

Abstract
Hypothalamic “metabolic-sensing” neurons sense glucose and fatty acids (FAs) and play an integral role in the regulation of glucose, energy homeostasis, and the development of obesity and diabetes. Using pharmacologic agents, we previously found that ∼50% of these neurons responded to oleic acid (OA) by using the FA translocator/receptor FAT/CD36 (CD36). For further elucidation of the role of CD36 in neuronal FA sensing, ventromedial hypothalamus (VMH) CD36 was depleted using adeno-associated viral (AAV) vector expressing CD36 short hairpin RNA (shRNA) in rats. Whereas their neuronal glucosensing was unaffected by CD36 depletion, the percent of neurons that responded to OA was decreased specifically in glucosensing neurons. A similar effect was seen in total-body CD36-knockout mice. Next, weanling rats were injected in the VMH with CD36 AAV shRNA. Despite significant VMH CD36 depletion, there was no effect on food intake, body weight gain, or total carcass adiposity on chow or 45% fat diets. However, VMH CD36–depleted rats did have increased plasma leptin and subcutaneous fat deposition and markedly abnormal glucose tolerance. These results demonstrate that CD36 is a critical factor in both VMH neuronal FA sensing and the regulation of energy and glucose homeostasis.
Several lines of evidence support the idea that specialized hypothalamic metabolic-sensing neurons can monitor peripheral fuel availability by altering their activity in response to ambient levels of glucose and fatty acids (FAs) as a means of regulating energy and glucose homeostasis in the body (1–8). Glucose is the primary energy substrate for neuronal metabolism (9), whereas astrocytes are the primary source of FA oxidation in the brain (10,11). However, metabolic-sensing neurons possess specialized pathways that allow them to use FA as a signaling molecule to regulate their activity (5,7,12–14). We previously showed that at least 50% of the FA sensing in ventromedial hypothalamic [VMH = arcuate (ARC) + ventromedial nuclei (VMN)] neurons is attributable to the interaction of long-chain FA (LCFA) with FA translocase/CD36 (CD36) acting as a receptor, whereas only ∼20% is attributable to intracellular metabolism of FA (5). In those studies, we used both fura-2 calcium imaging and fluorometric imaging plate reader (FLIPR) membrane potential dye, together with various pharmacological agents to identify some of the mechanisms by which individual VMN neurons responded to the LCFA oleic acid (OA) (5,15). In the present studies, we used these same techniques, together with molecular manipulations of CD36 expression, to determine the importance of CD36 in the sensitivity of VMN neurons to OA in vitro and the overall effect of depleting CD36 in the VMH on food intake, body weight and adipose gain, and glucose tolerance in vivo. We confirmed our prior studies showing that CD36 is a critical mechanism for neuronal sensing of LCFA and demonstrate that depletion of VMH CD36 alters the distribution of adiposity and glucose homeostasis in outbred rats.
RESEARCH DESIGN AND METHODS
Experiment 1: assessment of glucose and OA sensing in dissociated VMH neurons
Animals.
Animals were housed at 23–24°C on a 12:12-h light-dark cycle (lights on at 1000 h). Male Sprague-Dawley rats and C57/Bl6 mice were purchased from Charles River Laboratories. Total-body CD36-knockout mice bred on a C57BL/6J background (CD36KO) (16) were provided by Dr. Maria Febbraio (Lerner Research Institute, Cleveland, OH). All work was in compliance with the Animal Care and Use Committee of the East Orange VA Medical Center.
Adeno-associated viral vectors.
Murine CD36 open reading frame was PCR amplified from brain cDNA library with a COOH-terminal FLAG tag and subcloned into an adeno-associated viral (AAV) expression plasmid to generate AAV.CD36. A control vector (AAV.mCherry) was designed to express mCherry-FLAG. The expression of both transgenes is regulated by a hybrid cytomegalovirus/chicken β-actin promoter. Virus stocks were prepared by packaging the vector plasmids into AAV serotype 2 particles using a helper-free plasmid transfection system. The vectors were purified using heparin affinity chromatography, dialyzed against PBS supplemented with 2 mmol/L MgCl2, and diluted to 1012 genomic particles per milliliter.
Stereotaxic surgery.
Cold-anesthetized, postnatal day 5 (P5) rats were stereotaxically injected bilaterally in the border between the VMN and ARC over 10 min with 0.4 μL of saline containing 2 × 108 genomic particles of AAV per side containing either CD36 short hairpin RNA (shRNA) + mCherry or a control sequence + mCherry. With level bregma and lambda, coordinates were as follows: 2.6 mm caudal to bregma, 0.40 mm lateral to midline, and 6.6 mm down from dural surface as previously described (3).
Measurement of OA- and glucose-induced changes in intracellular Ca2+ oscillations in dissociated VMN neurons.
Rats were injected in the VMH at P5 with AAV expressing CD36 shRNA. At P21–28, they were perfused, the VMN was bilaterally punched from VMH slices, and neurons were dissociated as previously described (5,17). Evaluation of glucose- and OA-induced alterations in intracellular Ca2+ ([Ca2+]i) oscillations in individual VMN neurons that expressed mCherry (see below) were assessed using fura-2 acetoxy-methyl ester (Molecular Probes, Eugene, OR), as previously described (5,17). Neurons were classified as glucose excited (GE), glucose inhibited (GI), and nonglucosensing (NG) and as OA excited (OAE), OA inhibited (OAI), or nonresponsive using previously established criteria for changes in [Ca2+]i area under the curve (AUC) (5,17). The lipophilic, anionic bis-oxonol dye from the FLIPR membrane potential assay kit (Molecular Devices, Sunnyvale, CA) was also used in dissociated VMN neurons to assess OA- and glucose-induced changes in membrane potential as previously described (1). Studies began with neurons held at 2.5 mmol/L glucose unless otherwise specified. Changes in [Ca2+]i fluctuations in response to glucose and OA were assessed over 10-min periods after the addition of each substance. All neurons were incubated with 20 nmol/L glutamate terminally to ensure that they were functionally viable.
mRNA expression in dissociated VMH neurons and VMN and ARC micropunches.
After determination of glucose- and FA-sensing properties, 120 µL of lysis buffer (Ambion, Austin, TX) was added to the dissociated neurons, and mRNA was quantified by real-time quantitative PCR as previously described (1,5,18). The resultant cDNA was analyzed using TAQman MGB primer/probe sets targeting genes involved in glucose and FA metabolism (Supplementary Table 1). Similar techniques were used to extract and assay mRNA in ARC and VMN micropunches. Data were expressed as the ratio of the gene of interest relative to the housekeeping gene, cyclophilin (1,5,18).
Experiment 2: in vivo effects of VMH CD36 depletion
Animals and diet.
P21 rats were housed at 23–24°C on a 12:12-h light-dark cycle (lights on at 1000 h) and injected with AAV expressing CD36 shRNA or control sequence + mCherry bilaterally in the VMH (see below). Rats were fed Purina rat chow (4.5% fat, 3.75 kcal/g) for 6 weeks followed by 9 weeks on 45% fat diet (high-fat diet [HFD], 4.75 kcal/g, Research Diet D12541). Food and water were available ad libitum. Food intake and body weight were monitored biweekly. During the last week on HFD, rats underwent an oral glucose tolerance test (OGTT). Terminally, rats were decapitated and the brains were harvested for quantitative PCR. Trunk blood was collected for glucose, insulin, leptin, free FA, and β-hydroxybutyrate (β-OHB). Retroperitoneal, perirenal, mesenteric, and epididymal fat pads were weighed as representative of visceral fat, and inguinal pads were taken as representative of subcutaneous fat.
Stereotaxic surgery.
Chloropent (chloral hydrate + nembutal)-anesthetized P21 rats were stereotaxically injected bilaterally with 0.4 μL containing 2 × 108 genomic particles of AAV expressing CD36 shRNA or control sequence + mCherry into both the VMN and ARC (n = 8–10/group). With bregma and lambda level, coordinates were as follows: 2.7 mm caudal to bregma, 0.6 mm lateral to midline, and 8.4 mm below dura (VMN), and 0.3 mm lateral to midline and 8.9 mm below dura (ARC).
OGTT.
During the last week on HFD, rats were gavaged with 2 g/kg glucose, and blood was sampled by tail nip at baseline and 15, 30, 60, 90, and 120 min for glucose and insulin.
Assays of insulin, leptin, free FA, β-hydroxybutyrate, and glucose.
Plasma insulin and leptin levels were analyzed with radioimmunoassay kits (Linco, Carlsbad, CA). Plasma-free FA and β-OHB levels were analyzed using a colorimetric assay (Wako, Richmond, VA). Plasma blood glucose was measured using a glucose Analox instrument.
Statistics.
Responses of neurons to changes in glucose, OA, or drug concentrations were compared using the Student t test for nonparametric statistics (GraphPad Prism, La Jolla, CA). One-way and two-way ANOVA and ANOVA for repeated measures with post hoc Bonferroni corrections were used for the in vivo studies. Outliers were removed if necessary (Systat, Chicago, IL).
RESULTS
Experiment 1
Effect of in vivo CD36 VMH depletion on neuronal glucose and FA sensing.
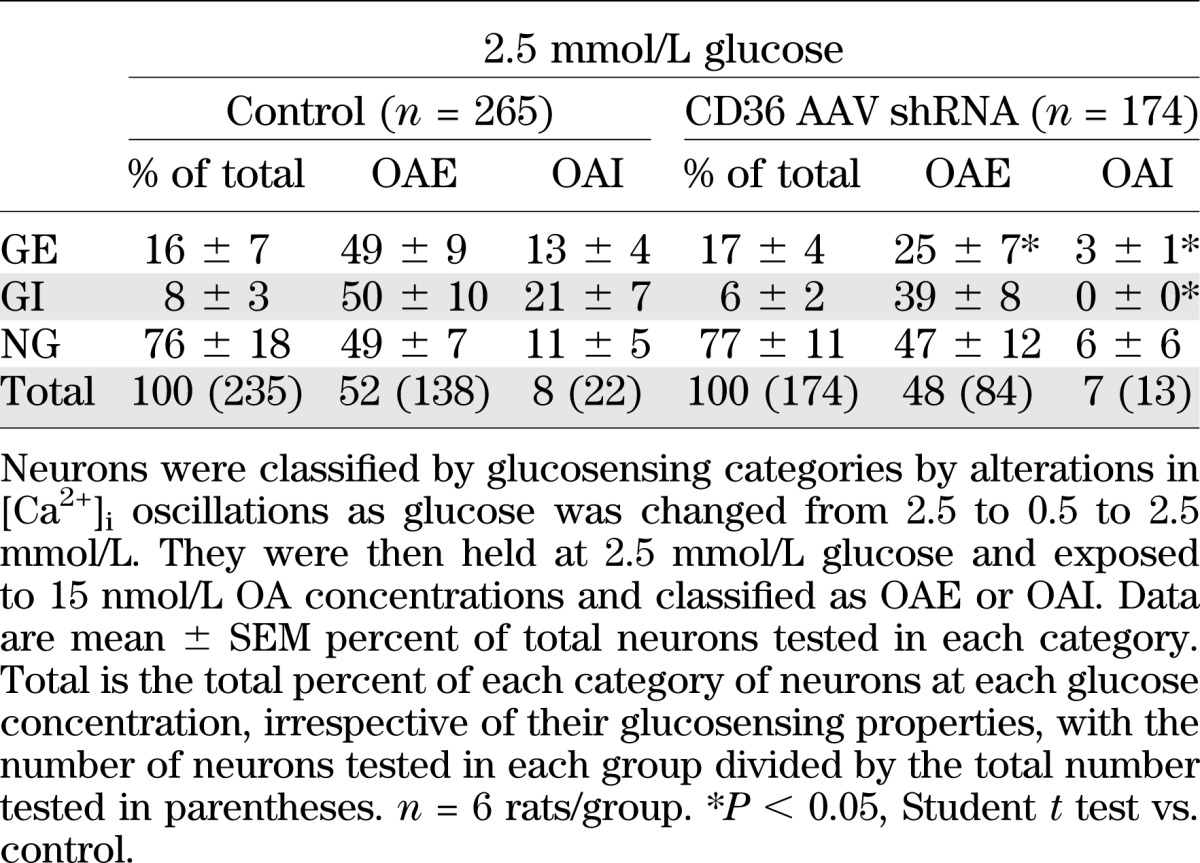
VMH injection of AAV CD36 shRNA at P5 reduced neuronal CD36 mRNA expression by 45% as compared with controls (control = 0.91 ± 0.09, CD36 AAV = 0.50 ± 0.10; P < 0.05) at P21–28, but had no effect on body weight gain from P5 to P21 (Supplementary Fig. 1). This reduction in VMH CD36 did not affect the proportion of GE or GI neurons (Table 1). When considered independently of their glucosensing status, there was no effect of VMH CD36 depletion on the proportion of VMH neurons that was excited or inhibited by 15 nmol/L OA. However, as a function of their glucosensing properties, the proportion of CD36 shRNA GE neurons that was further excited by 15 nmol/L OA was reduced by 49%, whereas the percent of GE neurons inhibited by OA was decreased by 77% versus control neurons (Table 1). VMH CD36 depletion had no effect on the percent of GI neurons excited by OA. However, whereas 21% of control GI neurons were inhibited by OA, no CD36 shRNA neurons were inhibited by OA (Table 1).
TABLE 1.
Effect of OA on VMH neurons from 3-week-old male Sprague-Dawley rats injected at P5 with CD36 AAV shRNA in the VMH
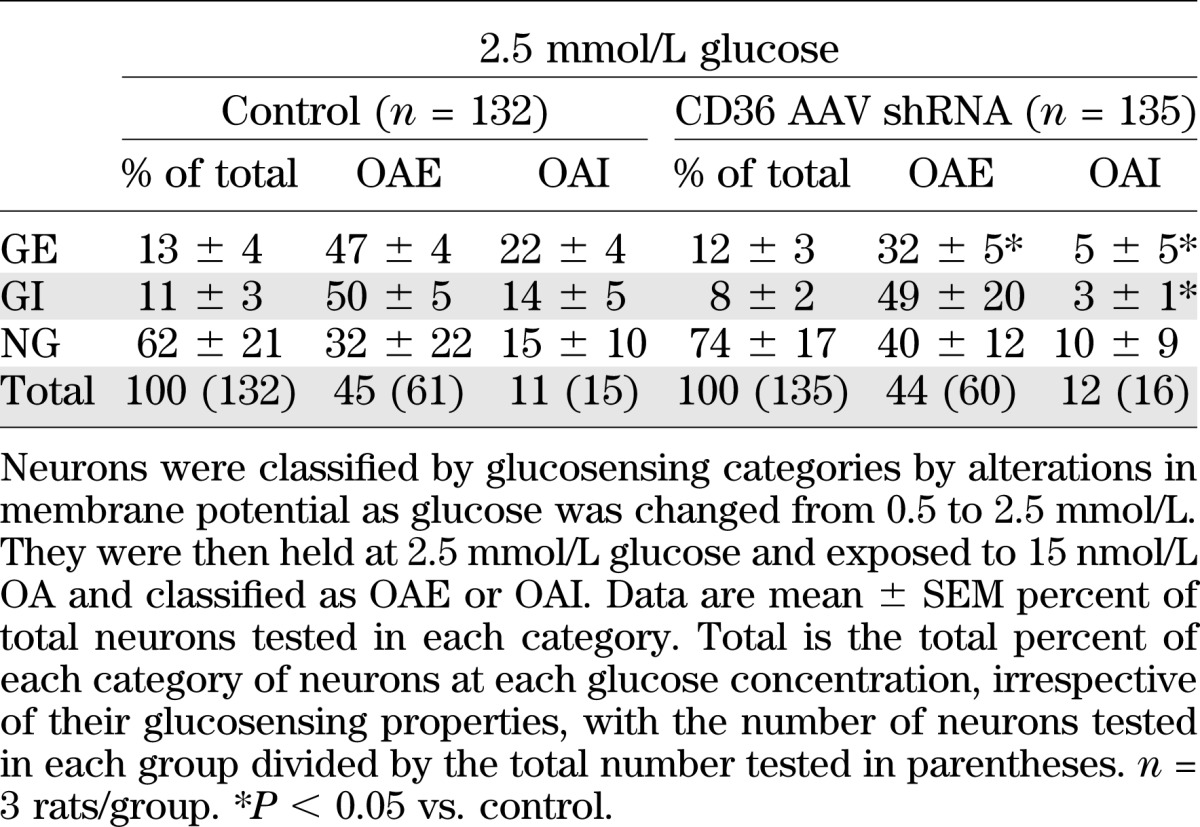
Since it is possible that OA might alter [Ca2+]i oscillations without necessarily altering membrane potential or neuronal activity, neurons were also imaged with FLIPR membrane potential dye for changes in membrane potential induced by glucose and OA (Table 2). Similar to calcium imaging, CD36 depletion did not alter the proportion of GE or GI neurons using FLIPR dye. Also, whereas CD36 depletion reduced the proportion of GE neurons excited by OA using calcium imaging by 49%, these OAE GE neurons were decreased by 32% using FLIPR dye (Tables 1 and 2). With calcium imaging and FLIPR dye, the number of CD36 shRNA GE neurons inhibited by OA was reduced by 77%, but there was no effect on the number of OAE GI neurons. On the other hand, whereas CD36 depletion abolished OA inhibition of GI neurons using calcium imaging, there was a 79% decrease in the number of these neurons using FLIPR. Therefore, although there were some differences, the relative proportions of OAE to OAI neurons identified by the two techniques were quite comparable.
TABLE 2.
Effect of OA on VMH neurons from 3-week-old male Sprague-Dawley rats injected at P5 with CD36 AAV shRNA in the VMH using FLIPR membrane potential
Effect of total-body CD36KO on mouse VMH neuronal glucose and FA sensing.
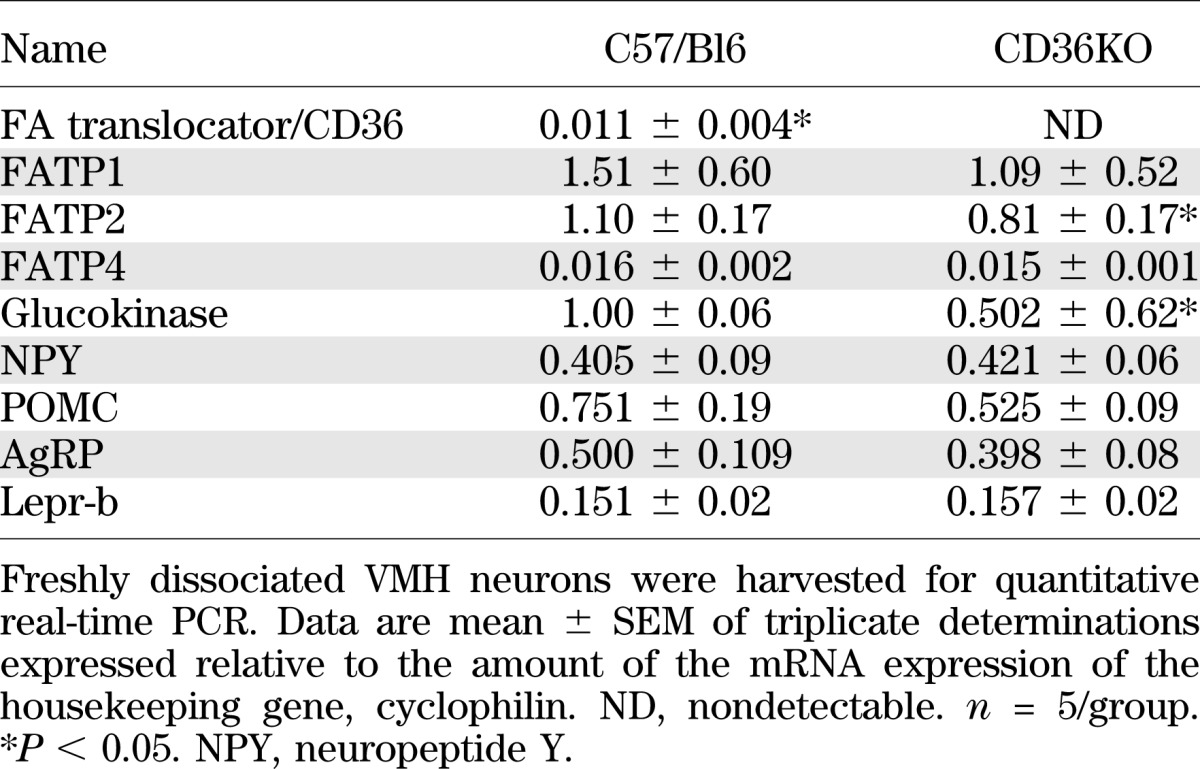
VMH neurons were dissociated from P21–28 CD36KO and C57/BL6 wild-type (WT) control mice. In control mice, 18% of VMH neurons were GE and 11% were GI, whereas only 11% of CD36KO neurons were GE and 5% were GI (*P < 0.001; data not shown). Thus, CD36KO mice had 39% fewer GE and 55% fewer GI neurons than control mice. Using OA concentrations from 10 to 1,000 nmol/L at 2.5 and 0.5 mmol/L glucose, neurons from both genotypes demonstrated similar, but somewhat flat, concentration-dependent responses for excitation and inhibition (Fig. 1). When considered independently of their glucosensing properties and over the entire range of OA concentrations, there were no significant differences in the percentages of neurons excited or inhibited by OA (Table 3). Also, whereas there were equivalent numbers of OAI VMH neurons in the Sprague-Dawley rat (Table 1) and the C57/BL6 mouse (Table 3), the mouse had ∼50% more neurons that were excited by OA at 2.5 mmol/L glucose, regardless of whether VMH CD36 expression had been decreased or not. As in the rat with VMH knockdown of CD36 (Table 1), CD36KO mice had significant alterations in OA responsiveness when considered with respect to their glucosensing properties (Table 3). In CD36KO mouse GE neurons, there were 53% fewer OAI GE neurons held at 2.5 mmol/L glucose versus WT mice. As opposed to the outbred rat, the control mouse had no GI neurons that were inhibited by OA in 2.5 mmol/L glucose, whereas 13% were inhibited by OA in CD36KO mice (Table 3). At 0.5 mmol/L glucose, CD36KO mouse GE and GI neurons were 30 and 37% more excited by OA than in WT mice (P < 0.05) (Table 3). As opposed to control mice where OA inhibited 12 and 26% of the GE and GI neurons, respectively, no inhibition was induced by OA in CD36KO mice. Overall, in C57/Bl6 mice, more neurons were responsive to OA when held at 2.5 than 0.5 mmol/L glucose, whereas in CD36KO mice, no inhibition induced by OA was observed in 0.5 mmol/L glucose as opposed to 2.5 mmol/L glucose. Finally, total body knockout of CD36 was associated with a 26% decrease in FA transport protein 2 (FATP2) and a 50% decrease in glucokinase mRNA expression (Table 4). Overall, these data suggest that germ cell deletion of CD36 has a major effect on both VMN neuronal glucose and CD36-mediated FA-sensing, which represents compensation for loss of the major FA-sensing regulatory pathway.
FIG. 1.

VMN neurons from C57/Bl6 and CD36KO mice were held at 2.5 (A and B) or 0.5 mmol/L (C and D) glucose and exposed to three concentrations of OA: 10, 100, and 1,000 nmol/L. They were then classified as OAE (A and C), OAI (B and D), or nonresponsive (OAN, not shown). Data are mean ± SEM percent of total neurons tested in each category. There were no significant intergroup differences.
TABLE 3.
Effect of OA on VMH GE, GI, and NG neurons in C57/Bl6 WT and C57/Bl6 CD36KO mice
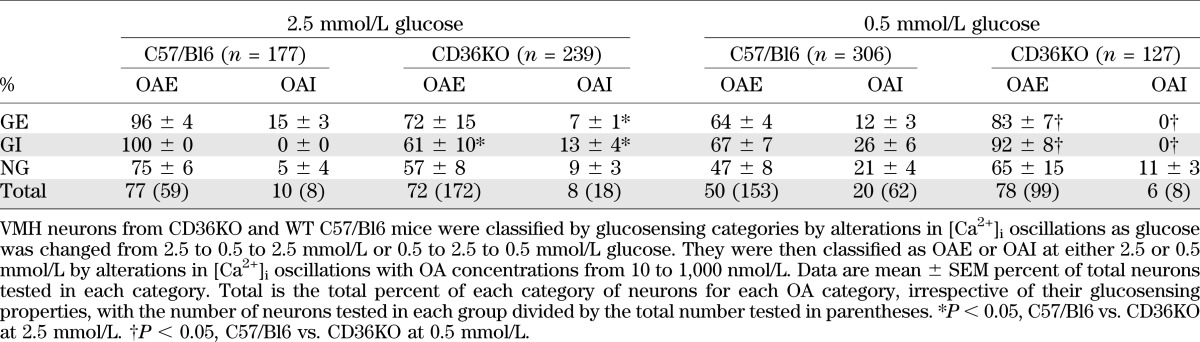
TABLE 4.
mRNA expression in VMN freshly dissociated neurons from CD36KO and C57/Bl6 mice
Experiment 2
In vivo effects of VMH CD36 depletion in rats.
Bilateral VMH (ARC and VMN) injections of AAV expressing CD36 shRNA into 3-week-old rats produced no significant changes in food intake or body weight gain over 6 weeks of chow intake (Table 5 and Fig. 2A and B). However, they did have 122, 12, and 66% higher plasma leptin, glucose, and insulin levels, respectively (Table 5), suggesting that they were both fatter and significantly less glucose tolerant than control rats. VMH CD36 depletion also did not significantly alter food intake or weight gain over 9 weeks on HFD, although feed efficiency (body weight gain/caloric intake) was 16% higher and leptin levels were 54% higher than controls (Table 5). Although total adiposity (total weight of five fat pads as a percent of carcass weight) did not differ, inguinal fat pads, representative of subcutaneous depots, were 60% heavier in the VMH CD36 shRNA rats, suggesting a possible redistribution of fat tissue from visceral to subcutaneous depots in these rats. The most striking effect of reducing VMH CD36 expression was a 232% increase in plasma insulin levels in association with a 98% increase in glucose and 244% increase in insulin AUC during an OGTT after 9 weeks on HFD (Table 5 and Fig. 2C and D).
TABLE 5.
Morphometric and biochemical data for 19-week-old male Sprague-Dawley rats injected at 3 weeks old with control AAV shRNA or CD36 AAV shRNA in the VMH
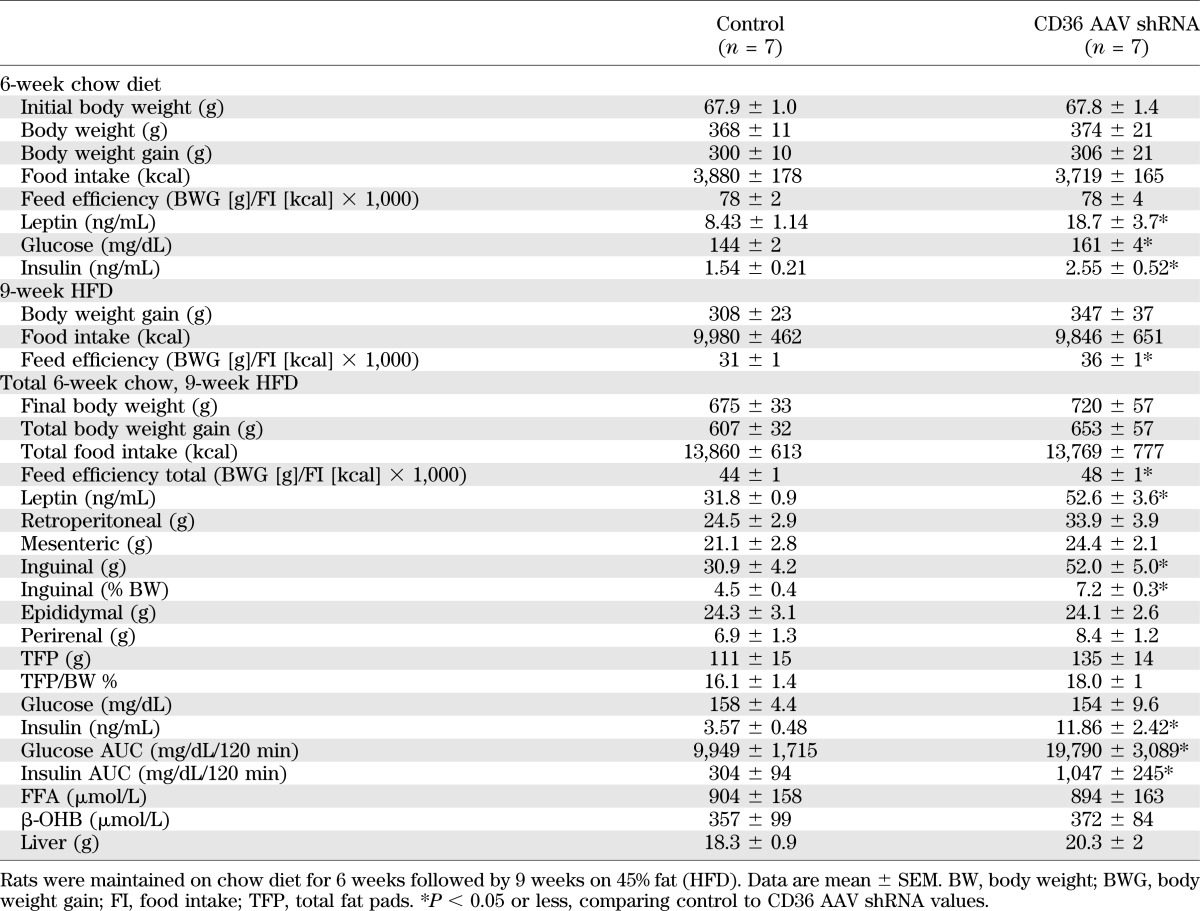
FIG. 2.

Food intake (A) and body weight gain (B) in 3-week-old rats injected in the VMH with either control AAV or AAV expressing CD36 shRNA. C: OGTT (2 g/kg) was performed in control (n = 7) and CD36 AAV shRNA rats (n = 6) after 6 weeks on chow diet and 9 weeks on HFD; glucose concentration in mg/dL over 120 min. D: Insulin concentration in ng/mL over 120 min. *P ≤ 0.05.
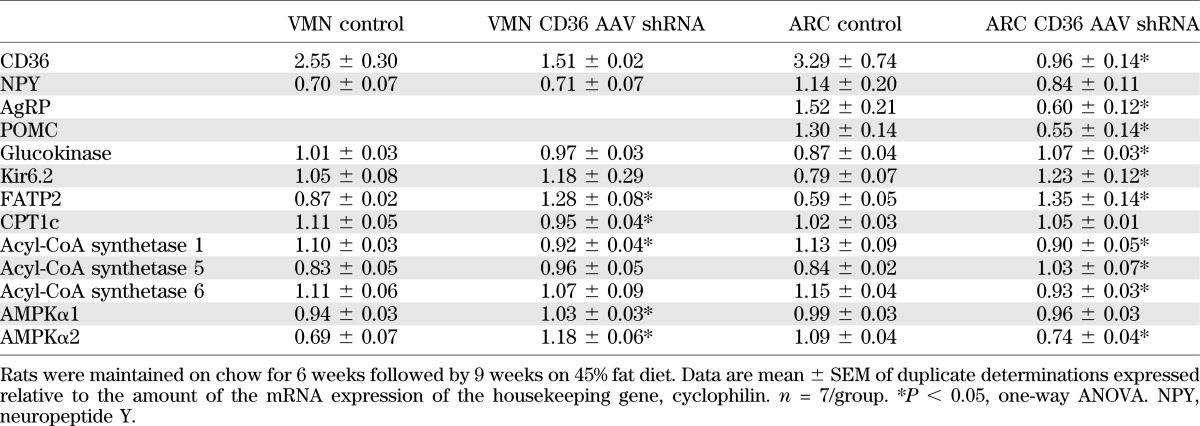
Terminally, VMN and ARC micropunches were assessed for alterations in gene expression due to CD36 depletion (Table 6). In the VMN, there was a nonsignificant 40% decrease in CD36 expression that was associated with increases of 48% in FATP2, 10% in AMP-activated protein kinase α1 (AMPKα1), and 70% in AMPKα2 mRNA expression with 15% decreases in carnitine palmitoyltransferase 1c (CPT1c) and acyl-CoA synthase 1 expression (Table 6). In the ARC, where CD36 expression was decreased by 71%, agouti-related peptide (AgRP), proopiomelanocortin (POMC), acyl-CoA synthase 5 and 6, and AMPKα2 were reduced by 60, 58, 21, 19, and 32%, respectively, whereas glucokinase, Kir6.2, and FATP2 expressions were increased by 23, 56, and 127% as compared with controls, respectively (Table 6). Other ARC and/or VMN genes involved in FA and glucose metabolism (FA synthase; GLUT4; FATP1, -4, and -6; CPT1a; acyl-CoA synthase 3 and 4; and malonyl-CoA decarboxylase) were measured but did not differ between groups (data not shown).
TABLE 6.
mRNA expression in VMN and ARC micropunches harvested from male Sprague-Dawley rats injected at 3 weeks old with control AAV shRNA or CD36 AAV shRNA in the VMH
DISCUSSION
The current studies were undertaken to examine the importance of FAT/CD36 in neuronal FA and glucose sensing in freshly dissociated VMH neurons from rats and mice and the effects of reducing VMH CD36–mediated FA sensing on long-term energy and glucose homeostasis. Using pharmacological methods, we previously demonstrated that CD36 accounted for at least 50% of the excitatory and inhibitory effects of OA in VMN neurons, whereas inhibiting any of the steps of neuronal formation of LCFA acyl-CoA, oxidation, formation of reactive oxygen species, or activation of the KATP channel accounted for no more than 20% of these effects of OA (5). Here, using in vivo AAV shRNA to deplete VMH CD36 in rats and mice with total body depletion of CD36, we show a similar effect of interfering with CD36 signaling, although the results varied somewhat depending upon the method and species used. Importantly, in both rats and CD36KO mice, the reduction in FA sensing was completely dependent upon the underlying glucosensing status of the neurons assessed. In rats injected at P5 with AAV CD36 shRNA, resulting in a 45% depletion of VMN CD36 mRNA at P21–28, there was no effect on the percentage of glucosensing neurons, but there was a 49% reduction in OA excitation, a 77% reduction in OA inhibition in GE neurons, and a complete loss of OA inhibition in GI neurons held at 2.5 mmol/L glucose. In the CD36KO mouse, there were 40–55% fewer VMH glucosensing neurons in association with a 50% decrease in glucokinase, a critical regulator of glucosensing (1,19,20). The KO mouse also had 53% fewer OAI GE neurons but no differences in OAE GE neurons. However, as opposed to the rat, the KO mouse actually had more GI neurons that were inhibited by OA at 2.5 mmol/L glucose, and, at 0.5 mmol/L glucose, the CD36KO mouse had 30 and 37% more GE and GI neurons that were excited by OA than control mice. These differences between the rat and mouse data are likely due to both species differences and the germ cell deletion of CD36 where both early plastic changes (as witnessed by the loss of glucosensing neurons) and altered energy and glucose homeostasis can occur (21,22). Despite these differences, our cumulative present and previous data (5) strongly suggest that CD36 plays a critical role in the responsiveness of VMH neurons to the LCFA, OA.
This conclusion differs from a previous study demonstrating an important role for the KATP channel in mediating the effects of LCFA in ARC POMC neurons under high (2.5 to 5 mmol/L) glucose concentrations (23). However, our data clearly demonstrate that at 2.5 mmol/L glucose, which is comparable to hypothalamic levels seen during the postingestive state (24,25), approximately fourfold more GE neurons were excited than inhibited in VMH CD36–depleted rats than control rats. Since glucokinase and the KATP channel are the dominant mediators of activation of VMN GE neurons (1,19,20), these data and our previous studies (5) suggest that in those GE neurons that were further excited by OA, this effect is mediated by an alternate pathway that is not dependent upon this mechanism or other pathways involving FA oxidation. In fact, data in FA-sensing taste buds suggest that their LCFA-induced activation is dependent upon CD36 acting as a receptor to activate store-operated calcium channels (26). Similarly, our present and previous (5) data support the contention that CD36-mediated FA sensing is similar to that seen in taste buds and is largely independent of intracellular metabolism of LCFA. However, our current studies demonstrate that CD36-mediated FA sensing occurs specifically in VMH neurons that are also responsive to glucose. This supports the idea that such neurons are metabolic sensors that integrate signals from metabolic substrates, as well as hormones and neural inputs from the periphery, to regulate their activity (9,27–29). As opposed to GE neurons, GI neurons are largely inhibited at 2.5 mmol/L glucose and appear to use AMPK, nitric oxide, and a nonspecific cation channel to mediate this effect (30,31). The fact that 50% of rat GI neurons held at 2.5 mmol/L glucose (where they are largely, but not totally, inactive) (1,20) were excited and 21% were inhibited by OA also suggests that such FA sensing occurs by mechanisms that are largely independent of those used by GI neurons to sense glucose. One possibility is that the increase in neuronal FATP2 gene expression in rats with VMH CD36 depletion might be associated with increased LCFA transport into the cell and a switch to a dependence on LCFA metabolism in the face of decreased availability of CD36-mediated FA sensing.
Having established a crucial role for CD36 in mediating neuronal FA sensing in the VMH in vitro, we next assessed the importance of VMH CD36–mediated neuronal FA sensing on long-term energy and glucose homeostasis. Depletion of VMH CD36 expression using AAV shRNA at 3 weeks of age had no significant effects on either food intake or body weight gain when rats were fed low-fat chow from weaning for 6 weeks, or after an additional 9 weeks on a 45% fat diet. However, although they had no increase in the percent of total carcass adiposity (using five pad weights as surrogates), VMH CD36–depleted rats did have a 20% increase in feed efficiency, a 125 and 65% increase in plasma leptin levels after 6 weeks of chow and 9 weeks of HFD, and a 60% increase in inguinal fat pad weights relative to carcass weights terminally. These differences from control rats suggest that VMH CD36–depleted rats had a redistribution of fat from visceral to subcutaneous (inguinal) depots, the predominant source of circulating leptin (32,33). Assuming this is so and that sparing of visceral depots by shunting fat to subcutaneous depots should improve insulin sensitivity, it was surprising to find that CD36 shRNA rats had markedly elevated plasma insulin levels and impaired glucose tolerance after both chow and HFD. Since visceral adiposity is usually associated with such impairments in insulin sensitivity (34), these results suggest that VMH CD36–mediated neuronal FA sensing is responsible for both the distribution of carcass adiposity and peripheral insulin sensitivity. These differences could not be attributed to changes in ARC neuropeptide expression since there were comparable decreases in AgRP and POMC expression observed in CD36 shRNA rats. The parallel decrease in the expression of both these neuropeptides suggests that this might be due to a reduction in their FA sensing ability rather than elevated leptin levels, which should decrease AgRP but increase POMC expression. Although we made no measures of energy expenditure or motor activity, it seems unlikely that either of these could account for differences in fat deposition, although decreased activity might contribute to reduced insulin sensitivity. In fact, despite the fact that CD36 was depleted only in the VMH in rats, total-body CD36KO mice also have a similar increase in plasma insulin levels and an impaired whole-body glucose tolerance, as well as impaired FA oxidation, when fed an HFD for several months (21,22).
One important caveat is that assessment of OA-induced changes in [Ca2+]i using calcium imaging does not provide a direct measure of changes in membrane potential or neuronal activity, per se. In gustatory cells, occupation of the CD36 receptor leads to activation of store-operated calcium channels via phosphorylation of Src protein–tyrosine kinases and subsequent release of serotonin and norepinephrine. CD36-mediated release of these neurotransmitters suggests a parallel between CD36-mediated increases in intracellular calcium and neuronal activity produced by exposure to LCFA (26). We previously showed that ∼50% of the OA-induced alterations in [Ca2+]i oscillations were paralleled by alterations in membrane potential (5). Here we confirm these findings in CD36 shRNA rats. Taken together with studies in FA-sensing taste buds (26), our data support a role for LCFA acting on CD36 as a receptor to alter neuronal activity in VMH FA-sensing neurons. However, only direct measurements of neuronal activity will settle this issue.
In conclusion, using a variety of molecular manipulations to deplete the FA receptor CD36 from the VMH or whole animal, we have demonstrated its prominence as a mediator of VMH neuronal LCFA sensing specifically in glucosensing neurons in vitro and as a regulator of adipose deposition and glucose tolerance in vivo. Thus, whereas VMH glucosensing appears to be most important as a detector and regulator of feeding and neurohumoral responses during states of low glucose availability (3,25), CD36-mediated VMH neuronal FA sensing appears to play an important role in the physiological regulation of both energy and glucose homeostasis.
ACKNOWLEDGMENTS
This work was supported by the Research Service of the Department of Veterans Affairs (to B.E.L. and A.D.-M.) and the National Institute of Diabetes and Digestive and Kidney Diseases (DK-53181 to B.E.L.).
No potential conflicts of interest relevant to this article were reported.
C.L.F. performed the research, designed the experiments, and wrote the manuscript. A.D.-M. performed all the virus surgeries. S.M. provided the CD36 AAV shRNA. C.M. contributed to the design of the experiments. B.E.L. helped design the experiments and write the manuscript. C.L.F. and B.E.L. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Sunny Lee, Antoinette Moralishvili, and Charlie Salter (all VA Medical Center) for their technical assistance. Dr. Maria Febbraio (Lerner Research Institute, Cleveland, Ohio) kindly supplied the CD36KO mice.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1689/-/DC1.
REFERENCES
- 1.Kang L, Dunn-Meynell AA, Routh VH, et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 2006;55:412–420 [DOI] [PubMed] [Google Scholar]
- 2.Lam TK, Pocai A, Gutierrez-Juarez R, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med 2005;11:320–327 [DOI] [PubMed] [Google Scholar]
- 3.Levin BE, Becker TC, Eiki J, Zhang BB, Dunn-Meynell AA. Ventromedial hypothalamic glucokinase is an important mediator of the counterregulatory response to insulin-induced hypoglycemia. Diabetes 2008;57:1371–1379 [DOI] [PubMed] [Google Scholar]
- 4.Pocai A, Lam TK, Obici S, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest 2006;116:1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Foll C, Irani BG, Magnan C, Dunn-Meynell AA, Levin BE. Characteristics and mechanisms of hypothalamic neuronal fatty acid sensing. Am J Physiol Regul Integr Comp Physiol 2009;297:R655–R664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clément L, Cruciani-Guglielmacci C, Magnan C, et al. Intracerebroventricular infusion of a triglyceride emulsion leads to both altered insulin secretion and hepatic glucose production in rats. Pflugers Arch 2002;445:375–380 [DOI] [PubMed] [Google Scholar]
- 7.Migrenne S, Cruciani-Guglielmacci C, Kang L, et al. Fatty acid signaling in the hypothalamus and the neural control of insulin secretion. Diabetes 2006;55(Suppl. 2):S139–S144 [Google Scholar]
- 8.Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes 2002;51:271–275 [DOI] [PubMed] [Google Scholar]
- 9.Sokoloff L, Reivich M, Kennedy C, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 1977;28:897–916 [DOI] [PubMed] [Google Scholar]
- 10.Escartin C, Pierre K, Colin A, et al. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci 2007;27:7094–7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 1987;18:551–561 [DOI] [PubMed] [Google Scholar]
- 12.Levin BE. Metabolic sensors: viewing glucosensing neurons from a broader perspective. Physiol Behav 2002;76:397–401 [DOI] [PubMed] [Google Scholar]
- 13.Oomura Y, Nakamura T, Sugimori M, Yamada Y. Effect of free fatty acid on the rat lateral hypothalamic neurons. Physiol Behav 1975;14:483–486 [DOI] [PubMed] [Google Scholar]
- 14.Wang R, Liu X, Hentges ST, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004;53:1959–1965 [DOI] [PubMed] [Google Scholar]
- 15.Le Foll C, Irani BG, Magnan C, Dunn-Meynell AA, Levin BE. Effects of maternal genotype and diet on offspring glucose and fatty acid-sensing ventromedial hypothalamic nucleus neurons. Am J Physiol Regul Integr Comp Physiol 2009;297:R1351–R1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laugerette F, Passilly-Degrace P, Patris B, et al. CD36 involvement in orosensory detection of dietary lipids, spontaneous fat preference, and digestive secretions. J Clin Invest 2005;115:3177–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 2004;53:549–559 [DOI] [PubMed] [Google Scholar]
- 18.Levin BE, Magnan C, Migrenne S, Chua SC, Jr, Dunn-Meynell AA. The F-DIO obesity-prone rat is insulin resistant prior to obesity onset. Am J Physiol Regul Integr Comp Physiol 2005;289:R704–R711 [DOI] [PubMed] [Google Scholar]
- 19.Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 2002;51:2056–2065 [DOI] [PubMed] [Google Scholar]
- 20.Kang L, Dunn-Meynell AA, Routh VH, Liu X, Levin BE. Knockdown of GK mRNA with GK RNA interference (RNAi) blocks ventromedial hypothalamic (VMH) neuronal glucosensing (Abstract). Diabetes 2004;53:A43 [Google Scholar]
- 21.Koonen DP, Sung MM, Kao CK, et al. Alterations in skeletal muscle fatty acid handling predisposes middle-aged mice to diet-induced insulin resistance. Diabetes 2010;59:1366–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonen A, Han XX, Habets DD, Febbraio M, Glatz JF, Luiken JJ. A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin- and AICAR-stimulated fatty acid metabolism. Am J Physiol Endocrinol Metab 2007;292:E1740–E1749 [DOI] [PubMed] [Google Scholar]
- 23.Jo YH, Su Y, Gutierrez-Juarez R, Chua S., Jr Oleic acid directly regulates POMC neuron excitability in the hypothalamus. J Neurophysiol 2009;101:2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 1994;14:5068–5076 [DOI] [PMC free article] [PubMed]
- 25.Dunn-Meynell AA, Sanders NM, Compton D, et al. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci 2009;29:7015–7022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Yassimi A, Hichami A, Besnard P, Khan NA. Linoleic acid induces calcium signaling, Src kinase phosphorylation, and neurotransmitter release in mouse CD36-positive gustatory cells. J Biol Chem 2008;283:12949–12959 [DOI] [PubMed] [Google Scholar]
- 27.Edmond J. Energy metabolism in developing brain cells. Can J Physiol Pharmacol 1992;70(Suppl.):S118–S129 [DOI] [PubMed] [Google Scholar]
- 28.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 2004;53:2521–2528 [DOI] [PubMed] [Google Scholar]
- 29.Pellerin L, Pellegri G, Martin JL, Magistretti PJ. Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc Natl Acad Sci USA 1998;95:3990–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol 2009;297:C750–C758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fioramonti X, Marsollier N, Song Z, et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010;59:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barichello T, Milioli G, Generoso JS, et al. Imipramine reverses depressive-like parameters in pneumococcal meningitis survivor rats. J Neural Transm 2012;119:653–660 [DOI] [PubMed] [Google Scholar]
- 33.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997;387:903–908 [DOI] [PubMed] [Google Scholar]
- 34.Gastaldelli A, Cusi K, Pettiti M, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007;133:496–506 [DOI] [PubMed] [Google Scholar]