

Abstract
Krüppel-like factor 3 (KLF3) is a transcriptional regulator that we have shown to be involved in the regulation of adipogenesis in vitro. Here, we report that KLF3-null mice are lean and protected from diet-induced obesity and glucose intolerance. On a chow diet, plasma levels of leptin are decreased, and adiponectin is increased. Despite significant reductions in body weight and adiposity, wild-type and knockout animals show equivalent energy intake, expenditure, and excretion. To investigate the molecular events underlying these observations, we used microarray analysis to compare gene expression in Klf3+/+ and Klf3−/− tissues. We found that mRNA expression of Fam132a, which encodes a newly identified insulin-sensitizing adipokine, adipolin, is significantly upregulated in the absence of KLF3. We confirmed that KLF3 binds the Fam132a promoter in vitro and in vivo and that this leads to repression of promoter activity. Further, plasma adipolin levels were significantly increased in Klf3−/− mice compared with wild-type littermates. Boosting levels of adipolin via targeting of KLF3 offers a novel potential therapeutic strategy for the treatment of insulin resistance.
The mechanisms linking obesity and the development of insulin resistance are not well understood despite intensive research (1,2). An improved understanding of the molecular control of metabolism, and how nutrition and environmental factors can influence gene expression to modulate metabolic pathways, is critical for the development of successful therapeutics to treat metabolic disorders, such as type 2 diabetes. Numerous transcriptional regulators have been implicated in physiological responses to metabolic stimuli, and the pathways and downstream effectors involved in these processes are now beginning to be elucidated. One family of regulators that play diverse roles in metabolism is the Krüppel-like factor (KLF) family of transcription factors (3–5).
KLFs are a family of evolutionarily conserved, zinc-finger transcription factors, which recognize and bind to GC-rich sequences and CACCC boxes in the promoters and enhancers of their target genes (3). With the exception of KLF1, family members are widely expressed and have diverse biological roles (6,7). Several KLFs have been implicated in adipogenesis; KLF4, KLF5, KLF6, KLF9, and KLF15 (8–12) all positively regulate, while KLF2, KLF3, and KLF7 (13–15) inhibit adipogenesis. KLFs also regulate other aspects of metabolism. KLF15 has been shown to induce Glut4 expression in 3T3-L1 cells (16), and Klf15−/− mice demonstrate abnormal gluconeogenesis, leading to severe hypoglycemia after fasting (17). KLF11 can bind and regulate expression of the insulin promoter in pancreatic β-cells, and a number of KLF11 single nucleotide polymorphisms show significant association with susceptibility to type 2 diabetes (18). A Japanese study linked a KLF7 variant to type 2 diabetes risk (19), while a different polymorphism has been shown to protect against obesity in a Danish population (20). Single nucleotide polymorphisms in the maternally imprinted KLF14 gene locus have been shown to affect expression of numerous adipocyte genes and are also associated with a number of metabolic disorders, including type 2 diabetes and elevated HDL cholesterol (21). Collectively, numerous studies place KLFs as important regulators of metabolism.
We have been studying the role of KLF3/BKLF as a regulator of adipocyte biology (22). We have previously reported that Klf3−/− mice are lean, partly due to reduced adipose tissue mass and adipocyte size (14). As adipose is a focal point for a number of mechanisms linking obesity and insulin resistance, including altered adipocyte secretory profile, we set out to consider the role of KLF3 on whole-body metabolism and insulin action. We used a microarray-based approach to compare tissues and cells derived from Klf3−/− mice and wild-type littermates and identified Fam132a as a highly upregulated KLF3 target gene in a number of tissues. Very recently, two independent laboratories identified FAM132A, also known as C1QDC2 (C1q domain-containing protein 2), CTRP12 (C1q/tumor necrosis factor–related protein 12) and adipolin (adipose-derived insulin-sensitizing factor), as a new factor positively influencing glucose homeostasis (23,24).
In this study, we verified a direct in vivo interaction between KLF3 and the Fam132a promoter by chromatin immunoprecipitation (ChIP) studies and show systemic upregulation of Fam132a mRNA expression and plasma adipolin levels in mice that lack KLF3. We have also further characterized the metabolic phenotype of Klf3−/− mice on chow and high-fat diets to understand the role of KLF3 in the regulation of body weight, composition, and energy metabolism. Importantly, we show that KLF3-null mice are protected from diet-induced obesity and have improved insulin resistance. These data suggest a molecular mechanism whereby KLF3 may orchestrate effects on metabolism via regulation of factors such as the insulin-sensitizing hormone adipolin and imply that the improved metabolic profile in the absence of KLF3 may result, at least in part, from a significant elevation in Fam132a transcription and circulating adipolin levels.
RESEARCH DESIGN AND METHODS
Approval for the use of animals was from the University of Sydney Animal Care and Ethics Committee (protocol: L02/7–2009/3/5054). Klf3−/− mice on an FVB/NJ background were generated as previously described (14). Mice were weaned at 3 weeks and fed standard chow (6% kcal from fat, 14.3 MJ/kg; Specialty Feeds, Glen Forest, Western Australia, Australia) or high-fat diet (45% kcal from fat [mainly lard], 21.8 MJ/kg; made in house) until age 12 weeks.
Adipocyte histology.
Epididymal adipose tissue (n = 3–6 per group) was fixed in 4% paraformaldehyde in PBS for 48 h at 4°C. Samples were washed in PBS, dehydrated through graded ethanol solutions, and paraffin embedded. Sections (5 μm) were stained with hematoxylin-eosin (Sigma-Aldrich). The size of 200–300 adipocytes was measured per mouse at ×20 magnification (AnalySIS FIVE; Olympus).
Glucose and insulin tolerance tests.
Glucose tolerance tests were performed on overnight-fasted mice. After determination of fasting blood glucose, mice were given an intraperitoneal injection of 50% glucose solution (2 g/kg). For insulin tolerance tests, mice were fasted 4 h and given an intraperitoneal injection of 1 unit/kg insulin (Actrapid; Novo Nordisk). For both tests, tail blood glucose was measured using an Accu-chek Performa glucometer (Roche).
Energy metabolism.
Food intake was measured for mice housed individually with a custom-made cage insert (City West Plastics, Rydalmere, Australia) designed to catch spilled food. Mice were given 24 h to acclimatize, after which food was weighed daily for 3 days.
For calculation of total energy expenditure, whole-body lean and fat mass was determined by dual-energy X-ray absorptiometry (Lunar PIXImus2 mouse densitometer; GE Healthcare). Vo2 was measured at 22°C using an indirect calorimetry system (Oxymax series; Columbus Instruments, Columbus, OH). Studies commenced after 2 h of acclimation to the metabolic chamber. Vo2 was measured in individual mice at 20- to 30-min intervals over a 48-h period. Energy excreted in feces was measured using a bomb calorimeter (model 1356; PARR Instrument Company). Feces was compacted and dried in a freeze dryer overnight before combustion.
Plasma biochemistry.
Plasma leptin, insulin, and adiponectin were measured by ELISA according to the manufacturer’s guidelines (Quantikine Mouse Leptin and Adiponectin, R&D Systems, Minneapolis, MN; Ultrasentivite Mouse Insulin ELISA, Mercodia, Uppsala, Sweden). Plasma nonesterified fatty acid (NEFA), glycerol, and triglyceride were determined by colorimetric assay per the manufacturer’s instructions (NEFA-C assay kit; Wako Chemicals, Osaka, Japan; serum triglyceride (and glycerol) determination kit; Sigma-Aldrich, Sydney, NSW, Australia).
Electrophoretic mobility shift assays.
Electrophoretic mobility shift assays (EMSAs) were carried out as previously described (25). Oligonucleotides used in the synthesis of radiolabeled probes were as follows: mouse Fam132a promoter probe A, 5-TGCTCCGCCCCGCCCCGCCCCGCCCTGCTCC-3 and 5-GGAGCAGGGCGGGGCGGGGCGGGGCGGAGCA-3; Fam132a probe B, 5-GGTCCCGCCCCGCCCTGCCCCGCCCCGCCCTGCTCC-3 and 5-GGAGCAGGGCGGGGCGGGGCAGGGCGGGGCGGGACC-3; Fam132a probe C, 5-TGCCCCGCCCCGCCCTGCTCCGCCCCGCCCCGCCCC-3 and 5- GGGGCGGGGCGGGGCGGAGCAGGGCGGGGCGGGGCA-3; and Klf8 promoter, 5- GGGCCCGCCCCACCCCCTCCT-3 and 5-AGGAGGGGGTGGGGCGGGCCC-3.
Cloning of the Fam132a promoter for transactivation assays.
Plasmid pGL4.10[luc2]-Fam132a promoter contained the sequence from −150 to +100 of the Fam132a promoter; for pGL4.10[luc2]-Fam132a-ΔCACCC, the sequence from −101 to −29 was deleted.
Transactivation assays.
SL-2 cells were transfected in sixwell plates using the calcium phosphate method (26), and 3T3-L1 cells were transfected using Fugene 6 (Promega). For SL2 cells, 250 ng pPac-KLF1 and 0, 25, 50, and 250 ng pPac-KLF3 were transfected along with 1 μg pGL4.10[luc2], pGL4.10[luc2]-Fam132a promoter, or pGL4.10[luc2]-Fam132a-ΔCACCC. For 3T3-L1 cells, 0, 50, 250, and 500 ng pCDNA3-KLF3 was transfected along with 1 μg pGL4.10[luc2], pGL4.10[luc2]-Fam132a promoter, or pGL4.10[luc2]-Fam132a-ΔCACCC. For all transfections, 0.1 μg pGL4.74[hRluc/TK] was included as a transfection control. Forty-eight hours posttransfection, the cells were lysed and assayed for luciferase activity by using a dual luciferase reporter assay system (Promega). In all cases, reporter activity was normalized with respect to Renilla luciferase levels.
Western blotting.
Diluted plasma (5 μL; 1:20 in PBS), equivalent to 0.25 μL total plasma, was subjected to SDS-PAGE. Proteins were transferred to polyvinylidene fluoride membranes and incubated with primary antibody (anti-Fam132a, Santa Cruz sc-241304) followed by horseradish peroxidase–conjugated anti-goat IgG secondary. Signals were visualized using Immobilon Western Chemiluminescent horseradish peroxidase substrate (Millipore). Membranes were exposed to film and scanned on a densitometer and band densities quantitated with IPLab software (IPLab Gel H; Signal Analytics).
ChIP assay.
ChIP assays were preformed as previously described (27), using a Biorupter for chromatin sonication (Diagenode, Liège, Belgium). Chromatin was immunoprecipitated from murine erythroleukemia cells after induction of erythroid differentiation in 1.8% DMSO (28), using either the Pierce anti-KLF3 antibody (PA5-18030) or rabbit polyclonal anti-KLF3 serum (29). Real-time PCR primers were as follows: Fam132a promoter, 5-GATTCGCTTCCCTGGAGGTGTGG-3 and 5-GCCCAGTCTCTGGTCTCCTCTCT-3; C/ebpα promoter, 5-AGGAGAAGGCGGGCTCTAAG-3 and 5-ATCGAAGGCGCCAGTAGGA-3; C/ebpα negative control (5 kb upstream of C/ebpα exon 1), 5-CCCAGGCAGAACAAAACATAGG-3 and 5-GGGCAGGCCATTGTTTTGTA-3; Klf8 promoter 1a, 5-CCAGCTCGTGCACACTGAA-3 and 5-GAAGCCTTAACATCAGGAGTGGAA-3; Klf8 negative control 1 (4.5 kb upstream of Klf8 exon 1a), 5-GGTTTCTGAGACCTAACACTTCACACT-3 and 5-CCATTTAGTCATCCAGCGAACAA-3; and Klf8 negative control 2 (33 kb downstream of Klf8 exon 1a), 5-AACCTGGGTGCCTCCTTGTA-3 and 5-TCATGCCTTTGACTTTAGTGCTTT-3;
Determination of mRNA expression.
RNA extraction, cDNA synthesis, and quantification were performed as previously described (28,30). Oligonucleotides were as follows: ribosomal 18S: 5-GACGGACCAGAGCGAAAGC-3 and 5-AACTCCGACTTTCGTTCTTGATT-3, and Fam132a, 5-GATTCACAGCCCCAGTCTCT-3 and 5-ACAGATGAGAACACGGACCA-3 or 5-GGTCTTCACAGTGCAGGTTCAG-3 and 5-GACTGCCCCAGAACTGTTGTC-3.
Statistical analysis.
Data are presented as means ± SEM. One-way ANOVA was used to compare data from different genotypes and diet groups followed by pairwise comparisons using t tests with a Bonferroni correction to identify differences between groups. For energy expenditure data, ANCOVA analysis was performed using lean body mass as a covariate. All statistical analyses were completed using R (version 2.13.1; R Foundation for Statistical Computing, Vienna, Austria), and P < 0.05 was considered statistically significant.
RESULTS
Klf3−/− mice are resistant to diet-induced obesity.
In agreement with our previous observations (14), male Klf3−/− mice are shorter in length than their wild-type littermates and maintained a lower body weight and white adipose tissue (WAT) mass when fed a chow diet (Fig. 1A, B, and D). On a high-fat diet, Klf3−/− mice were significantly and consistently lighter than either Klf3+/+ or Klf3+/− littermates (Fig. 1C). Both subcutaneous (inguinal) and visceral (epididymal and retroperitoneal) WAT mass increased in mice in response to the high-fat diet, but Klf3−/− mice maintained less adiposity than Klf3+/+ mice (Fig. 1E). The reduction in adiposity in Klf3−/− mice was selective for WAT. Subscapular brown adipose tissue depots from Klf3−/− mice weighed the same as brown adipose tissue from wild-type or heterozygote littermates despite their lower body weight (Fig. 1D and E). Liver weight was significantly lower in Klf3−/− mice on both the chow and high-fat diet (Fig. 1F and G). White adipocytes of Klf3−/− male mice were visibly smaller than those of Klf3+/+ mice on both chow and high-fat diets (Fig. 1H). We also compared the size distribution of fat cells in Klf3+/+ and Klf3−/− male mice by measuring the diameter of epididymal adipocytes (Fig. 1I). We found that high-fat feeding noticeably increased the maximum and average adipocyte size in Klf3+/+ mice but had little effect in Klf3−/− animals (Fig. 1I).
FIG. 1.

Klf3−/− mice are lean and resistant to diet-induced obesity. A: Body length of 9-week-old male mice on chow diet (Klf3+/+, n = 8; Klf3−/−, n = 5). Data are means ± SEM. ***P < 0.001. B and C: Male body weight with respect to age. Mice were placed on chow diet (Klf3+/+, n = 48; Klf3+/−, n = 57; and Klf3−/−, n = 11) (B) or high-fat diet (C) from age 3–12 weeks (Klf3+/+, n = 20; Klf3+/−, n = 24; and Klf3−/−, n = 6). D–G: Tissue weights as determined by dissection and individual weighing of tissues in chow diet (Klf3+/+, n = 31; Klf3+/−, n = 53; and Klf3−/−, n = 11) (D and F) and high-fat diet (Klf3+/+, n = 25; Klf3+/−, n = 20; and Klf3−/−, n = 6) (E and G). BAT, brown adipose tissue; EPI, epididymal; ING, inguinal subcutaneous; RP, retroperitoneal. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Klf3+/+ vs. Klf3−/−; #P < 0.05, ##P < 0.01, ###P < 0.001, Klf3+/− vs. Klf3−/−. H: Representative photographs of hematoxylin-eosin stained epididymal WAT (×20 magnification) from Klf3+/+ and Klf3−/− male mice fed either chow or high-fat diet. I: Size distribution of epididymal adipocytes. Left panel: Adipocytes from male mice on chow diet. Right panel: Adipocytes from male mice on high-fat diet. Klf3+/+, n = 2–7; Klf3−/−, n = 3–6 (200–300 cells measured per mouse).
Loss of KLF3 does not alter energy intake or expenditure.
To investigate the lean phenotype of Klf3−/− mice, we compared energy intake and expenditure between wild-type mice and knockout littermates. Klf3−/− mice consumed the same amount of food as wild-type littermates on both chow and high-fat diets when expressed as kJ/day (Fig. 2A) or when corrected for body weight (data not shown). Similarly, loss of KLF3 did not appear to alter fecal weight or energy content on either diet (Fig. 2B). Furthermore, total energy expenditure was not affected by Klf3 deficiency when expressed per mouse or per gram of lean body mass, as determined by dual-energy X-ray absorptiometry (Fig. 2C–E). High-fat feeding significantly increased energy expenditure in wild-type and Klf3−/− mice (Fig. 2F).
FIG. 2.

No differences were detected in energy intake or output between Klf3+/+ and Klf3−/− mice. A: Food intake was measured daily over 3 days in male 12-week Klf3+/+ (n = 7–9) and Klf3−/− (n = 5–6) mice fed either chow or high-fat diet. B: Energy in feces was determined by bomb calorimetry and data corrected for daily fecal output. Klf3+/+, n = 10; Klf3−/−, n = 7–8. C and D: Total energy expenditure determined from Vo2 and expressed as kJ/day per mouse and per gram of lean body mass (n = 7–10 per group). Data are means ± SEM. **P < 0.01, ***P < 0.001, chow vs. high-fat diet by ANCOVA. E and F: Relationship between individual energy expenditure data calculated as heat production and lean body mass; diet, but not genotype, significantly affects energy expenditure.
Klf3−/− mice are protected from diet-induced glucose intolerance.
For assessment of the effect of KLF3 deficiency on systemic insulin action, glucose and insulin tolerance tests were performed. Klf3−/− mice had better glucose tolerance than wild-type littermates (Fig. 3A). After high-fat feeding, glucose tolerance in wild-type mice was markedly reduced, as expected; however, Klf3−/− mice maintained an improved glucose tolerance, and the calculated area under the curve was not different from chow-fed Klf3−/− mice (Fig. 3B). We observed comparable insulin tolerance between genotypes on a chow or high-fat diet (Fig. 3C). While previous reports have indicated that elevated adipolin improves insulin sensitivity (23,24), it is possible that the inability to detect a change in Klf3−/− mice may be due to phenotypic effects resulting from the deregulation of KLF3 target genes other than Fam132a. High-fat feeding increased the fasting insulin-to-glucose ratio, a marker of insulin resistance (31) in wild-type mice but not Klf3−/− mice (Fig. 3D). KLF3-deficient mice tended to have a reduced insulin-to-glucose ratio, but this was not significantly different from wild-type controls on either diet.
FIG. 3.

Klf3−/− mice are protected from diet-induced glucose intolerance and insulin resistance. A: Glucose tolerance test (2 g glucose/kg) in mice fasted for 15 h. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Klf3+/+ vs. Klf3−/− ; #P < 0.05, ##P < 0.01, chow vs. high-fat diet (n = 7–19). B: Area under the curve for glucose tolerance test. *Effect of genotype. #Effect of diet. *P < 0.05, ***P < 0.001, Klf3+/+ vs. Klf3−/− ; ##P < 0.001 chow vs. high-fat diet (n = 7–19). C: Change in blood glucose levels during an insulin tolerance test (0.5 units insulin/kg) in mice fasted for 4 h (n = 8–17). D: Insulin resistance index calculated from fasting plasma insulin and blood glucose levels. Data are means ± SEM. *P < 0.05, chow-fed Klf3+/+ vs. high-fat diet–fed Klf3+/+.
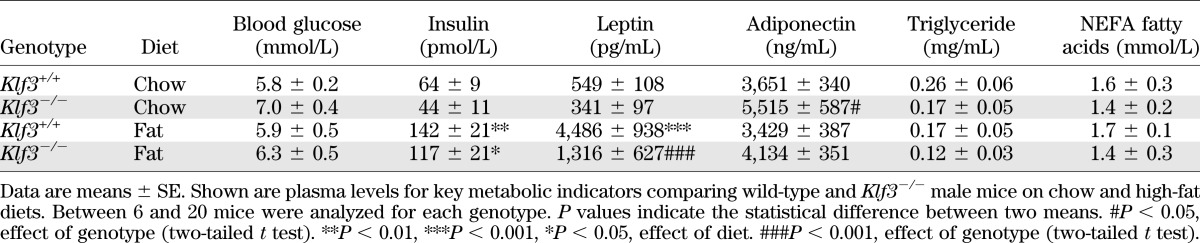
Klf3−/− mice blood biochemistry.
To further investigate the metabolic phenotype of Klf3−/− mice, we examined the plasma levels of a number of key metabolic indicators (Table 1). We noted that despite improved glucose tolerance, Klf3−/− mice have a slight, but nonsignificant, elevation in fasting blood glucose (Table 1, chow-fed Klf3+/+ vs. Klf3−/−). High-fat feeding increased plasma insulin and leptin levels in wild-type and Klf3−/− mice; however, the increase in leptin levels in the fat-fed Klf3−/− mice was significantly lower than in wild-type. On a chow diet, KLF3 deficiency increased plasma adiponectin levels, while high-fat feeding did not significantly reduce levels in either genotype. Plasma triglyceride and NEFA fatty acid levels were unchanged between genotypes and diets.
TABLE 1.
Summary of plasma metabolic parameters for Klf3+/+ and Klf3−/− mice
Fam132a, the gene coding for adipolin, is upregulated in KLF3-deficient tissue.
Having found no measurable differences in acute energy balance between wild-type and lean Klf3−/− mice, we explored alternative mechanisms to determine an explanation for this beneficial phenotype. We revisited previous microarray analysis, in which we had examined gene expression in erythrocytes that lack KLF3 (32), and also carried out further arrays to compare gene expression in several wild-type and Klf3−/− metabolic tissues (white adipose, red skeletal muscle, and liver). When comparing gene expression between wild-type and Klf3−/− tissues, we found that in a number of samples Fam132a was one of the most highly upregulated genes in Klf3−/− cells. This was an intriguing finding, as Fam132a is the gene that codes for the recently identified insulin-sensitizing factor adipolin, shown to promote glucose tolerance and insulin sensitivity in animal models of obesity and diabetes (23,24). In order to validate this observation, we examined Fam132a gene expression using real-time RT-PCR in several tissues isolated from wild-type and Klf3−/− mice. We found that expression of the Fam132a gene is significantly upregulated in heart, lung, gut, bone marrow, and spleen in the absence of its repressor KLF3 (Fig. 4A). In particular, our examination of bone marrow and spleen revealed that Fam132a mRNA is readily detectable in wild-type erythroid tissue and that this high-level expression is significantly increased in cells lacking KLF3. This derepression is also observed in epididymal WAT of KLF3-deficient mice but not in brown adipose tissue (Fig. 4B and C). In agreement with this, Klf3−/− mice also show significantly elevated plasma adipolin levels on both chow and high-fat diets (Fig. 4D).
FIG. 4.

Fam132a gene expression and adipolin protein levels are increased in selected tissues in Klf3−/− mice. A: Fam132a mRNA expression in various tissues, as determined by real-time quantitative PCR (n = 3 for each genotype). B: Fam132a mRNA expression in epididymal WAT (n = 6 for each genotype). C: Fam132a mRNA expression in brown adipose tissue (n = 6 for each genotype). For panels A, B, and C, the relative expression of Fam132a mRNA was normalized against 18S rRNA, and the expression level of the lowest sample was set to 1.0. Error bars represent SEM. P values indicate the statistical difference between means (Klf3+/+ vs. Klf3−/−): *P < 0.05, **P < 0.01 (two-tailed t test). D: Adipolin protein levels in plasma of Klf3+/+ and Klf3−/− mice. Equal aliquots of diluted total plasma (1:20 in PBS) were resolved by SDS-PAGE and Western blotted with an anti-adipolin antibody to determine the relative concentration of plasma adipolin. The positions of molecular weight standards in kDa are indicated on the right-hand side of the blot. ***P < 0.001, Klf3+/+ vs. Klf3−/− (n = 11–29). BAT, brown adipose tissue.
KLF3 binds and represses the Fam132a promoter in vitro and in vivo.
Having determined that Fam132a gene expression is significantly upregulated in KLF3-null tissue (32), we investigated whether KLF3 can directly bind the Fam132a promoter by EMSA. An inspection of the Fam132a proximal promoter revealed the presence of several consensus KLF3-binding sites (25) (Fig. 5A). Three probes were designed to provide full coverage of these sites, and the binding of KLF3 to each of these probes was assessed by EMSA (Fig. 5B). KLF3 bound robustly to all three probes in comparison with a control sequence derived from the Klf8 proximal promoter, which has previously been validated as a KLF3 target both in vitro by EMSA and in vivo by ChIP assays (29). The addition of an anti-KLF3 antibody resulted in supershift of the most prominent probe complexes, confirming the presence of KLF3 (Fig. 5B). In addition to the major bands, slower migrating complexes were also observed, in particular for probes B and C, which could be supershifted by the addition of anti-KLF3 antibody. The presence of these higher-order complexes raises the possibility that precise regulation of the Fam132a promoter may depend upon multiple KLF3 occupancy and is concordant with the presence of multiple CACCC-box binding sites.
FIG. 5.

KLF3 binds the Fam132a promoter in vitro and in vivo and is able to repress its activity. A: Probe design to assess binding of KLF3 to the Fam132a proximal promoter by EMSA: three probes (probes A, B, and C) were designed to provide coverage of major CACCC boxes identified in the promoter. Black arrows indicate CACCC boxes; numbering is with respect to the transcriptional start site. B: EMSA; KLF3 was expressed in COS-7 cells and binding of nuclear extracts to probe A (lanes 4 and 5), probe B (lanes 6 and 7), and probe C (lanes 8 and 9) assessed. Also shown is binding of KLF3 to a previously validated consensus sequence in the Klf8 promoter (29) (lanes 2 and 3). Binding of untransfected COS nuclear extract to the Klf8 probe is shown in lane 1. α-KLF3 indicates an anti-KLF3 antibody used in supershift to validate KLF3 specific binding. Arrows show the positions of probe complexes bound by KLF3. *KLF3, higher-order multimeric complexes. **KLF3, supershift complexes. C: SL-2 cells were transiently transfected with either pGL4.10[luc2], pGL4.10[luc2] containing a region of the wild-type Fam132a proximal promoter, or pGL4.10[luc2] containing a mutant version of the promoter lacking major CACCC boxes (Fam132aΔCACCC). pPac-KLF1 (250 ng) was used to drive activation of the reporter constructs. Increasing amounts of pPac-KLF3 were included to assess repression: 0 ng (lanes 1, 5, and 9), 25 ng (lanes 2, 6, and 10), 50 ng (lanes 3, 7, and 11), and 250 ng (lanes 4, 8, and 12). Renilla vector pGL4.74[HRluc/TK] (1 μg) was used as a transfection control. Shown is the average of three replicates, with error bars representing SEM. Also included is a cartoon summarizing the luciferase reporter assay strategy. White arrows represent Fam132a promoter CACCC boxes, which can be bound by either KLF1 to drive luciferase activity or by KLF3 to repress expression. Luc, luciferase reporter gene. D: 3T3-L1 cells were transiently transfected with either pGL4.10[luc2], pGL4.10[luc2] containing a region of the wild-type Fam132a proximal promoter, or pGL4.10[luc2] containing a mutant version of the promoter lacking major CACCC boxes (Fam132aΔCACCC). Cells were transfected with increasing amounts of pCDNA3-KLF3 to assess repression: 0 ng (lanes 1, 5, and 9), 50 ng (lanes 2, 6, and 10), 250 ng (lanes 3, 7, and 11), and 500 ng (lanes 4, 8, and 12). Renilla vector pGL4.74[HRluc/TK] (1 μg) was used as a transfection control. Shown is the average of three replicates, with error bars representing SEM. E and F: In vivo binding of KLF3 to the endogenous Fam132a promoter was assessed in 3T3-LI (E) and murine erythroleukemia (F) cells by ChIP. Assays were carried out in duplicate for 3T3-L1 cells and in triplicate for murine erythroleukemia cells. Enrichment was determined by real-time PCR and is relative to input signal and IgG precipitated controls. The lowest signals for anti-KLF3 precipitated chromatin were each set to 1.0. Also shown is KLF3 enrichment at previously reported positive control regions in the Klf8 and C/ebpα promoters and additional negative control regions in the Klf8 and C/ebpα locus, where KLF3 has been shown not to bind (14,29). Error bars show SEM; P values indicate the statistical difference between means for Fam132a, C/ebpα, or Klf8 promoters and negative control region 1 in the Klf8 gene locus. **P < 0.01, *P < 0.05.
To demonstrate that KLF3 regulates Fam132a promoter activity, we performed transient transactivation assays using a luciferase reporter gene under the control of the Fam132a proximal promoter. We first tested the ability of KLF3 to repress promoter activity in Drosophila SL2 cells (Fig. 5C), which are conventionally used for such assays, as they lack the ubiquitously expressed CACCC-box binding proteins found in mammalian cells (26). In these reporter assays, we used KLF1, a transcriptional activator closely related to KLF3 (33), to drive the Fam132a promoter and found that including increasing doses of KLF3 in the assay resulted in strong downregulation of the active promoter. These effects were not observed for a control reporter that lacked the Fam132a promoter or a mutant version of the promoter from which the major KLF3 binding sites had been deleted, thereby confirming that KLF3 can repress Fam132a gene expression. To address whether KLF3 can repress the Fam132a promoter in a physiologically relevant mammalian cell line, we next repeated our reporter assays in 3T3-L1 cells (Fig. 5D). We found that the Fam132a promoter is robustly activated by endogenous factors when transfected into 3T3-L1s and addition of KLF3 again resulted in dose-dependent repression of this activity. Mutation of the CACCC boxes in the promoter resulted in weaker activation by endogenous factors and noticeably reduced KLF3-mediated repression.
Finally, we used ChIP to show that endogenous KLF3 binds to the Fam132a promoter in vivo (Fig. 5E). We first examined 3T3-L1 cells and found significant enrichment of KLF3 at the Fam132a proximal promoter. In agreement with a previous study (14), we also detected binding of KLF3 at the C/ebpα promoter but observed only background levels of KLF3 at known negative control regions in the C/ebpα and Klf8 gene loci (14,29). As our previous microarray studies had shown that Fam132a expression is most notably derepressed in erythroid tissue (32), we decided to investigate binding of KLF3 to the Fam132a promoter in murine erythroleukemia cells, an erythroid cell line particularly amenable to ChIP analysis (Fig. 5F). Our analysis revealed significant enrichment of KLF3 at the Fam132a gene, using primers covering a region of the proximal promoter rich in KLF3 consensus binding sites (Fig. 5A). This enrichment was greater than that seen at a site in the Klf8 proximal promoter, which has previously been confirmed to be bound by KLF3 both in vitro and in vivo (29). Only background binding of KLF3 was observed at negative control regions in the Klf8 promoter, and no significant precipitation of chromatin was seen for immunoglobulin controls.
DISCUSSION
Given the current worldwide epidemic of diet-induced obesity, there is considerable interest in understanding the factors and molecular mechanisms that control physiological responses to metabolic stimuli and how dysregulation of these mechanisms can lead to the metabolic disturbances of glucose intolerance, insulin resistance, and type 2 diabetes. We have previously reported that mice deficient in the transcriptional regulator KLF3 are lean with disrupted adipogenesis (14). In this study, we show that this phenotype is the result of reduced adipocyte size and number and that deletion of the Klf3 gene in vivo results in reduction of WAT depots (subcutaneous and visceral) and liver weight. We also examined the effect of high-fat diet on body mass and found that the absence of KLF3 offers partial protection from obesity, despite no observable change in food intake, energy expenditure, or energy excretion. In addition, glucose tolerance is improved in Klf3−/− mice compared with Klf3+/+ littermates, and high-fat feeding has no significant effect on glucose clearance in knockout mice.
To further explore mechanisms underlying the lean phenotype of Klf3−/− mice, we examined previous microarray data to identify genes whose deregulation might explain the observed resistance to diet-induced obesity, improved glucose tolerance, and insulin sensitivity seen in the absence of KLF3 (32). This analysis revealed that the gene coding for FAM132A/CTRP12/adipolin is upregulated in tissues lacking KLF3. Adipolin has recently been described as an adipokine capable of improving glucose tolerance and insulin sensitivity in both cell lines and mouse models of obesity and diabetes (23,24). Having identified this factor as a potential KLF3 target gene, we confirmed in vitro binding to the Fam132a proximal promoter by EMSA and also demonstrated by ChIP that endogenous KLF3 can interact with the promoter in vivo. We were also able to show that KLF3 can repress Fam132a promoter activity and demonstrate that expression of the Fam132a gene is significantly derepressed in several Klf3−/− tissues, including white adipose but not brown adipose tissue. Moreover, circulating plasma adipolin is markedly elevated in the absence of KLF3 on both chow and high-fat diets.
Upregulation of adipolin potentiates insulin release and improves glucose clearance in fed wild-type mice. In the case of obese and leptin-deficient ob/ob mice, increased adipolin also drives improved glucose tolerance, which is associated with unchanged levels of insulin, suggesting improved insulin sensitivity (23,24). In addition, administration of adipolin in obese mice is reported to reduce body mass and adipocyte size. The effects of systemic upregulation of adipolin are therefore highly aligned to the metabolic phenotype of Klf3−/− mice.
Fam132a gene expression is increased in 3T3-L1 adipocytes treated with insulin or the insulin-sensitizing drug rosiglitazone, while levels are reduced in adipose tissue from ob/ob mice, diet-induced obese mice, and diabetic Zucker rats. Significantly, we found that elevated plasma adipolin levels are maintained in Klf3−/− obese mice. We also noted that serum fatty acids are unchanged in Klf3−/− mice, in agreement with previous observations that elevated adipolin does not affect their levels (23,24). Finally, in this work and previously (32), we observed high levels of Fam132a gene expression in erythroid tissue of Klf3−/− mice, suggesting that this is a potential additional source of the factor.
The discovery that KLF3 regulates expression of a systemic factor capable of significantly improving whole body metabolism also offers a potential explanation for a long-standing paradox in which loss of KLF3 can drive adipogenesis at the cellular level but promote leanness in the whole animal (14). However, the capacity of adipolin to restrict body weight is currently unclear. To date, there have been two short-term studies on the effect of elevating systemic adipolin on body weight in obese mice, with one study reporting a modest but significant reduction in weight, while the other found no difference (23,24). It is possible that persistent upregulation of adipolin and, indeed, other factors in Klf3−/− mice may have a more profound effect on body weight.
We have observed that Klf3−/− male mice are lean and resistant to diet-induced obesity, yet show no detectable changes in energy metabolism. It is possible that this leanness is initiated during embryonic development or early postnatal life, with loss of KLF3 leading to changes in gene expression that cumulatively result in differences in adiposity that are apparent by weaning. It has previously been shown that programming of adipocyte cell number is set before adulthood with implications for fat mass and obesity later in life (34), and the involvement of KLF3 in developmental programs such as this may help explain the early-onset leanness of Klf3−/− mice. Given that this Klf3−/− mouse model is a global knockout, it is quite possible that the complex phenotype of these mice is influenced by the absence of KLF3 in a variety of tissues at different developmental time points.
In summary, we have identified the transcriptional repressor KLF3 as a regulator of Fam132a, the gene coding for the insulin-sensitizing factor adipolin. We have demonstrated that mice deficient in KLF3 have significantly elevated plasma adipolin levels on both chow and high-fat diets and that this is associated with a lean phenotype and improved glucose tolerance and insulin sensitivity. Future studies aimed at modulating adipolin levels in Klf3−/− mice will reveal the extent to which the improved metabolic phenotype of these mice is dependent upon the systemic upregulation of plasma adipolin. In conclusion, regulating the activity of KLF3, potentially via targeting of upstream kinases, has the potential to inform future therapeutic strategies in the treatment of obesity and diabetes.
ACKNOWLEDGMENTS
This work is supported by funding from the Australian National Health and Medical Research Council and the Australian Research Council.
No potential conflicts of interest relevant to this article were reported.
K.S.B.-A. designed and carried out experiments, performed data analysis, and wrote the manuscript. A.P.F., H.W., H.M.J., T.S., W.F.L., J.G.B., K.S.M., and A.J.K. performed experiments. A.J.H., H.R.N., and A.S. reviewed and edited the manuscript. N.T. performed experiments. R.C.P. and M.C. designed experiments, analyzed data, and contributed to writing the manuscript. K.S.B.-A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes 2012;19:81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virtue S, Vidal-Puig A: Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome–an allostatic perspective. Biochimica et Biophysica Acta 2010;1801:338-349 [DOI] [PubMed]
- 3.Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. Krüppel-like transcription factors: a functional family. Int J Biochem Cell Biol 2008;40:1996–2001 [DOI] [PubMed] [Google Scholar]
- 4.Wu Z, Wang S. Role of kruppel-like transcription factors in adipogenesis. Dev Biol 2013;373:235–243 [DOI] [PubMed] [Google Scholar]
- 5.Lefterova MI, Lazar MA. New developments in adipogenesis. Trends Endocrinol Metab 2009;20:107–114 [DOI] [PubMed] [Google Scholar]
- 6.Yien YY, Bieker JJ. EKLF/KLF1, a tissue-restricted integrator of transcriptional control, chromatin remodeling, and lineage determination. Mol Cell Biol 2013;33:4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiol Rev 2010;90:1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab 2008;7:339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oishi Y, Manabe I, Tobe K, et al. Krüppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab 2005;1:27–39 [DOI] [PubMed] [Google Scholar]
- 10.Li D, Yea S, Li S, et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem 2005;280:26941–26952 [DOI] [PubMed] [Google Scholar]
- 11.Pei H, Yao Y, Yang Y, Liao K, Wu JR. Krüppel-like factor KLF9 regulates PPARγ transactivation at the middle stage of adipogenesis. Cell Death Differ 2011;18:315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori T, Sakaue H, Iguchi H, et al. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem 2005;280:12867–12875 [DOI] [PubMed] [Google Scholar]
- 13.Banerjee SS, Feinberg MW, Watanabe M, et al. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem 2003;278:2581–2584 [DOI] [PubMed] [Google Scholar]
- 14.Sue N, Jack BH, Eaton SA, et al. Targeted disruption of the basic Krüppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol Cell Biol 2008;28:3967–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawamura Y, Tanaka Y, Kawamori R, Maeda S. Overexpression of Kruppel-like factor 7 regulates adipocytokine gene expressions in human adipocytes and inhibits glucose-induced insulin secretion in pancreatic beta-cell line. Mol Endocrinol 2006;20:844–856 [DOI] [PubMed] [Google Scholar]
- 16.Gray S, Feinberg MW, Hull S, et al. The Krüppel-like factor KLF15 regulates the insulin-sensitive glucose transporter GLUT4. J Biol Chem 2002;277:34322–34328 [DOI] [PubMed] [Google Scholar]
- 17.Gray S, Wang B, Orihuela Y, et al. Regulation of gluconeogenesis by Krüppel-like factor 15. Cell Metab 2007;5:305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA 2005;102:4807–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanazawa A, Kawamura Y, Sekine A, et al. Single nucleotide polymorphisms in the gene encoding Krüppel-like factor 7 are associated with type 2 diabetes. Diabetologia 2005;48:1315–1322 [DOI] [PubMed] [Google Scholar]
- 20.Zobel DP, Andreasen CH, Burgdorf KS, et al. Variation in the gene encoding Krüppel-like factor 7 influences body fat: studies of 14 818 Danes. Eur J Endocrinol 2009;160:603–609 [DOI] [PubMed] [Google Scholar]
- 21.Small KS, Hedman AK, Grundberg E, et al. GIANT Consortium. MAGIC Investigators. DIAGRAM Consortium. MuTHER Consortium Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet 2011;43:561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearson RC, Funnell AP, Crossley M. The mammalian zinc finger transcription factor Krüppel-like factor 3 (KLF3/BKLF). IUBMB Life 2011;63:86–93 [DOI] [PubMed] [Google Scholar]
- 23.Enomoto T, Ohashi K, Shibata R, et al. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J Biol Chem 2011;286:34552–34558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Z, Peterson JM, Lei X, et al. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem 2012;287:10301–10315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin SH. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol Cell Biol 1996;16:1695–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perdomo J, Verger A, Turner J, Crossley M. Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol Cell Biol 2005;25:1549–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 2009;48:240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hancock D, Funnell A, Jack B, Johnston J. Introducing undergraduate students to real-time PCR. Biochem Mol Biol Educ 2010;38:309–316 [DOI] [PubMed] [Google Scholar]
- 29.Eaton SA, Funnell AP, Sue N, Nicholas H, Pearson RC, Crossley M. A network of Krüppel-like Factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J Biol Chem 2008;283:26937–26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell-Anderson KS, Aouad L, Williams H, et al. Coordinated improvement in glucose tolerance, liver steatosis and obesity-associated inflammation by cannabinoid 1 receptor antagonism in fat Aussie mice. Int J Obes (Lond) 2011;35:1539–1548 [DOI] [PubMed] [Google Scholar]
- 31.Lam YY, Ha CW, Campbell CR, et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 2012;7:e34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funnell AP, Norton LJ, Mak KS, et al. The CACCC-binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol Cell Biol 2012;32:3281–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funnell AP, Maloney CA, Thompson LJ, et al. Erythroid Krüppel-like factor directly activates the basic Krüppel-like factor gene in erythroid cells. Mol Cell Biol 2007;27:2777–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature 2008;453:783–787 [DOI] [PubMed] [Google Scholar]