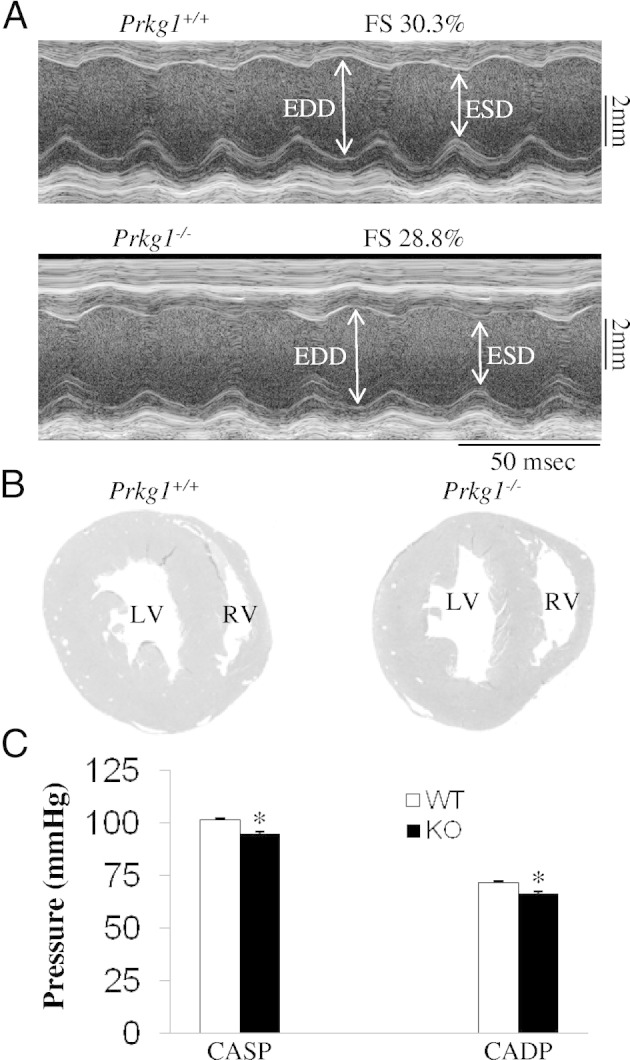
Normal left-sided heart function in Prkg1−/− mice. A: Transthoracic M-mode echocardiographic tracings in a WT and Prkg1−/− mouse (45-day-old littermate). Double-sided arrows, LV dimensions. EDD, end-diastolic dimension; ESD, end-systolic dimension; FS, fraction shortening, indicating cardiac contractility. B: Histological sections of hearts from 45-day-old littermates. Hearts were fixed in formalin and stained with H&E. Data are representative of three independent experiments with nearly identical results. C: Measurement of carotid artery pressure demonstrating no systemic hypertension in Prkg1−/− mice. Data are expressed as mean ± SD (n = 3). *P > 0.05. CADP, carotid artery diastolic pressure; CASP, carotid artery systolic pressure; KO, knockout.
Table 1.
Analysis of in Vivo Cardiac Size and Function by Echocardiography in WT and Prkg1−/− Mice
| Mouse type | LVEDD (mm) | LVESD (mm) | FS (%) | EF (%) | SEPth (mm) | PWth (mm) | BW (g) |
|---|---|---|---|---|---|---|---|
| WT | 3.93 ± 0.07 | 2.84 ± 0.04 | 28.9 ± 2.1 | 56.1 ± 3.5 | 0.75 ± 0.06 | 0.75 ± 0.07 | 24.7 ± 1.5 |
| Prkg1−/− | 3.87 ± 0.05 | 2.82 ± 0.03 | 28.5 ± 1.5 | 54.4 ± 2.1 | 0.73 ± 0.04 | 0.71 ± 0.03 | 20.0 ± 0.8⁎ |
Data are expressed as the mean ± SD (n = 3). Echocardiography was performed in anesthetized sex-matched 45-day-old mice.
BW, body weight; EF, ejection fraction; FS, fractional shortening; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; PWth, left ventricular posterior wall thickness; SEPth, intraventricular septal wall thickness.
P < 0.05.
PKG-I Deficiency Results in Decreased Phosphorylation of Rho A Ser188 and Increased Activation of Rho A
Previous pharmacological studies13,29,30 have demonstrated that PKG phosphorylates Rho A at Ser188 and subsequently inhibits its membrane association, which prevents activation of its downstream targets, such as Rho kinase. To assess whether PKG-I deficiency results in decreased phosphorylation of Rho A, we performed Western blot analysis on lung tissues from WT or Prkg1−/− mice using antibody against phosphorylated Ser188 of Rho A. As shown in Figure 5, A and B, a marked decrease of phosphorylation of RhoA Ser188 in Prkg1−/− lungs was observed compared with WT lungs. To determine whether PKG-I deficiency promotes Rho A membrane association and activation, we performed a pull-down assay with glutathione-Sepharose beads, coupled with glutathione-S-transferase–rhotekin fusion protein.28 As shown in Figure 5, C and D, there was a more than twofold increase in GTP-bound Rho A in Prkg1−/− lungs compared with WT lungs, whereas total Rho A expression was similar in both WT and Prkg1−/− lungs, indicating Rho A activation in Prkg1−/− lungs. Rho A activation was also markedly increased in SMCs isolated from Prkg1−/− lungs compared with WT SMCs (Figure 6, A and B).
Figure 5.
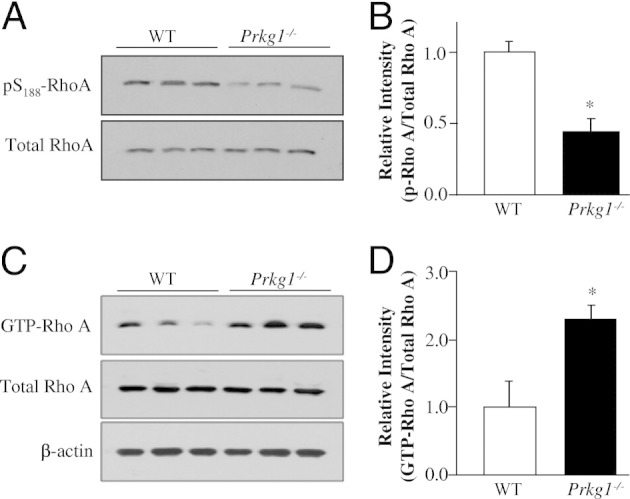
PKG-I deficiency activates Rho A signaling in Prkg1−/− lungs. A: Western blot analysis demonstrating decreased phosphorylation of Rho A at Ser188 (pS188-RhoA). Lung lysates, 30 μg, from 6-week-old mice per lane were loaded and immunoblotted with an anti-phosphorylated Ser188 of Rho A antibody. The experiment was repeated twice with similar data. B: Densitometric analysis of Rho A phosphorylation (p-Rho A) at Ser188. The intensity of each band of phosphorylated Ser–Rho A was normalized to the intensity of the respective total Rho A band. Data are expressed as mean ± SD (n = 3). *P < 0.05 versus WT. C: PKG-I deficiency results in decreased phosphorylation of Rho A at Ser188. Prominent Rho A activation in Prkg1−/− lungs. Lung lysates, 300 μg, from 6-week-old WT or Prkg1−/− mice were used for pull-down assays to determine the GTP-bound active RhoA (GTP-Rho A). Each lung lysate, 30 μg, was loaded per lane for detection of total Rho A with an anti-Rho A antibody. Anti-β-actin immunoblotting was used as a loading control. D: Densitometric analysis of Rho A activation. The intensity of each band of GTP–Rho A was normalized to the intensity of the respective total Rho A band. Data are expressed as mean ± SD (n = 3). *P < 0.01 versus WT. Rho A activity in Prkg1−/− lungs was more than twofold greater than in WT lungs.
Figure 6.
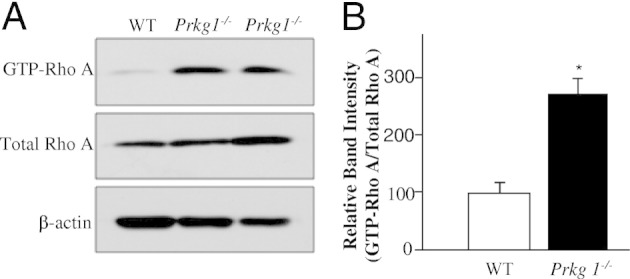
PKG-I deficiency induces Rho A activation in Prkg1−/− pulmonary vascular SMCs. A: Representative Western blot analyses demonstrating Rho A activation in Prkg1−/− pulmonary vascular SMCs. Pulmonary vascular SMCs isolated from 6-week-old mouse lungs were expanded in culture and lysed at passage 2 for detection of GTP–Rho A by pull-down assays. Total Rho A was detected by direct immunoblotting with an anti-Rho A antibody. β-Actin immunoblotting was used as a loading control. B: Densitometric analysis of Rho A activation. The intensity of each band of GTP–Rho A was normalized to the intensity of the respective total Rho A band. Data are expressed as mean ± SD (n = 3 independent experiments). Rho A activity in Prkg1−/− SMCs was threefold greater than in WT SMCs. *P < 0.01 versus WT.
PKG-I Deficiency–Induced Vasoconstriction through Activation of Rho A–Rho Kinase Signaling Is a Critical Component of the Pathogenesis of PH
We next addressed the mechanistic role of PKG-I deficiency–induced activation of Rho A–Rho kinase signaling in the pathogenesis of PH. After measurement of RVSP at the basal level, the mice were immediately treated with fasudil, a Rho kinase inhibitor, and RVSP was continuously monitored for an additional 30 minutes. There was a marked increase of RVSP in Prkg1−/− mice compared with WT mice under basal conditions; however, fasudil treatment resulted in a significant decrease of RVSP in Prkg1−/− mice but not in WT mice (Figure 7, A–C). These data suggest that Rho kinase–mediated vasoconstriction is an important component of the pathogenesis of PH in Prkg1−/− mice.
Figure 7.
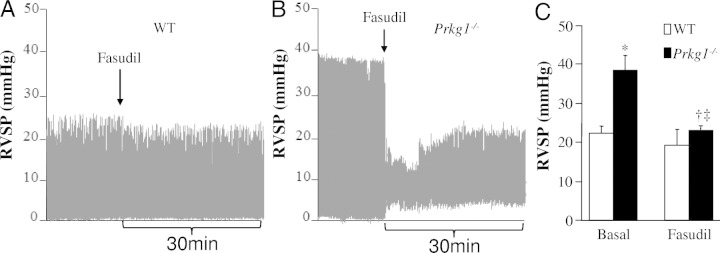
The causal role of Rho kinase activation, secondary to PKG-I deficiency, in the pathogenesis of PH in Prkg1−/− mice. Representative RVSP tracings at the basal level and after fasudil treatment in WT (A) and Prkg1−/− mice (B). After measurement of RVSP under basal conditions, the 7-week-old mice were treated with Rho kinase inhibitor fasudil (60 mmol/L in 25 μL of saline/mouse, i.t.), and then RVSP was continuously monitored for 30 minutes. C: Normalized RVSP in Prkg1−/− mice after fasudil treatment. Data are expressed as mean ± SD (n = 3 to 5). *P < 0.01 versus WT at the basal level, †P < 0.05 versus Prkg1−/− at the basal level, and ‡P > 0.05 versus WT treated with fasudil.
Discussion
To our knowledge, this is the first study to demonstrate that genetic disruption of Prkg1 causes PH in mice. Our data have shown that Prkg1−/− lungs exhibit severe pulmonary vascular remodeling, as evidenced by muscularization of distal pulmonary vessels and impaired vascular integrity. PKG-I deficiency results in activation of Rho A–Rho kinase signaling, which causes vascular remodeling and vasoconstriction. Our data demonstrate that PKG-I deficiency–induced vasoconstriction is the critical component of the pathogenesis of PH in Prkg1−/− mice.
The biological importance of NO/cGMP/PKG signaling was first appreciated for promoting vascular smooth muscle relaxation.31,32 NO stimulates cGMP synthesis and activates PKG in vascular SMCs, thereby relaxing small arteries and arterioles, resulting in a decreased blood pressure.33 Prkg1−/− mice exhibit defective relaxation of vascular, visceral, and penile smooth muscle17–20; thus, Prkg1−/− mice show impaired cGMP-dependent relaxation of large and small arteries and develop systemic hypertension as early as the age of 4 weeks.17 However, the elevated blood pressure in Prkg1−/− mice is returned to a normal level at an older age (eg, 6 weeks old).17,34 The normalization of blood pressure in older Prkg1−/− mice may be the result of cross activation of protein kinase A by the high cGMP levels that are potentially generated in these mice.35,36 Herein, we show that Prkg1−/− mice exhibit hypertensive pulmonary phenotypes at the age of 6 to 8 weeks, independent of systemic hypertension and LV dysfunction. The persistent hypertensive pulmonary phenotypes suggest that vasorelaxation via the cGMP/PKG-I pathway is essential to the regulation of basal pulmonary vascular pressure, which is in contrast to its role in regulation of basal blood pressure.17,34 Our recent studies26,37 have also shown that tyrosine nitration-mediated impairment of PKG-I activity, secondary to loss of caveolin-1, results in the development of PH in Cav1−/− mice. Taken together, these data demonstrate the essential role of normal PKG-I activity in regulating the pulmonary vascular tone. Impaired PKG activity, through either decreased expression or post-translational modification, leads to PH.
Lincoln and colleagues38,39 first described a role for PKG in modulation of aortic SMC phenotype and implicated its role in vascular disease. They observed that constitutive expression of PKG-Iα restores the contractile phenotype in cultured aortic SMCs, whereas inhibition of PKG reverses the phenotype.39 Overexpression of PKG-I increases the sensitivity of cultured vascular SMCs to the anti-proliferative and pro-apoptotic effects induced by NO/cGMP,23 whereas down-regulation of PKG-I induces the change of SMC phenotype from a contractile, differentiated to a synthetic, dedifferentiated state,24 which is the characteristic feature of pulmonary vascular remodeling. We have observed a >10-fold increase of muscularization of distal pulmonary vessels, a drastic loss of microvascular perfusion, and widespread pre-capillary occlusion in Prkg1−/− lungs. These data suggest that PKG-I deficiency induces pulmonary microvascular remodeling and pre-capillary occlusion, resulting in impaired pulmonary vascular integrity, and thereby contributes to the pathogenesis of PH in Prkg1−/− mice.
Recent studies14,40–42 have shown an important role of the PKG-regulated Rho A/Rho kinase signaling pathway in modulating pulmonary vascular remodeling and vasoconstriction. PKG-I phosphorylates Rho A at Ser188 and inhibits its membrane association, thereby preventing Rho kinase activation.13,15 Consistently, we observed decreased phosphorylation of Rho A Ser188 and resultant activation of Rho A in Prkg1−/− lungs. PKG-I deficiency also leads to marked Rho A activation in primary cultures of pulmonary vascular SMCs isolated from Prkg1−/− lungs. However, the total protein levels of Rho A in Prkg1−/− lungs and isolated pulmonary vascular SMCs are the same as in WT lungs. In contrast, other studies have shown a critical role of PKG in mediating NO-stimulated Rho A expression. NO from pulmonary artery SMCs of rat or human induces Rho A expression via an increase in Rho A protein stability and Rho A gene transcription through PKG.43 Long-term inhibition of NO synthesis induces a significant decrease in Rho A mRNA and protein expression in nitro-L-arginine–treated rat aorta and pulmonary artery.44 Rho A has played a major role in the regulation of multiple cellular functions, including actin cytoskeleton organization, contraction, gene expression, differentiation, and proliferation.14,45–47 Thus, Rho A activation, secondary to PKG-I deficiency, may be the major contributor of the severe pulmonary vascular remodeling leading to PH in Prkg1−/− mice.
We have shown that the Rho A/Rho kinase signaling pathway plays a key role in the mechanisms of the pathogenesis of PH in Prkg1−/− mice. Short-term inhibition of Rho kinase by i.t administration of fasudil completely inhibited the increase of RVSP in Prkg1−/− mice. These data demonstrate that vasoconstriction secondary to impairment of PKG activity is an important component of the pathogenesis of PH. Several pharmacological studies have also suggested a role for Rho kinase in the development of PH. The i.v. or oral treatment with Rho kinase inhibitor (Y-27632 or fasudil) normalizes the high pulmonary arterial pressure in chronically hypoxic rats, attenuates the development of chronic hypoxia-induced PH in mice, and reduces pulmonary arterial lesions in monocrotaline-induced PH in rats.40,41,43 Inhaled Y-27362 or fasudil causes sustained and selective pulmonary vasodilation in monocrotaline-induced PH and in spontaneous PH in fawn-hooded rats, as well as in chronically hypoxic rats.48 Rho kinase is one of the main downstream effectors of Rho A. On activation, Rho A activates Rho kinase, which phosphorylates the myosin phosphatase target subunit 1 of smooth muscle myosin phosphatase at Thr696 and leads to inhibition of its activity, an increased sensitivity of myosin light chain to calcium, and contraction.15,49 These data collectively suggest that activation of the Rho A/Rho kinase signaling pathway secondary to decreased PKG activity is a critical component of the underlying mechanisms of PH in Prkg1−/− mice.
Acknowledgments
We thank Dr. Xiaoping Du (University of Illinois College of Medicine) for providing the Prkg1−/− mice and Jun Yuan for his expert technical support.
Footnotes
Supported by grants from the NIH (R01HL085462 and R01HL085462-3S1 to Y.Y.Z.).
Contributor Information
Yidan D. Zhao, Email: yidanzhao@gmail.com.
You-Yang Zhao, Email: yyzhao@uic.edu.
References
- 1.Rubin L.J. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G., Robbins I.M., Beghetti M., Channick R.N., Delcroix M., Denton C.P., Elliott C.G., Gaine S.P., Gladwin M.T., Jing Z.C., Krowka M.J., Langleben D., Nakanishi N., Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Rich S. The current treatment of pulmonary arterial hypertension: time to redefine success. Chest. 2006;130:1198–1202. doi: 10.1378/chest.130.4.1198. [DOI] [PubMed] [Google Scholar]
- 4.Farber H.W. The status of pulmonary arterial hypertension in 2008. Circulation. 2008;117:2966–2968. doi: 10.1161/CIRCULATIONAHA.108.782979. [DOI] [PubMed] [Google Scholar]
- 5.Lucas K.A., Pitari G.M., Kazerounian S., Ruiz-Stewart I., Park J., Schulz S., Chepenik K.P., Waldman S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 6.Hofmann F., Feil R., Kleppisch T., Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 7.Xu J., Liu L. The role of calcium desensitization in vascular hyporeactivity and its regulation after hemorrhagic shock in the rat. Shock. 2005;23:576–581. [PubMed] [Google Scholar]
- 8.Nishimura J., Setoguchi H., Kanaide H. Regulation of vascular tone by the modulation of Ca2+ sensitivity [Japanese] Clin Calcium. 2001;11:411–417. [PubMed] [Google Scholar]
- 9.Wernet W., Flockerzi V., Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- 10.Feil R., Lohmann S.M., de Jonge H., Walter U., Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907–916. doi: 10.1161/01.RES.0000100390.68771.CC. [DOI] [PubMed] [Google Scholar]
- 11.Stephens R.S., Rentsendorj O., Servinsky L.E., Moldobaeva A., Damico R., Pearse D.B. cGMP increases antioxidant function and attenuates oxidant cell death in mouse lung microvascular endothelial cells by a protein kinase G-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2010;299:L323–L333. doi: 10.1152/ajplung.00442.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMichael R.W., Jr, Lagarias J.C. Phosphopeptide mapping of Avena phytochrome phosphorylated by protein kinases in vitro. Biochemistry. 1990;29:3872–3878. doi: 10.1021/bi00468a011. [DOI] [PubMed] [Google Scholar]
- 13.Sawada N., Itoh H., Yamashita J., Doi K., Inoue M., Masatsugu K., Fukunaga Y., Sakaguchi S., Sone M., Yamahara K., Yurugi T., Nakao K. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem Biophys Res Commun. 2001;280:798–805. doi: 10.1006/bbrc.2000.4194. [DOI] [PubMed] [Google Scholar]
- 14.Sauzeau V., Le Jeune H., Cario-Toumaniantz C., Smolenski A., Lohmann S.M., Bertoglio J., Chardin P., Pacaud P., Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2000;275:21722–21729. doi: 10.1074/jbc.M000753200. [DOI] [PubMed] [Google Scholar]
- 15.Feng J., Ito M., Ichikawa K., Isaka N., Nishikawa M., Hartshorne D.J., Nakano T. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- 16.Sausbier M., Schubert R., Voigt V., Hirneiss C., Pfeifer A., Korth M., Kleppisch T., Ruth P., Hofmann F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res. 2000;87:825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 17.Pfeifer A., Klatt P., Massberg S., Ny L., Sausbier M., Hirneiss C., Wang G.X., Korth M., Aszodi A., Andersson K.E., Krombach F., Mayerhofer A., Ruth P., Fassler R., Hofmann F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persson K., Pandita R.K., Aszodi A., Ahmad M., Pfeifer A., Fassler R., Andersson K.E. Functional characteristics of urinary tract smooth muscles in mice lacking cGMP protein kinase type I. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1112–R1120. doi: 10.1152/ajpregu.2000.279.3.R1112. [DOI] [PubMed] [Google Scholar]
- 19.Ny L., Pfeifer A., Aszodi A., Ahmad M., Alm P., Hedlund P., Fassler R., Andersson K.E. Impaired relaxation of stomach smooth muscle in mice lacking cyclic GMP-dependent protein kinase I. Br J Pharmacol. 2000;129:395–401. doi: 10.1038/sj.bjp.0703061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedlund P., Aszodi A., Pfeifer A., Alm P., Hofmann F., Ahmad M., Fassler R., Andersson K.E. Erectile dysfunction in cyclic GMP-dependent kinase I-deficient mice. Proc Natl Acad Sci U S A. 2000;97:2349–2354. doi: 10.1073/pnas.030419997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamahara K., Itoh H., Chun T.H., Ogawa Y., Yamashita J., Sawada N., Fukunaga Y., Sone M., Yurugi-Kobayashi T., Miyashita K., Tsujimoto H., Kook H., Feil R., Garbers D.L., Hofmann F., Nakao K. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc Natl Acad Sci U S A. 2003;100:3404–3409. doi: 10.1073/pnas.0538059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z., Xi X., Gu M., Feil R., Ye R.D., Eigenthaler M., Hofmann F., Du X. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell. 2003;112:77–86. doi: 10.1016/s0092-8674(02)01254-0. [DOI] [PubMed] [Google Scholar]
- 23.Chiche J.D., Schlutsmeyer S.M., Bloch D.B., de la Monte S.M., Roberts J.D., Jr, Filippov G., Janssens S.P., Rosenzweig A., Bloch K.D. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem. 1998;273:34263–34271. doi: 10.1074/jbc.273.51.34263. [DOI] [PubMed] [Google Scholar]
- 24.Zhou W., Negash S., Liu J., Raj J.U. Modulation of pulmonary vascular smooth muscle cell phenotype in hypoxia: role of cGMP-dependent protein kinase and myocardin. Am J Physiol Lung Cell Mol Physiol. 2009;296:L780–L789. doi: 10.1152/ajplung.90295.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W., Dasgupta C., Negash S., Raj J.U. Modulation of pulmonary vascular smooth muscle cell phenotype in hypoxia: role of cGMP-dependent protein kinase. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1459–L1466. doi: 10.1152/ajplung.00143.2006. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y.Y., Zhao Y.D., Mirza M.K., Huang J.H., Potula H.H., Vogel S.M., Brovkovych V., Yuan J.X., Wharton J., Malik A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y.D., Courtman D.W., Deng Y., Kugathasan L., Zhang Q., Stewart D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96:442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y.D., Ohkawara H., Rehman J., Wary K.K., Vogel S.M., Minshall R.D., Zhao Y.Y., Malik A.B. Bone marrow progenitor cells induce endothelial adherens junction integrity by sphingosine-1-phosphate-mediated Rac1 and Cdc42 signaling. Circ Res. 2009;105:696–704. doi: 10.1161/CIRCRESAHA.109.199778. 8 p following 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolli-Derkinderen M., Sauzeau V., Boyer L., Lemichez E., Baron C., Henrion D., Loirand G., Pacaud P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–1160. doi: 10.1161/01.RES.0000170084.88780.ea. [DOI] [PubMed] [Google Scholar]
- 30.Kitazawa T., Semba S., Huh Y.H., Kitazawa K., Eto M. Nitric oxide-induced biphasic mechanism of vascular relaxation via dephosphorylation of CPI-17 and MYPT1. J Physiol. 2009;587:3587–3603. doi: 10.1113/jphysiol.2009.172189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walter U. Physiological role of cGMP and cGMP-dependent protein kinase in the cardiovascular system. Rev Physiol Biochem Pharmacol. 1989;113:41–88. doi: 10.1007/BFb0032675. [DOI] [PubMed] [Google Scholar]
- 32.Warner T.D., Mitchell J.A., Sheng H., Murad F. Effects of cyclic GMP on smooth muscle relaxation. Adv Pharmacol. 1994;26:171–194. doi: 10.1016/s1054-3589(08)60054-x. [DOI] [PubMed] [Google Scholar]
- 33.Garbers D.L., Dubois S.K. The molecular basis of hypertension. Annu Rev Biochem. 1999;68:127–155. doi: 10.1146/annurev.biochem.68.1.127. [DOI] [PubMed] [Google Scholar]
- 34.Koeppen M., Feil R., Siegl D., Feil S., Hofmann F., Pohl U., de Wit C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension. 2004;44:952–955. doi: 10.1161/01.HYP.0000147661.80059.ca. [DOI] [PubMed] [Google Scholar]
- 35.Cornwell T.L., Arnold E., Boerth N.J., Lincoln T.M. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol. 1994;267:C1405–C1413. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- 36.Soff G.A., Cornwell T.L., Cundiff D.L., Gately S., Lincoln T.M. Smooth muscle cell expression of type I cyclic GMP-dependent protein kinase is suppressed by continuous exposure to nitrovasodilators, theophylline, cyclic GMP, and cyclic AMP. J Clin Invest. 1997;100:2580–2587. doi: 10.1172/JCI119801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y.Y., Malik A.B. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc Med. 2009;19:238–242. doi: 10.1016/j.tcm.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lincoln T.M., Dey N.B., Boerth N.J., Cornwell T.L., Soff G.A. Nitric oxide–cyclic GMP pathway regulates vascular smooth muscle cell phenotypic modulation: implications in vascular diseases. Acta Physiol Scand. 1998;164:507–515. doi: 10.1111/j.1365-201x.1998.tb10700.x. [DOI] [PubMed] [Google Scholar]
- 39.Dey N.B., Boerth N.J., Murphy-Ullrich J.E., Chang P.L., Prince C.W., Lincoln T.M. Cyclic GMP-dependent protein kinase inhibits osteopontin and thrombospondin production in rat aortic smooth muscle cells. Circ Res. 1998;82:139–146. doi: 10.1161/01.res.82.2.139. [DOI] [PubMed] [Google Scholar]
- 40.Abe K., Shimokawa H., Morikawa K., Uwatoku T., Oi K., Matsumoto Y., Hattori T., Nakashima Y., Kaibuchi K., Sueishi K., Takeshit A. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res. 2004;94:385–393. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- 41.Fagan K.A., Oka M., Bauer N.R., Gebb S.A., Ivy D.D., Morris K.G., McMurtry I.F. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–L664. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 42.Gao Y., Portugal A.D., Negash S., Zhou W., Longo L.D., Usha Raj J. Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high altitude hypoxia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L678–L684. doi: 10.1152/ajplung.00178.2006. [DOI] [PubMed] [Google Scholar]
- 43.Nagaoka T., Morio Y., Casanova N., Bauer N., Gebb S., McMurtry I., Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–L672. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 44.Sauzeau V., Rolli-Derkinderen M., Marionneau C., Loirand G., Pacaud P. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem. 2003;278:9472–9480. doi: 10.1074/jbc.M212776200. [DOI] [PubMed] [Google Scholar]
- 45.Mack C.P., Somlyo A.V., Hautmann M., Somlyo A.P., Owens G.K. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 46.Gudi T., Chen J.C., Casteel D.E., Seasholtz T.M., Boss G.R., Pilz R.B. cGMP-dependent protein kinase inhibits serum-response element-dependent transcription by inhibiting rho activation and functions. J Biol Chem. 2002;277:37382–37393. doi: 10.1074/jbc.M204491200. [DOI] [PubMed] [Google Scholar]
- 47.Loirand G., Rolli-Derkinderen M., Pacaud P. RhoA and resistance artery remodeling. Am J Physiol Heart Circ Physiol. 2005;288:H1051–H1056. doi: 10.1152/ajpheart.00710.2004. [DOI] [PubMed] [Google Scholar]
- 48.Nagaoka T., Fagan K.A., Gebb S.A., Morris K.G., Suzuki T., Shimokawa H., McMurtry I.F., Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 49.Somlyo A.P., Somlyo A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]