

Abstract
Rapamycin is used frequently in both transplantation and oncology. Although historically thought to have little diabetogenic effect, there is growing evidence of β-cell toxicity. This Review draws evidence for rapamycin toxicity from clinical studies of islet and renal transplantation, and of rapamycin as an anticancer agent, as well as from experimental studies. Together, these studies provide evidence that rapamycin has significant detrimental effects on β-cell function and survival and peripheral insulin resistance. The mechanism of action of rapamycin is via inhibition of mammalian target of rapamycin (mTOR). This Review describes the complex mTOR signaling pathways, which control vital cellular functions including mRNA translation, cell proliferation, cell growth, differentiation, angiogenesis, and apoptosis, and examines molecular mechanisms for rapamycin toxicity in β-cells. These mechanisms include reductions in β-cell size, mass, proliferation and insulin secretion alongside increases in apoptosis, autophagy, and peripheral insulin resistance. These data bring into question the use of rapamycin as an immunosuppressant in islet transplantation and as a second-line agent in other transplant recipients developing new-onset diabetes after transplantation with calcineurin inhibitors. It also highlights the importance of close monitoring of blood glucose levels in patients taking rapamycin as an anticancer treatment, particularly those with preexisting glucose intolerance.
The macrolide rapamycin is both an antiproliferative and potent immunosuppressant. It is produced commercially as sirolimus and its derivative, everolimus. Sirolimus is predominantly used as an immunosuppressant in transplantation, while everolimus is used mainly as an anticancer agent. Early data suggested little or no diabetogenic effects of rapamycin, particularly in comparison with other immunosuppressants, and it was this that prompted its use in islet cell transplantation. It was also for this reason that rapamycin has been promoted as a second-line therapy for recipients of solid organ transplants who have developed new-onset diabetes after transplantation (NODAT) while taking calcineurin inhibitors (CNIs). However, there is an increasing view that rapamycin has profound effects on pancreatic β-cells, as well as altering insulin sensitivity. Evidence for this arises from in vitro and in vivo experiments and clinical studies. This article will review this evidence and also explore the potential mechanisms of rapamycin toxicity, drawn from experiments of β-cell physiology.
Clinical evidence of rapamycin β-cell toxicity
Islet cell transplantation.
Rapamycin has been one of the primary immunosuppressants for islet cell transplantation since the publication of the landmark Edmonton study in 2000 (1). The Edmonton immunosuppression protocol was designed to avoid the diabetogenic effects of corticosteroids and to minimize the effects of tacrolimus. Rapamycin was the main immunosuppressant used, as it was thought to have little or no detrimental effects on islet survival or function. Initial results were promising, with seven consecutive islet transplant recipients attaining insulin independence over a median follow-up of 11.9 months. After the initial success of the Edmonton protocol, a number of other centers adopted a similar regimen for their islet transplant programs. However, enthusiasm was tempered when the 5-year results of the initial cohort of patients from Edmonton was reported, with only ∼10% maintaining insulin independence (2). Long-term results from other centers using the Edmonton protocol were equally disappointing, with insulin independence at 2 years ranging from 14 to 20% (3–5).
Although the cause of this decline in islet graft function is multifactorial, there is evidence that it is partly related to the toxicity of the immunosuppressive agents used. For example, pathological examination of one patient who died with a failed islet transplant performed under the Edmonton protocol and one who died with a functioning islet transplant showed no evidence of allo- or autoimmune damage to the transplanted islets (6,7). This provides evidence for predominantly nonimmunological causes for the chronic loss of intrahepatic islets, such as toxicity from immunosuppressive agents. Interestingly, in the context of this Review, arguably the most promising long-term survival data reported for islet transplantation to date was achieved with a regimen that avoided rapamycin after the first year posttransplant (8). This resulted in 80% insulin independence at over 3 years posttransplant.
Solid organ transplantation.
Rapamycin is used predominantly for immunosuppression after renal transplantation, although it is also given after pancreas, liver, and cardiac transplants. Evidence for rapamycin toxicity in β-cells can be obtained from studies of NODAT in patients receiving rapamycin. One study showed that tacrolimus to rapamycin conversion in renal transplant recipients was associated with a 30% increase in impaired glucose tolerance (9). Furthermore, a study of renal transplant recipients from the U.S. Renal Data System showed that patients treated with rapamycin in combination with either tacrolimus or cyclosporine had the highest incidence of NODAT (10). Other studies have found sirolimus, on multivariate analysis, to be a risk factor for NODAT in kidney transplant recipients (11–15). Furthermore, in a large-scale randomized control trial of immunosuppressive regimens in renal transplantation, sirolimus was associated with the highest incidence of hyperglycemia (5 vs. 4.7% low-dose tacrolimus vs. 4.4% high-dose cyclosporine vs. 2.9% low-dose cyclosporine), although the incidence of NODAT was higher in the tacrolimus group (16). However, as patients in these studies also received other immunosuppressants, including corticosteroids, it is not possible to determine the exact influence of rapamycin on the development of NODAT. However, as part of their U.S. Renal Data System study, Johnston et al. (10) analyzed the risk of NODAT in renal transplant recipients receiving tacrolimus in combination with mycophenolate mofetil (MMF) or azathioprine versus those receiving tacrolimus and sirolimus. This demonstrated a hazard ratio of 1.25 (95% CI 1.03–1.52), suggesting an increased risk for NODAT from sirolimus independent of any effect of tacrolimus.
The data on the risk of NODAT with sirolimus use after liver transplantation are more sparse. However, in one study the incidence of NODAT in liver transplant recipients receiving sirolimus without CNIs was 10.5% compared with 29.4% in a historical control group receiving only CNIs (17).
Further insights into the effects of rapamycin on glucose homeostasis can be drawn from studies of its use in whole-organ pancreas transplantation. Reports of sirolimus use in combination with CNIs as part of steroid-free (18) or rapid steroid elimination (19) regimens after pancreas transplantation have shown equivalent endocrine graft function compared with patients receiving mycophenolate, CNIs, and steroids. The greater exposure to corticosteroids in the comparator groups does, however, bias the results in favor of sirolimus. More robust data come from a randomized trial of rapamycin versus mycophenalate after simultaneous kidney pancreas transplantation, with otherwise identical immunosuppressive regimens. This demonstrated significantly lower HbA1c levels in the mycophenalate group, while 6% of patients on rapamycin developed NODAT compared with 3% in the mycophenalate group (20). A further nonrandomized comparison of rapamycin with mycophenalate in pancreas transplant recipients also receiving tacrolimus demonstrated significantly lower plasma insulin levels during an intravenous glucose tolerance test with rapamycin (21).
Anticancer agent.
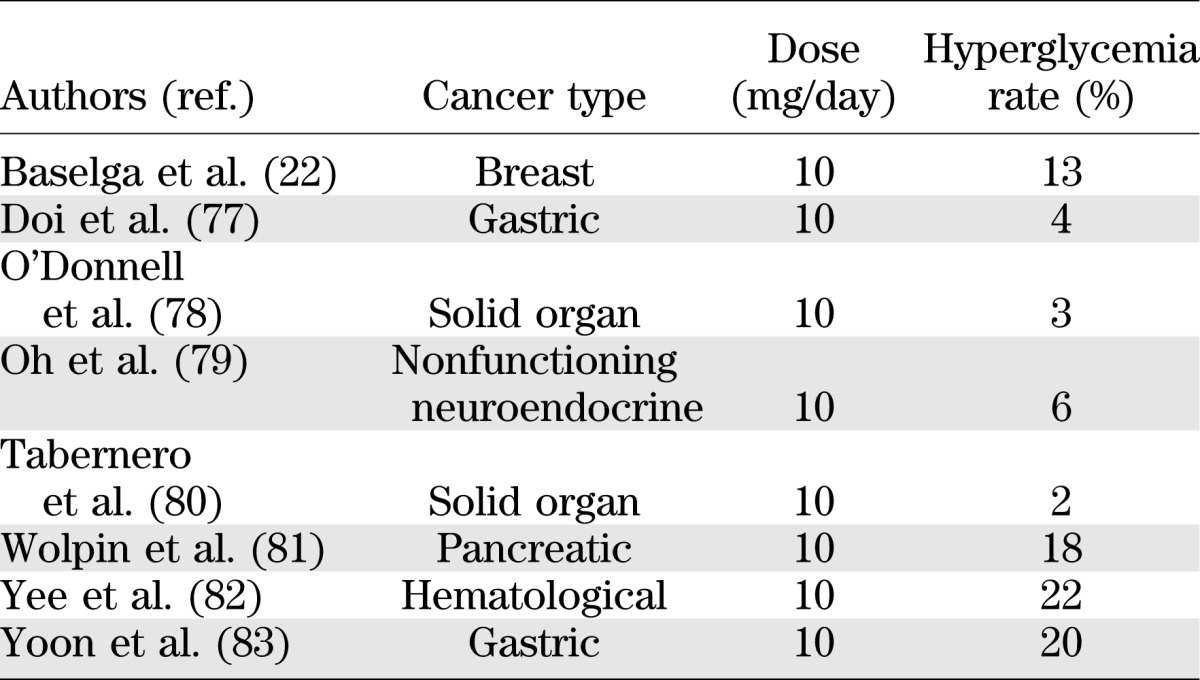
The rapamycin derivative everolimus is licensed for the treatment of advanced renal cell, neuroendocrine, and breast cancers. Evidence for pancreatic β-cell toxicity can be gleaned from rates of hyperglycemia reported in studies of everolimus monotherapy given for various advanced cancers. Reported rates of severe hyperglycemia are shown in Table 1. There are difficulties in interpreting such data, such as lack of information on the number of patients with preexisting diabetes and the potential tumor effects contributing to the hyperglycemia. However, the overall inference is that everolimus does have a detrimental effect on pancreatic β-cells. The randomized control trial by Baselga et al. (22) provides more persuasive evidence. Patients with advanced breast cancer received an aromatase inhibitor alone or in combination with everolimus. The incidence of hyperglycemia was 13% in the everolimus group against 2% in the noneverolimus group. As a randomized control group, this study has fewer of the confounding factors seen in the studies above and provides more robust evidence of everolimus toxicity in β-cells. It is important to note that the dose of everolimus (10 mg/day) used in the above oncological studies is higher than that used after transplantation (1.5 mg/day) and, as such, the relevance of these findings to a transplant setting is reduced.
TABLE 1.
Reported rates of severe hyperglycemia in oncological studies of everolimus monotherapy
Everolimus has also been used successfully as a treatment for refractory hypoglycemia in unresectable or metastatic insulinoma (23–25). Although some of this benefit may result from the antitumor effect of the drug, improvement in hypoglycemia has also been seen in patients with no radiological tumor regression. This suggests a direct effect of everolimus on glycemic control, which appears to be due to both a reduction in serum insulin levels and reduced peripheral uptake of glucose in muscles (23).
A further clinical study of interest is a phase I clinical trial of a combination of rapamycin and interleukin-2 treatment in type 1 diabetic patients (26). The hypothesis was that this treatment would reduce autoimmune damage to β-cells and improve function. Interestingly, the combination therapy resulted in transient impairment of β-cell function, as measured by C-peptide levels. However, what is not clear is whether this impairment was due to rapamycin, interleukin-2, or a combination of both. Furthermore, it is not possible to determine whether the impairment was due to direct effects on β-cells or due to indirect effects on immune regulatory cells.
Taken as the whole, the evidence from studies of rapamycin use for different clinical indications does point toward rapamycin having a detrimental effect on glucose homeostasis. However, the majority of these studies have significant confounding factors, precluding any definitive conclusion. Also, none of these studies are able to address whether the hyperglycemia seen is predominantly due to a result of direct effects on β-cells or due to peripheral effects on glucose metabolism, such as increases in insulin resistance.
Direct effects of rapamycin on pancreatic β-cells.
There have been a number of studies investigating the direct effects of rapamycin on pancreatic β-cell function, survival, and proliferation, which are summarized in Tables 2–4. This research is heterogeneous, using a variety of cell types and rapamycin concentrations. There is some disparity in the results, which likely reflects differing rapamycin doses and treatment periods. It may also reflect species-specific differences in rapamycin sensitivity. A further explanation for the disparities is that it is a reflection of islet purity. A study comparing the effects of rapamycin on pure (>90%) and impure (40–60%) islets found glucose-stimulated insulin secretion (GSIS) to be significantly higher in impure islets treated with rapamycin but saw no difference in pure islets (27). Overall, the majority of these studies demonstrate significant effects of rapamycin on glucose homeostasis, and the combined evidence strongly suggests that rapamycin adversely affects GSIS from β-cells.
TABLE 2.
Summary of studies investigating the effects of rapamycin on pancreatic β-cell function and insulin sensitivity
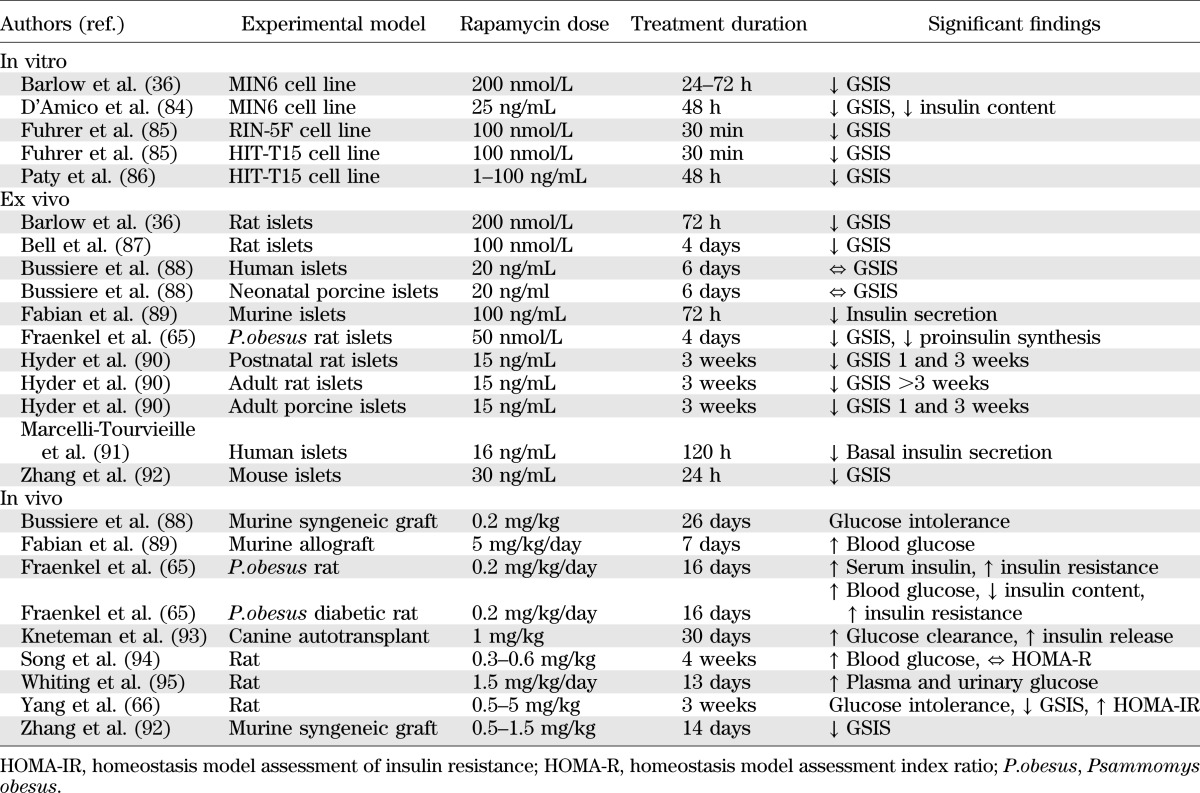
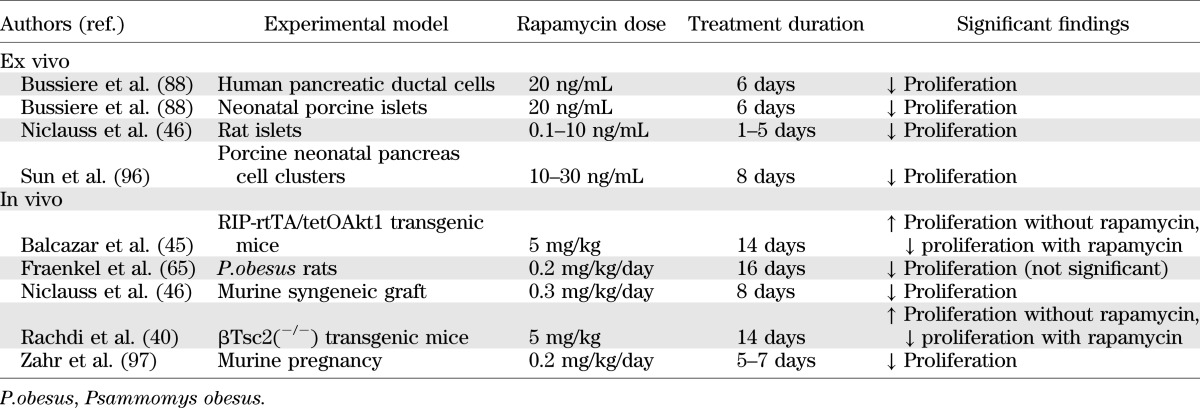
TABLE 4.
Summary of studies investigating the effects of rapamycin on pancreatic β-cell proliferation
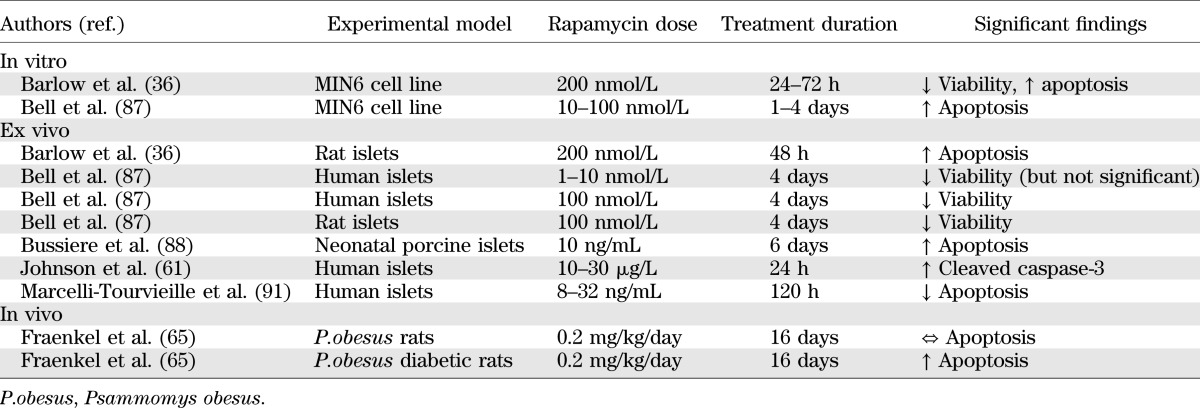
Viability studies in murine and human β-cells treated with rapamycin conclusively demonstrate significant detrimental effects on cell survival, with increased levels of apoptosis (Table 3). Importantly, this includes studies in human islets at clinically relevant rapamycin doses similar to those measured in the portal vein of islet transplant recipients (28).
TABLE 3.
Summary of studies investigating the effects of rapamycin on pancreatic β-cell survival
One downside of in vitro studies is that only the acute effects of rapamycin over a few days can be studied because of the difficulties in maintaining islets in culture for extended time periods. Of greater clinical importance is the effect over the months and years patients will be taking rapamycin. Yet, even after a relatively short period of incubation with rapamycin cultured islets show signs of decreased function and viability (Tables 2 and 3).
Mechanism of action of rapamycin.
In order to appreciate the possible mechanisms of rapamycin toxicity, it is important to understand its mechanism of action. Rapamycin binds to the immunophilin FK506-binding protein 12 (FKBP12) to form a complex that binds to and inhibits the serine/threonine kinase mammalian target of rapamycin (mTOR) (29). This kinase is a key regulator of cell metabolism, growth, and proliferation. Importantly, the inhibition of mTOR by rapamycin results in cell cycle arrest in mid- to late G1 phase and thus has the potential to repress tumor cell growth and, importantly with respect to its immunosuppressive function, inhibit T- and B-cell proliferation. However, FKBP12 and mTOR are ubiquitously expressed. Thus, there is the potential for possible “off target” effects on cells other than tumor and immunoregulatory cells.
mTOR.
The mTOR kinase exists in two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). These are differentially regulated and have distinct substrates (Fig. 1). Although they share some core components such as mTOR, mLST8, and DEPTOR, they also contain additional proteins that are distinct and define them. For example, a unique component of mTORC1 is RAPTOR (regulatory associated protein of mTOR), which serves as a scaffold binding both mTOR and its downstream effectors (30,31). An essential component of mTORC2 is the protein Rictor (rapamycin-insensitive companion of mTOR), which is required for both mTORC2 complex formation and its kinase activity (32,33). Importantly, mTORC1 is acutely sensitive to inhibition by rapamycin, whereas mTORC2, although originally thought to be resistant to rapamycin (32,33), is in fact sensitive to prolonged rapamycin treatment in certain cell types (34–36). Thus, both complexes could potentially play a role in both the immunosuppressive and toxic effects of rapamycin.
FIG. 1.

mTOR signaling pathways. After stimulation by insulin and other growth factors, phosphatidylinositol 3-kinase (PI3K) converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3), which localizes PKB to the membrane where it is activated by PDK1 and mTORC2. Activated PKB phosphorylates and inhibits tuberous sclerosis complex (TSC1/2). Rheb, a small GTPase that is inhibited by TSC2, positively modulates mTORC1 activity. mTORC1 phosphorylates S6 kinase 1/2 and 4EBP1, resulting in increased mRNA translation. Amino acid sufficiency activates mTORC1 via Rag A/B and C/D. Under low-energy conditions, the AMP-to-ATP ratio rises and activates AMP kinase (AMPK), which phosphorylates and activates the TSC1/2 complex, resulting in mTORC1 inhibition. mTORC2 activity is mediated via predominantly unknown pathways. mTORC2 phosphorylates and activates PKB, serum- and glucocorticoid-induced protein kinase 1 (SGK1), and PKC. Arrows, stimulatory effects; block ends, inhibitory effects; solid lines, direct effects, dashed lines, indirect effects. Atg13, autophagy-related protein 13; DAP1, death-associated protein 1; Deptor, DEP domain–containing mTOR-interacting protein; 4EBP, eIF4E-binding protein; GβL- G, protein β-subunit-like protein; HIF1, hypoxia-induced factor 1; IMP2, insulin-like growth factor 2 mRNA-binding protein; mLST8, mammalian lethal with Sec13 protein 8; PDK1, phosphoinositide-dependent protein kinase 1; Protor, protein observed with Rictor; Raptor, regulatory associated protein of mTOR; Rictor, rapamycin-insensitive companion of mTOR; Sin1, stress-activated protein kinase–interacting protein 1; TFIIIC, transcription factor 3C; ULK1, Unc-51–like kinase 1.
mTORC1.
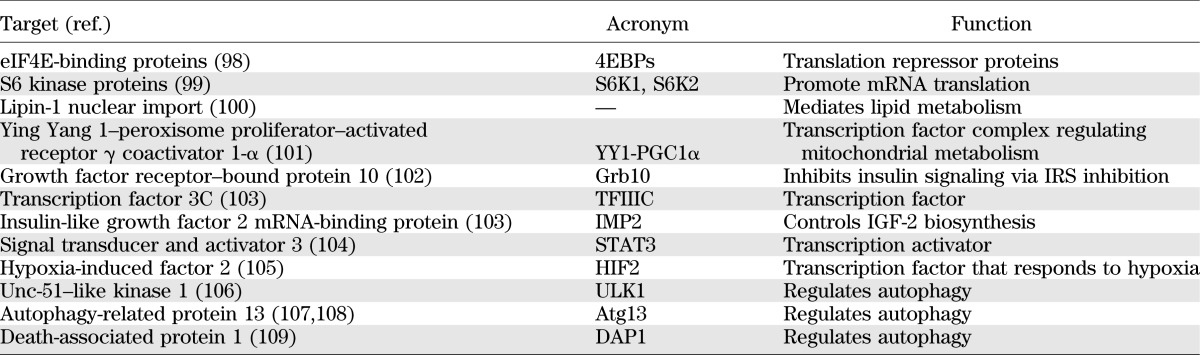
Consistent with its role as a key regulator of cell metabolism, proliferation, and growth, mTORC1 activity is regulated by nutrients, growth factors, and cellular energy levels (Fig. 1). The best characterized targets of mTORC1 are the eIF4E-binding proteins (4E-BPs) and the S6 kinase proteins (S6K), both of which play important roles in the regulation of protein synthesis. However, a number of other downstream targets have been identified (Table 5).
TABLE 5.
Downstream targets of mTORC1
Role of mTORC1 in β-cell function.
A vital aspect in the preservation of glucose homeostasis is the maintenance of pancreatic β-cell mass and also the ability for β-cell mass to increase in response to insulin resistant states such as obesity. This increase in β-cell mass results from increases in neogenesis (generation from progenitor cells) and proliferation (hyperplasia), hypertrophy, and reductions in apoptosis.
There is evidence from a number of studies that rapamycin significantly reduces proliferation of both β-cells and progenitor cells (Table 4), with consequences for the maintenance of β-cell mass.
Although in vitro studies provide important insights into the role of mTORC1 in the regulation of β-cell mass, more compelling evidence comes from in vivo transgenic mouse models (rev. in 37). Hyperactivation of mTORC1 by overexpression of Rheb (38) or deletion of TSC1 (39) or TSC2 (40,41), selectively in β-cells, leads to increases in β-cell size and mass, coincident with improvements in insulin secretion and glucose tolerance. These effects may be mediated, at least in part, through S6K, as mice deficient in S6K1 or rpS6 are also hypoinsulinemic and glucose intolerant with diminished β-cell size (42,43). Furthermore, transgenic mice overexpressing constitutively active S6K exhibit improved glucose tolerance and enhanced insulin secretion, with increased β-cell size (44). Although these studies are strongly suggestive of a key role of mTORC1, it is possible that manipulation of upstream regulators of mTOR such as Rheb may affect pathways other than mTORC1, and therefore a causative effect cannot be categorically proven.
There is an immense body of work investigating the role of mTOR in the regulation of cell proliferation for certain cell types but much less knowledge on the exact mechanisms by which mTORC1 signaling regulates β-cell cycle progression. However, mTORC1 is known to modulate the synthesis and stability of cyclin D2 and D3 in β-cells (45). These cyclins form a complex with cyclin-dependent kinase 4, which controls cell cycle progression. Interestingly, reduced cyclin D1 and D2 levels have been observed in rat islets treated with rapamycin, with associated reduction in β-cell proliferation (46).
mTORC1 also appears to play a role in insulin secretion in pancreatic β-cells. Knockdown of TSC1 in mice results in significant increases in insulin production, independent of β-cell number (39). In addition, chronic treatment with rapamycin inhibits GSIS in clonal β-cell lines as well as rodent and human islets (Table 1). However, whether this effect is via mTORC1 or mTORC2 is unclear. The control of insulin secretion in β-cells involves a number of complex signaling pathways, and as such the mechanisms by which rapamycin may regulate insulin secretion remain unknown. One proposed mechanism is that inhibition of mTORC1 decreases mitochondrial function, specifically, the activity of α-ketoglutarate dehydrogenase. This results in reduced carbohydrate metabolism and therefore reduced mitochondrial ATP production (47), which is known to regulate insulin secretion in β-cells (48). Other explanations are that rapamycin promoted autophagy, a process primarily controlled by mTORC1 rather than mTORC2, or that the intracellular degradation of cytoplasmic proteins involved in insulin production results in the inhibition of insulin secretion (49).
mTORC2.
It is not entirely clear how mTORC2 activity is regulated, but there is evidence that it can be stimulated by amino acids and growth factors (50,51). Downstream targets of mTORC2 include protein-kinase C (PKC)-α (44–46) and protein kinase B (PKB) (52)—both serine/threonine kinases that play roles in the regulation of a number of key cellular processes including apoptosis, proliferation, motility, and differentiation—and serum- and glucocorticoid-induced protein kinase 1 (53), which has a role in control of ion transport (54).
Role of mTORC2 in pancreatic β-cell homeostasis.
β-cell–specific deletion of Rictor, an essential component of mTORC2, in mice is associated with reduced plasma insulin levels due to reduced insulin secretion from islets with resultant hyperglycemia (55). This was associated with reductions in β-cell mass and proliferation but no increase in β-cell apoptosis. Our group has demonstrated that knockdown of Rictor by small interfering RNA in rat islets results in increases in β-cell apoptosis and reduced GSIS (36). These studies specifically demonstrate that mTORC2 activity plays a leading role in both β-cell survival and function. Importantly, prolonged rapamycin treatment (24 h) of MIN6 cells, rat islets, or human islets (36) results in the inhibition of mTORC2 through the dissociation of mTORC2. This precedes the toxic effects of rapamycin on both function and viability and is coincident with a decrease in PKB phosphorylation and downstream signaling. Interestingly, the expression of constitutive active PKB in MIN6 cells and rat islets can ameliorate the detrimental effects of rapamycin on GSIS and cell viability (36). Taken together, this suggests that rapamycin β-cell toxicity may be predominantly mediated via inhibition of mTORC2 and its subsequent effect on PKB signaling. However, this is based on in vitro experiments in β-cells and requires further confirmation in vivo.
A large number of studies have demonstrated that PKB, a key downstream effector of mTORC2, plays an important role in both survival and function of β-cells. These studies provide further insight into the possible role of mTORC2 in β-cell homeostasis. For example, transgenic mice expressing a constitutively active PKB in β-cells exhibit significant increases in β-cell mass resulting from an increase in both β-cell number and size (56,57). This manifested in significantly higher plasma insulin levels, improved glucose tolerance, and resistance to streptozotocin-induced diabetes. Expression of constitutively active PKB has also been shown to protect against fatty acid–mediated (58), cytokine-mediated (59), and AMP kinase–mediated (60) cytotoxicity in INS-1 cells, a rat β-cell line, and primary rat β-cells, respectively. Conversely, studies in transgenic mice lacking PKB show significantly higher blood glucose levels, lower insulin levels, and impaired glucose tolerance.
These studies raise the question of whether in vivo activation of PKB might improve the outcome of islet transplantation by improving the function and survival of transplanted β-cells or, indeed, provide some protection against rapamycin-induced NODAT. There are a number of potential pharmacological mechanisms by which PKB can be activated in β-cells in vivo. Experimental data highlight GLP-1 (61), erythropoietin, and statins (62,63) as promising agents that warrant clinical investigation.
Effects of rapamycin on insulin resistance.
In addition to the direct effects of rapamycin on β-cells described above, there is evidence that rapamycin has effects on peripheral insulin resistance (64–66), which could also contribute to the changes in glucose homeostasis seen in animal models and human studies. Rapamycin has been shown to inhibit phosphorylation of IRS-1 and IRS-2 in human adipocytes, mimicking changes in type 2 diabetes (67,68), and in human peripheral blood monocytes with associated insulin resistance (69). Interestingly, recent evidence suggests that this insulin resistance is mediated predominantly via mTORC2 rather than mTORC1 (70–72).
Conclusions.
Rapamycin is a key immunosuppressant, particularly in islet cell and kidney transplantation. However, there is a good body of both in vitro and in vivo evidence strongly suggesting that rapamycin has detrimental effects on pancreatic β-cells and peripheral insulin sensitivity. This has implications for islet transplant function and the development in NODAT in solid-organ transplantation. This toxicity is perhaps unsurprising, given that rapamycin inhibits mTOR, which via mTORC1 and mTORC2 is part of complex signaling pathways controlling a host of important cellular functions including mRNA translation, cell proliferation, cell growth, differentiation, protein synthesis, angiogenesis, and apoptosis.
This has important implications and brings into serious question the use of rapamycin as a primary immunosuppressant in islet transplantation and as a second-line agent in transplant recipients developing NODAT with calcineurin inhibitors. However, there are limited alternatives. One alternative is MMF, another antiproliferative agent that exerts its immunosuppressive effects via inhibition of inosine monophosphate dehydrogenase (73). However, studies in human islets have shown MMF treatment to result in significant reductions in GSIS (61). Despite this, MMF is now in increasing use in clinical islet programs, while the use of rapamycin has declined. Of note, this change in immunosuppressive strategy has coincided with an improvement in primary efficacy seen from the Collaborative Islet Transplant Registry (74). Indeed the Edmonton group, who was the original proponent of rapamycin in islet transplantation, no longer uses it, although this choice was because of the side effects of rapamycin rather than perceived toxicity to β-cells.
A promising new immunosuppressant is sotrastaurin, a PKC inhibitor, which has been used in phase II trials in kidney transplantation (75). Of particular note, sotrastaurin does not appear to have detrimental effects on cultured human islets or those transplanted into immunodeficient mice (76).
Given the suggestion that both rapamycin β-cell toxicity and insulin resistance may be mediated predominantly via mTORC2 rather than mTORC1 (36,55), the question arises whether an mTORC1-specific inhibitor would retain the immunosuppressive effects of rapamycin without any mTORC2-mediated toxicity. However, this involves the assumption that the immunosuppressive effects of rapamycin are indeed mediated predominantly via mTORC1, which is not currently known.
Further clinical investigation is also required regarding glucose homeostasis in patients receiving rapamycin to establish the relative influence of β-cell dysfunction and peripheral insulin resistance in the development of hyperglycemia.
In conclusion, the body of evidence from in vivo and in vitro studies strongly suggests that rapamycin has profound effects on glucose homeostasis. Potential mechanisms involved in this include reductions in β-cell size, mass, proliferation, and insulin secretion, alongside increases in apoptosis, autophagy, and peripheral insulin resistance.
ACKNOWLEDGMENTS
No potential conflicts of interest relevant to this article were reported.
A.D.B. conceived the manuscript, reviewed the literature, and wrote and edited the manuscript. M.L.N. conceived, wrote, and edited the manuscript. T.P.H. conceived the manuscript, reviewed the literature, and wrote and edited the manuscript.
REFERENCES
- 1.Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000;343:230–238 [DOI] [PubMed] [Google Scholar]
- 2.Ryan EA, Paty BW, Senior PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes 2005;54:2060–2069 [DOI] [PubMed] [Google Scholar]
- 3.Shapiro AM, Ricordi C, Hering BJ, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med 2006;355:1318–1330 [DOI] [PubMed] [Google Scholar]
- 4.Frank A, Deng S, Huang X, et al. Transplantation for type I diabetes: comparison of vascularized whole-organ pancreas with isolated pancreatic islets. Ann Surg 2004;240:631–640; discussion 640–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerber PA, Pavlicek V, Demartines N, et al. Simultaneous islet-kidney vs pancreas-kidney transplantation in type 1 diabetes mellitus: a 5 year single centre follow-up. Diabetologia 2008;51:110–119 [DOI] [PubMed] [Google Scholar]
- 6.Davalli AM, Maffi P, Socci C, et al. Insights from a successful case of intrahepatic islet transplantation into a type 1 diabetic patient. J Clin Endocrinol Metab 2000;85:3847–3852 [DOI] [PubMed] [Google Scholar]
- 7.Smith RN, Kent SC, Nagle J, et al. Pathology of an islet transplant 2 years after transplantation: evidence for a nonimmunological loss. Transplantation 2008;86:54–62 [DOI] [PubMed] [Google Scholar]
- 8.Bellin MD, Kandaswamy R, Parkey J, et al. Prolonged insulin independence after islet allotransplants in recipients with type 1 diabetes. Am J Transplant 2008;8:2463–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 2005;16:3128–3135 [DOI] [PubMed] [Google Scholar]
- 10.Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol 2008;19:1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Hutchinson II, Shah T, Min DI. Genetic and clinical risk factors of new-onset diabetes after transplantation in Hispanic kidney transplant recipients. Transplantation 2011;91:1114–1119 [DOI] [PubMed] [Google Scholar]
- 12.Van Laecke S, Van Biesen W, Verbeke F, De Bacquer D, Peeters P, Vanholder R. Posttransplantation hypomagnesemia and its relation with immunosuppression as predictors of new-onset diabetes after transplantation. Am J Transplant 2009;9:2140–2149 [DOI] [PubMed] [Google Scholar]
- 13.Roland M, Gatault P, Doute C, et al. Immunosuppressive medications, clinical and metabolic parameters in new-onset diabetes mellitus after kidney transplantation. Transpl Int 2008;21:523–530 [DOI] [PubMed] [Google Scholar]
- 14.Gyurus E, Kaposztas Z, Kahan BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc 2011;43:1583–1592 [DOI] [PubMed] [Google Scholar]
- 15.Bee YM, Tan HC, Tay TL, Kee TY, Goh SY, Kek PC. Incidence and risk factors for development of new-onset diabetes after kidney transplantation. Ann Acad Med Singapore 2011;40:160–167 [PubMed] [Google Scholar]
- 16.Ekberg H, Tedesco-Silva H, Demirbas A, et al. ELITE-Symphony Study Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007;357:2562–2575 [DOI] [PubMed] [Google Scholar]
- 17.Vivarelli M, Dazzi A, Cucchetti A, et al. Sirolimus in liver transplant recipients: a large single-center experience. Transplant Proc 2010;42:2579–2584 [DOI] [PubMed] [Google Scholar]
- 18.Rajab A, Pelletier RP, Ferguson RM, Elkhammas EA, Bumgardner GL, Henry ML. Steroid-free maintenance immunosuppression with rapamune and low-dose neoral in pancreas transplant recipients. Transplantation 2007;84:1131–1137 [DOI] [PubMed] [Google Scholar]
- 19.Kaufman DB, Leventhal JR, Koffron AJ, et al. A prospective study of rapid corticosteroid elimination in simultaneous pancreas-kidney transplantation: comparison of two maintenance immunosuppression protocols: tacrolimus/mycophenolate mofetil versus tacrolimus/sirolimus. Transplantation 2002;73:169–177 [DOI] [PubMed] [Google Scholar]
- 20.Ciancio G, Sageshima J, Chen L, et al. Advantage of rapamycin over mycophenolate mofetil when used with tacrolimus for simultaneous pancreas kidney transplants: randomized, single-center trial at 10 years. Am J Transplant 2012;12:3363–3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havrdova T, Saudek F, Boucek P, et al. Metabolic effect of sirolimus versus mycophenolate mofetil on pancreatic graft function in the early posttransplant period. Transplant Proc 2005;37:3544–3545 [DOI] [PubMed] [Google Scholar]
- 22.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiebrich HB, Siemerink EJ, Brouwers AH, et al. Everolimus induces rapid plasma glucose normalization in insulinoma patients by effects on tumor as well as normal tissues. Oncologist 2011;16:783–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med 2009;360:195–197 [DOI] [PubMed] [Google Scholar]
- 25.Bernard V, Lombard-Bohas C, Taquet MC, et al. French Group of Endocrine Tumors Efficacy of everolimus in patients with metastatic insulinoma and refractory hypoglycemia. Eur J Endocrinol 2013;168:665–674 [DOI] [PubMed] [Google Scholar]
- 26.Long SA, Rieck M, Sanda S, et al. Diabetes TrialNet and the Immune Tolerance Network Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs β-cell function. Diabetes 2012;61:2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mita A, Ricordi C, Miki A, et al. Anti-proinflammatory effects of sirolimus on human islet preparations. Transplantation 2008;86:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai NM, Goss JA, Deng S, et al. Elevated portal vein drug levels of sirolimus and tacrolimus in islet transplant recipients: local immunosuppression or islet toxicity? Transplantation 2003;76:1623–1625 [DOI] [PubMed] [Google Scholar]
- 29.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994;78:35–43 [DOI] [PubMed] [Google Scholar]
- 30.Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002;110:177–189 [DOI] [PubMed] [Google Scholar]
- 31.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002;110:163–175 [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004;14:1296–1302 [DOI] [PubMed] [Google Scholar]
- 33.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004;6:1122–1128 [DOI] [PubMed] [Google Scholar]
- 34.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006;22:159–168 [DOI] [PubMed] [Google Scholar]
- 35.Zeng Z, Sarbassov D, Samudio IJ, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007;109:3509–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barlow AD, Xie J, Moore CE, et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia 2012;55:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie J, Herbert TP. The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic β-cell mass: implications in the development of type-2 diabetes. Cell Mol Life Sci 2012;69:1289–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamada S, Hara K, Hamada T, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes 2009;58:1321–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori H, Inoki K, Opland D, et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am J Physiol Endocrinol Metab 2009;297:E1013–E1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rachdi L, Balcazar N, Osorio-Duque F, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci USA 2008;105:9250–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shigeyama Y, Kobayashi T, Kido Y, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 2008;28:2971–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruvinsky I, Sharon N, Lerer T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005;19:2199–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pende M, Kozma SC, Jaquet M, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000;408:994–997 [DOI] [PubMed] [Google Scholar]
- 44.Elghazi L, Balcazar N, Blandino-Rosano M, et al. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes 2010;59:2390–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balcazar N, Sathyamurthy A, Elghazi L, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem 2009;284:7832–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niclauss N, Bosco D, Morel P, Giovannoni L, Berney T, Parnaud G. Rapamycin impairs proliferation of transplanted islet β cells. Transplantation 2011;91:714–722 [DOI] [PubMed] [Google Scholar]
- 47.Shimodahira M, Fujimoto S, Mukai E, et al. Rapamycin impairs metabolism-secretion coupling in rat pancreatic islets by suppressing carbohydrate metabolism. J Endocrinol 2010;204:37–46 [DOI] [PubMed] [Google Scholar]
- 48.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature 2001;414:807–812 [DOI] [PubMed] [Google Scholar]
- 49.Tanemura M, Nagano H, Taniyama K, Kamiike W, Mori M, Doki Y. Role of rapamycin-induced autophagy in pancreatic islets. Am J Transplant 2012;12:1067. [DOI] [PubMed] [Google Scholar]
- 50.Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 2011;286:10998–11002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem 2011;286:6128–6142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 53.García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J 2008;416:375–385 [DOI] [PubMed] [Google Scholar]
- 54.Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J Biol Chem 2004;279:38134–38142 [DOI] [PubMed] [Google Scholar]
- 55.Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 2011;60:827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 2001;108:1631–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 2001;7:1133–1137 [DOI] [PubMed] [Google Scholar]
- 58.Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J Biol Chem 2002;277:49676–49684 [DOI] [PubMed] [Google Scholar]
- 59.Collier JJ, Fueger PT, Hohmeier HE, Newgard CB. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes 2006;55:1398–1406 [DOI] [PubMed] [Google Scholar]
- 60.Cai Y, Wang Q, Ling Z, et al. Akt activation protects pancreatic beta cells from AMPK-mediated death through stimulation of mTOR. Biochem Pharmacol 2008;75:1981–1993 [DOI] [PubMed] [Google Scholar]
- 61.Johnson JD, Ao Z, Ao P, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant 2009;18:833–845 [DOI] [PubMed] [Google Scholar]
- 62.Contreras JL, Smyth CA, Bilbao G, Young CJ, Thompson JA, Eckhoff DE. Simvastatin induces activation of the serine-threonine protein kinase AKT and increases survival of isolated human pancreatic islets. Transplantation 2002;74:1063–1069 [DOI] [PubMed] [Google Scholar]
- 63.Favaro E, Miceli I, Bussolati B, et al. Hyperglycemia induces apoptosis of human pancreatic islet endothelial cells: effects of pravastatin on the Akt survival pathway. Am J Pathol 2008;173:442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Houde VP, Brûlé S, Festuccia WT, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 2010;59:1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fraenkel M, Ketzinel-Gilad M, Ariav Y, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008;57:945–957 [DOI] [PubMed] [Google Scholar]
- 66.Yang SB, Lee HY, Young DM, et al. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med Berl 2012;90:575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Danielsson A, Ost A, Nystrom FH, Strålfors P. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J Biol Chem 2005;280:34389–34392 [DOI] [PubMed] [Google Scholar]
- 68.Pereira MJ, Palming J, Rizell M, et al. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol Cell Endocrinol 2012;355:96–105 [DOI] [PubMed] [Google Scholar]
- 69.Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: A crossroad between cancer and diabetes? J Am Soc Nephrol 2006;17:2236–2244 [DOI] [PubMed] [Google Scholar]
- 70.Ye L, Varamini B, Lamming DW, Sabatini DM, Baur JA. Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front Genet 2012;3:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012;335:1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar A, Lawrence JC, Jr, Jung DY, et al. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 2010;59:1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ransom JT. Mechanism of action of mycophenolate mofetil. Ther Drug Monit 1995;17:681–684 [DOI] [PubMed] [Google Scholar]
- 74.Barton FB, Rickels MR, Alejandro R, et al. Improvement in outcomes of clinical islet transplantation: 1999-2010. Diabetes Care 2012;35:1436–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friman S, Arns W, Nashan B, et al. Sotrastaurin, a novel small molecule inhibiting protein-kinase C: randomized phase II study in renal transplant recipients. Am J Transplant 2011;11:1444–1455 [DOI] [PubMed] [Google Scholar]
- 76.Merani S, McCall M, Pawlick RL, et al. AEB071 (sotrastaurin) does not exhibit toxic effects on human islets in vitro, nor after transplantation into immunodeficient mice. Islets 2011;3:338–343 [DOI] [PubMed] [Google Scholar]
- 77.Doi T, Muro K, Boku N, et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. J Clin Oncol 2010;28:1904–1910 [DOI] [PubMed] [Google Scholar]
- 78.O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 2008;26:1588–1595 [DOI] [PubMed] [Google Scholar]
- 79.Oh DY, Kim TW, Park YS, et al. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012;118:6162–6170 [DOI] [PubMed] [Google Scholar]
- 80.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008;26:1603–1610 [DOI] [PubMed] [Google Scholar]
- 81.Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol 2009;27:193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yee KW, Zeng Z, Konopleva M, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 2006;12:5165–5173 [DOI] [PubMed] [Google Scholar]
- 83.Yoon DH, Ryu MH, Park YS, et al. Phase II study of everolimus with biomarker exploration in patients with advanced gastric cancer refractory to chemotherapy including fluoropyrimidine and platinum. Br J Cancer 2012;106:1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.D’Amico E, Hui H, Khoury N, Di Mario U, Perfetti R. Pancreatic beta-cells expressing GLP-1 are resistant to the toxic effects of immunosuppressive drugs. J Mol Endocrinol 2005;34:377–390 [DOI] [PubMed] [Google Scholar]
- 85.Fuhrer DK, Kobayashi M, Jiang H. Insulin release and suppression by tacrolimus, rapamycin and cyclosporin A are through regulation of the ATP-sensitive potassium channel. Diabetes Obes Metab 2001;3:393–402 [DOI] [PubMed] [Google Scholar]
- 86.Paty BW, Harmon JS, Marsh CL, Robertson RP. Inhibitory effects of immunosuppressive drugs on insulin secretion from HIT-T15 cells and Wistar rat islets. Transplantation 2002;73:353–357 [DOI] [PubMed] [Google Scholar]
- 87.Bell E, Cao X, Moibi JA, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes 2003;52:2731–2739 [DOI] [PubMed] [Google Scholar]
- 88.Bussiere CT, Lakey JR, Shapiro AM, Korbutt GS. The impact of the mTOR inhibitor sirolimus on the proliferation and function of pancreatic islets and ductal cells. Diabetologia 2006;49:2341–2349 [DOI] [PubMed] [Google Scholar]
- 89.Fabian MC, Lakey JR, Rajotte RV, Kneteman NM. The efficacy and toxicity of rapamycin in murine islet transplantation. In vitro and in vivo studies. Transplantation 1993;56:1137–1142 [DOI] [PubMed] [Google Scholar]
- 90.Hyder A, Laue C, Schrezenmeir J. Effect of the immunosuppressive regime of Edmonton protocol on the long-term in vitro insulin secretion from islets of two different species and age categories. Toxicol In Vitro 2005;19:541–546 [DOI] [PubMed] [Google Scholar]
- 91.Marcelli-Tourvieille S, Hubert T, Moerman E, et al. In vivo and in vitro effect of sirolimus on insulin secretion. Transplantation 2007;83:532–538 [DOI] [PubMed] [Google Scholar]
- 92.Zhang N, Su D, Qu S, et al. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes 2006;55:2429–2436 [DOI] [PubMed] [Google Scholar]
- 93.Kneteman NM, Lakey JR, Wagner T, Finegood D. The metabolic impact of rapamycin (sirolimus) in chronic canine islet graft recipients. Transplantation 1996;61:1206–1210 [DOI] [PubMed] [Google Scholar]
- 94.Song HK, Han DH, Song JH, et al. Influence of sirolimus on cyclosporine-induced pancreas islet dysfunction in rats. Am J Transplant 2009;9:2024–2033 [DOI] [PubMed] [Google Scholar]
- 95.Whiting PH, Woo J, Adam BJ, Hasan NU, Davidson RJ, Thomson AW. Toxicity of rapamycin—a comparative and combination study with cyclosporine at immunotherapeutic dosage in the rat. Transplantation 1991;52:203–208 [DOI] [PubMed] [Google Scholar]
- 96.Sun CL, Ham DS, Park HS, et al. Rapamycin suppresses the expansion and differentiation of porcine neonatal pancreas cell clusters. Transplantation 2010;90:717–724 [DOI] [PubMed] [Google Scholar]
- 97.Zahr E, Molano RD, Pileggi A, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation 2007;84:1576–1583 [DOI] [PubMed] [Google Scholar]
- 98.Brunn GJ, Hudson CC, Sekulić A, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997;277:99–101 [DOI] [PubMed] [Google Scholar]
- 99.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA 1998;95:1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011;146:408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007;450:736–740 [DOI] [PubMed] [Google Scholar]
- 102.Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011;332:1322–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci USA 2010;107:11823–11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 2000;10:47–50 [DOI] [PubMed] [Google Scholar]
- 105.Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22:7004–7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009;284:12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol 2010;20:1093–1098 [DOI] [PubMed] [Google Scholar]