

Abstract
The dysregulation of receptor tyrosine kinases (RTKs) in multiple cell types during chronic inflammation is indicative of their pathogenic role in autoimmune diseases. Among the many RTKs, vascular endothelial growth factor receptor (VEGFR) stands out for its multiple effects on immunity, vascularization, and cell migration. Herein, we examined whether VEGFR participated in the pathogenesis of type 1 diabetes (T1D) in nonobese diabetic (NOD) mice. We found that RTK inhibitors (RTKIs) and VEGF or VEGFR-2 antibodies reversed diabetes when administered at the onset of hyperglycemia. Increased VEGF expression promoted islet vascular remodeling in NOD mice, and inhibition of VEGFR activity with RTKIs abrogated the increase in islet vascularity, impairing T-cell migration into the islet and improving glucose control. Metabolic studies confirmed that RTKIs worked by preserving islet function, as treated mice had improved glucose tolerance without affecting insulin sensitivity. Finally, examination of human pancreata from patients with T1D revealed that VEGFR-2 was confined to the islet vascularity, which was increased in inflamed islets. Collectively, this work reveals a previously unappreciated role for VEGFR-2 signaling in the pathogenesis of T1D by controlling T-cell accessibility to the pancreatic islets and highlights a novel application of VEGFR-2 antagonists for the therapeutic treatment of T1D.
In type 1 diabetes (T1D), genetic and environmental risk factors lead to immune dysregulation, provoking an autoimmune response directed toward insulin-producing β-cells of the islets of Langerhans. Previous investigations have estimated that β-cells or islets in nonobese diabetic (NOD) mice and humans are diminished to 10–30% of their initial mass (1,2), and the residual islets are largely dysfunctional when hyperglycemia is first detected (1,2). However, low levels of C-peptide can be detected in T1D patients as far out as 1–2 years postdiagnosis, indicating a window of opportunity for therapies that can restore or preserve islet mass and function (3).
Multitarget receptor tyrosine kinase inhibitors (RTKIs), such as sunitinib, were originally designed to target malignant tumors that express dysregulated tyrosine kinases, including platelet-derived growth factor (PDGF)-R, c-FMS, or c-Kit. However, these inhibitors also target vascular endothelial growth factor (VEGF) receptors (VEGFRs), which are elevated in the parenchyma and tissue vasculature in many tumor microenvironments and during chronic inflammation. VEGF regulates vasculogenesis and angiogenesis largely through activation of VEGFR-2 (4). In addition to stimulating endothelial cell mitogenesis and cell migration, VEGF also has effects on a limited number of other cell types, including stimulation of monocyte/macrophage migration. Studies of transgenic mice lacking VEGFR-1 (5) or that express VEGFR-1 with a “dead” kinase domain (6) reveal that VEGFR-1 functions as a negative regulator of vasculogenesis and angiogenesis. Similarly, VEGFR-2 deficiency is embryonically lethal in mice but is attributed to a nonfunctional and underdeveloped vascular system (7). The phenotypes of VEGFR-1 and VEGFR-2–null mice indicate that, although VEGF-A has limited function through VEGFR-1, the vascular remodeling functions of VEGF-A are largely mediated through the activation of VEGFR-2.
Tyrosine kinase inhibitors (TKIs) have shown efficacy in mouse models of muscular dystrophy (8), multiple sclerosis (9), rheumatoid arthritis (10–12), and psoriasis (13). TKI can prevent and reverse diabetes in NOD mice (14–16). Imatinib, which predominantly targets c-abl and PDGF, reversed diabetes in NOD mice (14), but other RTKIs with distinct inhibitory profiles (e.g., sunitinib) were even more effective, suggesting that the precise constellations of TK targets were critical for maximum efficacy. In this regard, the VEGF-A/VEGFR-2 pathway, a key target of sunitinib, stands out as a key kinase regulating the pathogenesis of several of these inflammatory disorders (17–19). Intriguingly, VEGF serum levels are elevated in T1D patients compared with healthy controls and positively correlate with increased HbA1c levels (20).
In this study, we determined whether VEGFR-2 might be involved in the pathogenesis of T1D and tested the therapeutic efficacy of VEGFR-2 inhibition in the NOD mouse model of T1D. We report that inhibition of VEGFR-2 by RTKIs or blocking antibodies rapidly reversed diabetes and maintains euglycemia with continued drug administration. Reversal of diabetes was attributed to an abrogation of vascular remodeling in the pancreatic islets, which impairs T-cell trafficking and the severity of insulitis, ultimately improving glucose tolerance. Histological analysis of human and mouse pancreata revealed a positive correlation between the severity of insulitis and islet vascularity, implicating inflammation as a major driving force in the vascular remodeling observed in the islets. Collectively, our findings suggest that VEGF/VEGFR-2 signaling serves a critical gatekeeper function by controlling essential remodeling of the vasculature that is necessary for T cells to gain access to tissues.
RESEARCH DESIGN AND METHODS
Animals.
Female NOD mice were purchased from Taconic. NOD.GREAT mice were derived in our laboratory (21). All mice were housed in a pathogen-free facility at the University of California, San Francisco. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Compounds and treatments.
Sunitinib (22), SU-9518 (23), and PF-337210 (24), provided by Pfizer, were resuspended in methylcellulose (MC) at 10 mg/mL. Inhibitory profiles and structure data are provided in Supplementary Tables 1 and 2. Female NOD mice with new-onset diabetes were treated with RTKI or MC only immediately on the day blood glucose levels were >250 but not >400 mg/dL. In some experiments, treatment was initiated 3 weeks after the onset of hyperglycemia (long-term diabetic mice). Diabetes reversal is defined as decline in mice of blood glucose levels to <250 mg/dL. NOD mice with new-onset diabetes were treated intraperitoneally three times per week for 3 weeks with 800 μg of DC101 (anti–VEGFR-2 antibody; BioXCell). Diabetic NOD mice were also treated with weekly intraperitoneal injection of 1 mg anti–PDGF-BB, anti–PDGF-DD mAb (provided by Pfizer), anti-PDGFRβ (eBioscience), or an isotype-matched, monoclonal rat IgG1 antibody. At the end of the dosing period, mice were killed and pancreases were dissected. Half the pancreas was embedded in optimum cutting temperature (OCT) compound and snap-frozen in liquid nitrogen; the remaining half was fixed in formalin and then embedded in paraffin for subsequent histological analysis.
Assessment of diabetes and insulitis.
Blood glucose levels were measured with a LifeScan glucose meter (OneTouch Ultra). Severity of insulitis was performed as previously described (25) on hematoxylin and eosin–stained sections separated by a 100-μm distance.
Flow cytometry analysis.
Single-cell suspensions were prepared from the lymph nodes and purified islets using standard procedures, followed by Fc receptor blocking with anti-CD16/32 (clone 2.4G2) prior to staining. Single-cell suspensions were stained with antibodies against the following cell surface antigens: CD4 and Thy1.2 or Thy1.1 (Biolegend); CD62L, CD8, CD11c, CD11b, CD25, CD44, and CD69 (BD Biosciences); and DAPI (Invitrogen Life Technologies) to assess cell viability. Analysis was performed on live cells (DAPI−) on a BD LSRII flow cytometer with FACSDiva software (BD Pharmingen). Postacquisition analysis was performed with Flowjo software version 9.1.
Histological staining of pancreatic tissue.
Frozen sections of human and mouse pancreas were analyzed using standard immunofluorescence staining procedures. The following antibodies were used: guinea pig anti-insulin (DAKO), goat anti-human or mouse VEGFR-2 (R&D Systems), mouse anti-human CD31 and CD45 (Biolegend), and rat anti-mouse CD31 and CD45 (Southern Biotech). Sections were subsequently washed and incubated with the species-appropriate, Alexa-conjugated secondary antibodies (Invitrogen). Sections were stained with DAPI for 5 min to stain nuclei and mounted with FluorSave reagent (Calbiochem).
Metabolic studies.
Intraperitoneal insulin (IPITT) and glucose tolerance testing (IPGTT) was performed on prediabetic NOD mice as described previously (26). In brief, NOD mice were treated with MC or PF-337210 (5–10 mg/kg) by gavage for 3 weeks, and IPITT or IPGTT were performed weekly. For IPITT, mice were intraperitoneally injected with regular human insulin (Novolin R) at 1.0 unit/kg after a 10-h fast. For IPGTT, mice were intraperitoneally injected with sterile 30% glucose solution (Sigma-Aldrich) at 1.5 g/kg after a 17-h fast. Blood glucose levels were sampled at the indicated time points after insulin or glucose administration. Insulin was measured by insulin ELISA (Exocell).
Statistical analysis.
Statistical analyses were performed using GraphPad Prism version 5.01 or Microsoft Office Excel 2007. Statistical comparisons between two groups were performed using an unpaired two-tailed Student t test or a nonparametric Mann-Whitney test. Comparisons between multiples groups were performed by one-way or two-way ANOVA, followed by a post hoc Bonferroni test to determine significance of differences between two groups. Values of P ≤ 0.05 were considered significant.
RESULTS
Inhibition of VEGFR-2 reverses T1D in NOD mice.
To delineate the specific contributions of PDGFR and VEGFR activity in the reversal of diabetes after RTKI treatment (14), we treated diabetic NOD mice at the onset of diabetes with oral doses of SU-9518 (40 mg/kg) and PF-337210 (10 mg/kg), RTKIs with varying affinity for VEGFRs (Supplementary Table 1). SU-9518, a strong PDGFR inhibitor with weak affinity for VEGFR-2, was ineffective at reversing hyperglycemia (Fig. 1A), suggesting a lack of efficacy in compounds that ineffectively block VEGFR activity. However, daily dosing of diabetic mice with the potent VEGFR inhibitor PF-337210 (5 mg/kg) reversed diabetes and reduced blood glucose levels in all mice by the first week of treatment (Fig. 1A and B). Despite 10 weeks of continuous dosing, all mice became diabetic within 3–5 weeks after drug administration was discontinued (Fig. 1C). All RTKI-mediated reversal of diabetes was dependent on viable islet mass, as we found no drug efficacy when treating mice that had no or very few remaining islets by dosing with streptozotocin or using long-standing diabetic mice (Supplementary Fig. 1A and B).
FIG. 1.
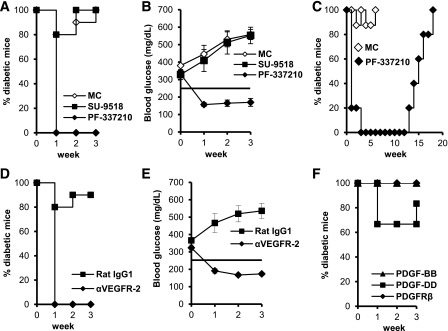
Inhibition of VEGFR-2 reverses diabetes at onset. A and B: NOD mice with new-onset diabetes (blood glucose levels ranging between 250 and 400 mg/dL) were treated daily (Monday–Friday) with SU-9518 (40 mg/kg), PF-337210 (PF; 10 mg/kg), or MC. C: Diabetic mice were treated with PF (5 mg/kg, n = 5) or MC (n = 10) for 10 weeks (Monday–Friday). D and E: Diabetic mice were treated with monoclonal antibodies specific for VEGFR-2 or a rat IgG1 isotype control (800 μg/mouse). F: Diabetic mice were treated with antibodies against PDGFRβ ligands (PDGF-BB or PDGF-DD) or PDGFRβ (1 mg/mouse). Five to ten mice were treated per group.
We tested whether an anti–VEGFR-2 blocking antibody, DC101, could also reverse diabetes (27–29). DC101 efficiently reversed diabetes in all mice treated for 3 weeks (Fig. 1D and E). Moreover, indirect blockade of VEGFR-2 activity through the use of a neutralizing anti–VEGF-A antibody (clone r84) (30) reversed diabetes in 70% of mice (Supplementary Fig. 1C). However, treatment of diabetic NOD mice with blocking antibodies against PDGFRβ-specific ligands (PDGF-BB and PDGF-DD) or PDGFRβ did not reverse diabetes (Fig. 1F). Although these data are in contrast with those made previously that soluble PDGFRβ receptor can reverse disease (14), this antagonist can also block elements of the VEGF pathway (31). Taken together, these data suggest that VEGFR-2 is a central target in the RTKI-mediated reversal of diabetes and reveal a previously unappreciated role for VEGFR-2 signaling in the pathogenesis of T1D.
VEGFR-2 expression in islets is confined to the vasculature of NOD mice with new-onset diabetes.
We next examined which cell types in the pancreas expressed VEGFR-2 to identify the key cellular target of RTKIs. Using immunofluorescence staining for CD45, a pan leukocyte marker, and VEGFR-2, we found that leukocytes did not express VEGFR-2 (Supplementary Fig. 2A). Flow cytometric analysis of CD11b+, CD8+, and naive or activated CD4+ immune cells isolated from the axillary or pancreatic lymph nodes of new-onset diabetic NOD mice confirmed our histological findings (Supplementary Fig. 2B). We also found that insulin-producing β-cells or CK19+ duct cells do not express VEGFR-2 (Supplementary Fig. 3). However, examination of the islet vasculature using coimmunofluorescence for CD31 (Fig. 2A and Supplementary Fig. 3E and F) and VEGFR-2 (Fig. 2B and Supplementary Fig. 3C and D) showed that CD31+ vessels distributed within the islets express significant levels of VEGFR-2 (overlay, Fig. 2C and Supplementary Fig. 3G and H). Quantitative PCR revealed that VEGFR-2 levels were not differentially regulated across the conditions we tested (Fig. 2D). Together, these results suggested that RTKI-mediated reversal of diabetes likely was not due to a direct effect on leukocytes or insulin-producing β-cells but, rather, occurred through a direct action on endothelial cells.
FIG. 2.

Insulitis promotes overexpression of VEGF-A. Immunofluorescence staining of CD31 (A, red), VEGFR-2 (B, green), and overlay (C). D: Quantification of VEGFR-2 transcripts from islet RNA at different stages of diabetes progression (n = 5 per group). NO-Db, new-onset diabetic; Pre-Db, prediabetic; RagKO, NOD RAG-deficient mice. Immunohistochemistry for VEGF-A performed on pancreatic sections from new-onset (E), prediabetic NOD (F), or NOD-RagKO female mice (G). H: Western analysis of whole-islet homogenates for VEGF-A from two mice per group. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. I–K: Representative coimmunofluorescence staining of pancreata from diabetic NOD mice for insulin (white), VEGF-A (green), CD4 (I, red), CD8 (J, red), CD11c (K, red), and DAPI (blue). White arrows highlight examples of CD4+, CD8+, and CD11c+ cells that express VEGF-A. Red arrows indicate β-cells that express VEGF-A. Images i and ii for I–K are the same section with different signals depicted to highlight the islet area (white, Ii–Ki) and the VEGF-A+ cells (green, Iii–Jii).
Next, we tested the hypothesis that receptor activity may be controlled by regulated expression of VEGF-A in the islets of new-onset diabetic NOD mice by immunohistochemistry and found increased VEGF-A expression in the islets (Fig. 2E). Similarly, we found increased VEGF-A staining in islets of prediabetic mice (Fig. 2F), whereas a low immune reactivity for VEGF-A was detected in β-cells of islets from NOD-RagKO mice that are devoid of inflammation (Fig. 2G). Western analysis of whole-islet homogenates substantiated these histological observations (Fig. 2H), suggesting that increased expression of VEGF-A is linked to inflammation rather than a secondary response to hyperglycemia.
We performed coimmunofluorescence for VEGF-A, immune cell markers, and insulin to determine the identity of the VEGF-A–expressing cells in the inflamed pancreas. We found that insulin+ β-cells expressed detectable levels of VEGF-A (Fig. 2I–K, red arrows) as well as multiple immune cell subsets, including CD4+ (Fig. 2Iii, white arrows), CD8+ (Fig. 2Jii, white arrows), and CD11c+ (Fig. 2Kii, white arrows) cells. Collectively, these results indicate that VEGFR activity in endothelial cells is regulated by a multicellular source of VEGF that is increased in inflamed islets and highlight a role for VEGF-A–expressing cell and islet endothelium interactions in the pathogenesis of T1D.
RTKIs prevent increases in islet vascular density associated with insulitis.
We next tested the hypothesis that the elevated expression of VEGF-A induces islet vascular remodeling, subsequently enhancing leukocyte migration and insulitis. Immunofluorescent staining of purified islets from NOD mice with new-onset diabetes revealed a more extensive and branched vascular network with an increased number of dilated vessels, compared with islets isolated at an age (4 weeks) preceding invasive insulitis (Fig. 3A and B). Islets of 18-week-old NOD mice were stained with anti-CD31 and anti-Thy1.2 antibodies to visualize the islet endothelium and infiltrating T cells, respectively. Linear regression analysis revealed a positive correlation between the CD31+ and Thy1.2+ islet area, indicating that increases in islet vascular area were partly attributed to increased insulitis (Fig. 3C). Moreover, the CD31+ islet area was increased threefold in islets isolated from 18-week-old NOD mice compared with islets isolated at 4 weeks of age (Fig. 3D). Treatment of NOD mice with new-onset diabetes with PF-337210 significantly decreased islet vascular density compared with control mice (Fig. 3E).
FIG. 3.

VEGFR inhibition prevents the increases in islet vascularity associated with insulitis. Representative micrographs of purified islets from 4-week-old (A) and new-onset (B) NOD females that were subjected to immunofluorescent staining with anti-CD31 (teal). C: Linear regression analysis shows a positive correlation between CD31+ and Thy1.2+ islet area in islets isolated from prediabetic mice. The micrograph depicts the cumulative data from 67 purified islets analyzed from five mice. D: The islet CD31+ area was increased in purified islets of 18-week-old, prediabetic mice relative to islets from 4-week-old NOD mice. E: A 5-day dosing period of mice with new-onset diabetes with PF-337210 (PF; 20 mg/kg) prevented further increases in islet vascular density. MC, 28 islets analyzed from two mice; PF-337210 (PF), 45 islets analyzed from three mice.
RTKI-mediated inhibition of VEGFR signaling impairs T-cell trafficking.
We next examined the effects of VEGFR inhibition on T-cell migration. To minimize variability in the absolute number and frequency of T cells isolated from purified islets of prediabetic NOD mice treated with PF-337210 (data not shown), we took advantage of an islet antigen–specific T-cell transgenic model (BDC2.5 T cells congenically marked with Thy1.1+) to synchronize the migration and tracking of T cells into the islet in an in vivo migration assay (32). A 2-week pretreatment of NOD-RagKO mice with PF-337210 resulted in a significant reduction in the proportion of islets infiltrated with Thy1.1+ T cells 48 h after transfer (Fig. 4A). The enumeration of Thy1.1+ T cells by immunofluorescence revealed that T cells in islets were reduced by twofold in PF-337210–treated animals compared with control-treated mice (Fig. 4B–D, red). The impaired T-cell migration was associated with a reduced vascularity in NOD-RagKO mice that received activated BDC2.5 T cells and were treated with PF-337210 (Fig. 4E).
FIG. 4.

VEGFR inhibition impedes T-cell migration by reducing islet vascularity. A: The proportion of islets infiltrated by BDC2.5 (Thy1.1+) cells 48 h after transfer in PF-337210 (PF)-treated or control mice. The micrograph depicts the cumulative data from 60–100 islets per mouse and 16–18 mice per group. Representative immunofluorescence images of islets purified from MC- (B) or PF-treated (C) mice stained for Thy1.1 (red) and CD31 (teal). D: Enumeration of the number of Thy1.1+ cells per islet in PF-treated mice versus control (MC) mice. E: Vascularity of PF-treated or control mice expressed as the percentage of CD31+ area of the total islet. PF was administered at a dose of 20 mg/kg.
The impairment in T-cell trafficking was not restricted to islets, as we noted reduced spleen mass, total splenocyte counts, and numbers of transferred Thy1.1+ T cells in the spleen (Supplementary Fig. 4A–C). However, the number of PBMCs or Thy1.1+ T cells in the blood was not affected by PF-337210 administration (Supplementary Fig. 4D and E), suggesting that VEGFR inhibition did not lead to T-cell toxicity or altered proliferation that could account for the reduced number of transferred BDC2.5 T cells in the tissues of PF-337210–treated mice. The expression of T-cell activation markers on transferred T cells in the blood and spleen was not affected by PF-337210 (Supplementary Fig. 5). Taken together, these data are consistent with our observations that leukocytes in NOD mice express insignificant levels of VEGFR-2 and confirm that PF-337210 does not directly affect T cells.
VEGFR-2 inhibition reduces insulitis and preserves islet function.
We next examined whether RTKI or the anti-VEGFR antibody (DC101) led to reduced insulitis and improved islet function in NOD mice. Examination of islet infiltration after drug administration showed that the percentage of severely infiltrated islets (score = 3) in diabetic mice treated for 3 weeks with RTKI or DC101 was significantly reduced compared with vehicle-treated (MC) mice (Fig. 5A–E). Furthermore, the decrease in severely infiltrated islets was accompanied by an increased proportion of immunologically spared islets (score = 0) and islets with noninvasive insulitis (score = 1). Similar to the transfer setting, inhibition of VEGFR-2 with PF-337210 had no effect on the expression of activation markers or interferon-γ (IFN-γ) by T cells in the islets (Fig. 5F). The reduced insulitis improved islet function based on an IPGTT assay in prediabetic mice, after a 3-week PF-337210 treatment. The RTKI treatment improved glucose tolerance, suggesting preservation of islet function (Fig. 5G). However, the improvement of glucose tolerance was not restored to the levels present in mice at 4 weeks of age, a stage preceding detrimental islet destruction in NOD mice (Fig. 5H). We also found that PF-337210 treatment reduced glucose levels of nonfasted diabetic mice (Fig. 5I) and retained pancreatic insulin content (Fig. 5J) but did not significantly alter serum insulin levels relative to vehicle (Fig. 5K). Measurements of serum insulin after a glucose challenge showed no difference between fasted mice treated with PF-337210 or vehicle, indicating that the first- and late-phase insulin release is not affected by inhibition of VEGFR (Fig. 5L). Collectively, the results indicate that the PF-337210–mediated increase in pancreatic insulin content is not attributed to defects in insulin secretion; they likely reflect a preservation of islet mass.
FIG. 5.

VEGFR-2 inhibition reduces insulitis and preserves islet function. A–D: Representative micrographs of new-onset pancreata used to score the severity of insulitis in VEGFR-2 antagonists and control (MC) mice. The red line in A depicts the approximated islet boundary. The red arrow in D highlights an islet with severe insulitis. Black arrows highlight islets without insulitis. E: Quantification of the severity of insulitis reported as the percentage of islets with a given disease score. The number within each bar represents the levels of significance determined by Student t test (the indicated value = P value). At least 100 islets were scored per mouse. Five to ten mice per group were analyzed. F: Measurement of the expression levels of T-cell activation markers by flow cytometry reveals no differential expression between MC and PF-337210 (PF)–treated prediabetic mice. uns, unstimulated. G: Glucose tolerance tests reveal that 14-week-old, prediabetic mice that were treated with PF (10 mg/kg) for 3 weeks had improved glucose tolerance relative to MC-treated mice. Five mice per group were used. #P < 0.0001; *P < 0.01, PF-treated relative to MC-treated mice. H: Quantification of area under the curve shows a significant decrease in PF-treated mice, but levels were still higher than 4-week-old NOD females. ♦P < 0.05, relative to 4-week NOD; *P < 0.05, relative to MC. I–K: Diabetic mice were treated daily for 3 days with PF (10 mg/kg) or MC. The red line in I indicates a blood glucose value of 250 mg/dL. A 3-day treatment significantly reduced blood glucose levels (I), retained insulin content (J), and did not significantly affect serum insulin levels (K) in PF-treated mice. ♦P < 0.05, relative to new-onset mice (NO). L: The insulin response after a fasting IPGTT in prediabetic NOD mice treated for 3 weeks with PF (10 mg/kg) or MC.
We tested whether the rapid effects of the RTKIs were attributed to augmented insulin sensitivity as seen in db/db mice or rats fed a high-fat diet treated with anti–VEGF-B antibodies (33). No difference was found in blood glucose clearance rates during an IPITT or in the levels of phosphorylated AKT, which is activated downstream of the insulin receptor, after an insulin bolus in PF-337210–treated mice versus controls (Supplementary Fig. 6A and B). Moreover, the improved glucose tolerance in PF-337210–treated mice was not attributed to differences in food consumption (Supplementary Fig. 6C) or weight loss (Supplementary Fig. 6D). These findings suggest that the enhanced glucose tolerance observed in PF-337210–treated mice is not attributed to improved insulin sensitivity in peripheral tissues. Instead, the data support our hypothesis that the RTKI-mediated improvement in glucose tolerance is due to impaired T-cell trafficking, which reduces insulitis and leads to improved islet function and glucose control.
Increase in islet vascularity in T1D patients is associated with increased insulitis.
To gain insight into the potential translation of our preclinical findings, we examined the vasculature features of islets from T1D patients and controls. The islet boundary was first approximated by insulin and DAPI staining (Fig. 6A, B, E, and F, white line). Concurrent staining of tissues with anti-CD31 and anti-CD45 antibodies to allow visualization of blood vessels and leukocytes, respectively, revealed that all CD31+ vessels in the islets of both T1D patients and controls were VEGFR-2+ (Fig. 6C and D). VEGFR-2 expression was not detected in CD45+ leukocytes in either normal or T1D patients, similar to what was observed in the mouse studies (Fig. 6G and H). In addition, we did not detect expression of VEGFR-2 in β-cells, unlike previous studies showing VEGFR expression in human islets (34). The images in Fig. 6I and J (which are from the same section shown in C and D, respectively) reveal that VEGFR-2+ cells (white arrows) are not β-cells. Instead these are the CD31+ cells in Fig. 6C and D (white arrows). Notably, the quantification of the CD45+ (insulitis, Fig. 6K) and VEGFR-2+ (vascularity, Fig. 6L) islet areas revealed that both are increased in T1D patients relative to controls. Collectively, our results suggest that inflammation and islet vascular remodeling are linked processes that are associated with the pathogenesis of T1D in humans.
FIG. 6.
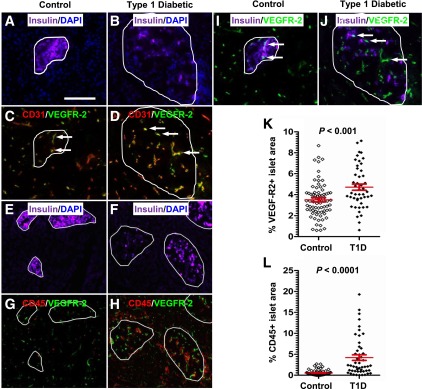
Insulitis in T1D patients with diabetes is associated with increased islet vascularity. The islet boundary (white line) was defined by insulin and DAPI staining (A, B, E, and F). Immunofluorescent staining of control (C) and T1D patients (D) reveals that all islet vessels (red) also expressed VEGFR-2 (green). Coexpression of both markers is indicated in yellow. Representative micrographs of control (G) and T1D patients (H) reveal that CD45+ leukocytes (red) do not express VEGFR-2 (green). We also found that insulin+ β-cells (purple) did not express VEGFR-2 (green) in control (I) or T1D patient (J) pancreata. The field of view depicted in I and J corresponds to the same images shown in C and D, respectively. The white arrows in I and J point to VEGFR-2+ cells, which according to C and D are CD31+ vessels. Quantification of the VEGFR-2+ (K) and CD45+ (L) islet area in control and T1D patient pancreata revealed that both were increased in T1D patients.
DISCUSSION
Herein, we demonstrate that VEGFR-2 is a central target in the RTKI-mediated reversal of diabetes in NOD mice. The pathogenesis of T1D requires infiltration of immune cells into the pancreatic islets, and this is dependent upon VEGF-mediated activation of VEGFR-2 within the islet endothelium. Inhibition of VEGFR-2 signaling impairs T-cell migration and reduces insulitis, restoring islet function and glucose control. Histological studies of human pancreata indicate that a similar pathogenic process occurs in T1D patients and suggests that activation of the VEGF/VEGFR-2 signaling axis is a key molecular event linking vascular remodeling and insulitis, processes that contribute to loss of glucose control and onset of hyperglycemia.
VEGFR-2 is a master regulator of angiogenesis and vascular permeability and contributes to the establishment of chronic inflammation in various pathological settings (35–37). We show that VEGFR-2 inhibitors abrogate increases in islet vascular density and subsequently impede the development of insulitis. Similar observations have been reported in streptozotocin-induced diabetes (38) and other inflammatory settings in which inhibition of VEGFR-2 signaling impairs vascular remodeling and reduces inflammation (19,28,39,40). In addition to promoting increased vascularity, VEGF-A contributes to inflammation and leukocyte recruitment by inducing the expression of adhesion molecules and chemokines by endothelial cells (19,41–43). Our findings support the conclusion that reduced insulitis is due to decreased islet vascularity. However, there is also the possibility that VEGFR-2 antagonists repress the expression of molecules that promote leukocyte trafficking/homing to the islet vasculature. Future studies will require the characterization of potential molecular changes in activated islet endothelial cells to fully understand the role of islet vascular remodeling in the pathogenesis of T1D.
Our results suggest that there may be distinct molecular and functional properties of islet endothelial cells in a pathogenic state versus those in homeostasis, rendering them more sensitive to RTKI treatment. We found that the islet vascularity showed obvious morphological changes indicative of pathological angiogenesis, which is induced by elevated expression of VEGF-A (36). Although Akirav et al. (44) previously reported that VEGF-A is increased in the inflamed islets of prediabetic NOD mice, they found that islet vascularity decreases with the progression of diabetes. The discrepancies between these studies may reflect a difference in the assays used to measure islet vascularity.
A pathogenic role for VEGF-A is supported by the observation that the overexpression of VEGF-A in β-cells led to increased islet vascularity, inflammation, and disruption in islet architecture, resulting in impaired glucose homeostasis and insulin secretion during a high-fat diet (45). However, a complete loss of VEGF signaling in islets is also detrimental. For instance, β-cell–specific ablation of VEGF-A resulted in reduced islet vascularity and endothelial cell fenestration, culminating in impaired insulin secretion and glucose intolerance. However, in the setting of autoimmune diabetes, this potential impairment in function is outweighed by the protection offered by preventing diabetogenic leukocytes from entering the islet and destroying β-cells. Collectively, our data and the altered islet function after perturbations in β-cell–specific VEGF-A expression indicate that VEGF-A functions in a narrow physiological range to maintain islet homeostasis and function.
We propose a model in which reversal of diabetes is delineated in three distinct stages, including 1) acute reversal, 2) maintenance of euglycemia, and 3) long-term remission of diabetes in the absence of continued drug dosing. The acute reversal and maintenance of euglycemia of diabetes is dependent on the inhibition of VEGFR-2. However, the lack of complete diabetes reversal in mice treated with anti–VEGF-A antibody indicates that other VEGFRs may also mediate the islet vascular remodeling. In this regard, inhibition of VEGFR-3 was shown to reduce insulitis in NOD mice (38). Alternatively, it is very likely that small molecule antagonists, such as the RTKIs used in this study, are able to penetrate the target tissue more effectively than mAbs such as r84. We cannot exclude the effect of RTKIs on other physiological systems that control glucose metabolism (e.g., liver or adipose tissue). For instance, RTKI treatment also enhances insulin-independent glucose metabolism in peripheral tissues. In this regard, recent studies have shown that JAK-STAT inhibitors increased peroxisome proliferator–activated receptor-γ (PPAR-γ), a critical regulator of fatty acid storage and glucose metabolism, expression in adipocytes and immune cells (46).
The discrepancy between the lack of reversal of diabetes with the PDGFR antagonists used in this study and the previously reported reversal using a soluble PDGFRβ-Ig fusion protein (14) may be explained by functional differences between the reagents used to inhibit PDGFR. Kietly and colleagues (31) recently demonstrated that PDGFR is able to functionally bind VEGF ligand, leading to PDGFR activation. Therefore, it is possible that the acute reversal of diabetes after soluble PDGFRβ-Ig may be attributed to the binding and neutralization of VEGF ligands in addition to PDGF ligands. The studies using Gleevec suggest that c-Abl also participates in reversal of hyperglycemia and long-term remission, but through a distinct mechanism that likely involves the inhibition of proapoptotic pathways in β-cells (15,47) and altered T-cell immunity (48), leading to islet mass preservation. Similarly, the current study suggests that RTKIs function by preserving islet mass. Although we cannot rule out the possibility of β-cell regeneration, previous studies have shown that the most likely effect of drugs on the islets is to reverse the metabolic stress caused by the immune response, leading to recovery of the insulin production by the remaining β-cells (49,50).
In summary, our findings that VEGFR-specific RTKIs and blocking anti–VEGFR-2 antibodies reverse diabetes by preserving islet function suggest that therapeutic modalities aimed at preventing islet vasculature activation may provide an alternative treatment strategy for T1D.
ACKNOWLEDGMENTS
This work was partially supported by a National Institutes of Health Diabetes and Endocrinology Research Center grant (P30-DK-63720-06A1). S.A.V. was supported by an American Diabetes Association Mentor-Based Minority Postdoctoral Fellowship and a National Research Service Award training grant.
This investigation was also supported by a Pfizer-QB3 grant and the Investigator-Initiated Study Program of LifeScan, Inc. R.H.A. and N.P. are employed by Pfizer, Inc. R.A.B. performed consultation services for Peregrine Pharmaceuticals Inc. for the development of r84. No other potential conflicts of interest relevant to this article were reported.
S.A.V. designed and performed research, analyzed data, and wrote the manuscript. J.L., S.K., B.K., G.L.S., and C.W. performed research. B.C. assisted in designing the in vivo migration assay used in this study. M.A.A. designed research. R.A.B. and N.P. contributed data as well as new reagents/analytic tools. R.H.A. designed research and contributed data as well as new reagents/analytic tools. J.A.B. designed research, analyzed data, and wrote the manuscript. J.A.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Michael DuPage and Mahesh Yadav, Diabetes Center of the University of California, San Francisco, for critical review of this manuscript; Gaurav Chopra, Diabetes Center of the University of California, San Francisco, for in silico analysis; Navdeep Grewal, Diabetes Center of the University of California, San Francisco, for the histological preparation of pancreatic sections; the Network for Pancreatic Organ Donors with Diabetes for providing pancreata tissue sections; Emil Unanue, Washington University School of Medicine, for assistance in designing the in vivo migration assay used in this study; and Fred Schaufele and the Diabetes and Endocrinology Research Center, University of California, San Francisco, for assistance in microscopy and quantitative image analysis.
Parts of this study were presented in abstract form at the Annual Meeting of the American Association of Immunologists, Honolulu, Hawaii, 3–7 May 2013.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1619/-/DC1.
REFERENCES
- 1.Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS. Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes 1999;48:989–996 [DOI] [PubMed] [Google Scholar]
- 2.Alanentalo T, Hörnblad A, Mayans S, et al. Quantification and three-dimensional imaging of the insulitis-induced destruction of beta-cells in murine type 1 diabetes. Diabetes 2010;59:1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherry NA, Tsai EB, Herold KC. Natural history of beta-cell function in type 1 diabetes. Diabetes 2005;54(Suppl. 2):S32–S39 [DOI] [PubMed] [Google Scholar]
- 4.Rossant J, Howard L. Signaling pathways in vascular development. Annu Rev Cell Dev Biol 2002;18:541–573 [DOI] [PubMed] [Google Scholar]
- 5.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995;376:66–70 [DOI] [PubMed] [Google Scholar]
- 6.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci USA 1998;95:9349–9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995;376:62–66 [DOI] [PubMed] [Google Scholar]
- 8.Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J 2009;23:2539–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uemura Y, Ohno H, Ohzeki Y, et al. The selective M-CSF receptor tyrosine kinase inhibitor Ki20227 suppresses experimental autoimmune encephalomyelitis. J Neuroimmunol 2008;195:73–80 [DOI] [PubMed] [Google Scholar]
- 10.Paniagua RT, Sharpe O, Ho PP, et al. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest 2006;116:2633–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosios K, Wood J, Esser R, Raychaudhuri A, Dawson J. Angiogenesis inhibition by the novel VEGF receptor tyrosine kinase inhibitor, PTK787/ZK222584, causes significant anti-arthritic effects in models of rheumatoid arthritis. Inflamm Res 2004;53:133–142 [DOI] [PubMed] [Google Scholar]
- 12.Milici AJ, Kudlacz EM, Audoly L, Zwillich S, Changelian P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther 2008;10:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Bassat H. Biological activity of tyrosine kinase inhibitors: novel agents for psoriasis therapy. Curr Opin Investig Drugs 2001;2:1539–1545 [PubMed] [Google Scholar]
- 14.Louvet C, Szot GL, Lang J, et al. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA 2008;105:18895–18900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hägerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J 2007;21:618–628 [DOI] [PubMed] [Google Scholar]
- 16.Davoodi-Semiromi A, Wasserfall CH, Xia CQ, et al. The tyrphostin agent AG490 prevents and reverses type 1 diabetes in NOD mice. PLoS ONE 2012;7:e36079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo SA, Kwok SK, Kim WU. Proinflammatory role of vascular endothelial growth factor in the pathogenesis of rheumatoid arthritis: prospects for therapeutic intervention. Mediators Inflamm 2008;2008:129873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chua RA, Arbiser JL. The role of angiogenesis in the pathogenesis of psoriasis. Autoimmunity 2009;42:574–579 [DOI] [PubMed] [Google Scholar]
- 19.Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009;136:585.e5–595.e5 [DOI] [PubMed]
- 20.Chiarelli F, Spagnoli A, Basciani F, et al. Vascular endothelial growth factor (VEGF) in children, adolescents and young adults with type 1 diabetes mellitus: relation to glycaemic control and microvascular complications. Diabet Med 2000;17:650–656 [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 2009;10:385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003;9:327–337 [PubMed] [Google Scholar]
- 23.Abdollahi A, Li M, Ping G, et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med 2005;201:925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marra MT, Khamphavong P, Wisniecki P, Gukasyan HJ, Sueda K. Solution formulation development of a VEGF inhibitor for intravitreal injection. AAPS PharmSciTech 2011;12:362–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bour-Jordan H, Salomon BL, Thompson HL, Santos R, Abbas AK, Bluestone JA. Constitutive expression of B7-1 on B cells uncovers autoimmunity toward the B cell compartment in the nonobese diabetic mouse. J Immunol 2007;179:1004–1012 [DOI] [PubMed] [Google Scholar]
- 26.Chaparro RJ, Konigshofer Y, Beilhack GF, Shizuru JA, McDevitt HO, Chien YH. Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proc Natl Acad Sci USA 2006;103:5–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang ES, Teruya-Feldstein J, Wu Y, Zhu Z, Hicklin DJ, Moore MA. Targeting autocrine and paracrine VEGF receptor pathways inhibits human lymphoma xenografts in vivo. Blood 2004;104:2893–2902 [DOI] [PubMed] [Google Scholar]
- 28.Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med 2010;207:2255–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung ES, Chauhan SK, Jin Y, et al. Contribution of macrophages to angiogenesis induced by vascular endothelial growth factor receptor-3-specific ligands. Am J Pathol 2009;175:1984–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan LA, Carbon JG, Roland CL, et al. r84, a novel therapeutic antibody against mouse and human VEGF with potent anti-tumor activity and limited toxicity induction. PLoS One 2010;5:e12031 [DOI] [PMC free article] [PubMed]
- 31.Ball SG, Shuttleworth CA, Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol 2007;177:489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci USA 2011;108:1561–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagberg CE, Mehlem A, Falkevall A, et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 2012;490:426–430 [DOI] [PubMed] [Google Scholar]
- 34.Cross SE, Richards SK, Clark A, et al. Vascular endothelial growth factor as a survival factor for human islets: effect of immunosuppressive drugs. Diabetologia 2007;50:1423–1432 [DOI] [PubMed] [Google Scholar]
- 35.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 2006;7:359–371 [DOI] [PubMed] [Google Scholar]
- 36.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol 2007;2:251–275 [DOI] [PubMed] [Google Scholar]
- 37.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond) 2005;109:227–241 [DOI] [PubMed] [Google Scholar]
- 38.Yin N, Zhang N, Lal G, et al. Lymphangiogenesis is required for pancreatic islet inflammation and diabetes. PLoS ONE 2011;6:e28023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schonthaler HB, Huggenberger R, Wculek SK, Detmar M, Wagner EF. Systemic anti-VEGF treatment strongly reduces skin inflammation in a mouse model of psoriasis. Proc Natl Acad Sci USA 2009;106:21264–21269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afuwape AO, Feldmann M, Paleolog EM. Adenoviral delivery of soluble VEGF receptor 1 (sFlt-1) abrogates disease activity in murine collagen-induced arthritis. Gene Ther 2003;10:1950–1960 [DOI] [PubMed] [Google Scholar]
- 41.Lee TH, Avraham H, Lee SH, Avraham S. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. J Biol Chem 2002;277:10445–10451 [DOI] [PubMed] [Google Scholar]
- 42.Goebel S, Huang M, Davis WC, et al. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol 2006;290:G648–G654 [DOI] [PubMed] [Google Scholar]
- 43.Boulday G, Haskova Z, Reinders ME, Pal S, Briscoe DM. Vascular endothelial growth factor-induced signaling pathways in endothelial cells that mediate overexpression of the chemokine IFN-gamma-inducible protein of 10 kDa in vitro and in vivo. J Immunol 2006;176:3098–3107 [DOI] [PubMed] [Google Scholar]
- 44.Akirav EM, Baquero MT, Opare-Addo LW, et al. Glucose and inflammation control islet vascular density and beta-cell function in NOD mice: control of islet vasculature and vascular endothelial growth factor by glucose. Diabetes 2011;60:876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agudo J, Ayuso E, Jimenez V, et al. Vascular endothelial growth factor-mediated islet hypervascularization and inflammation contribute to progressive reduction of β-cell mass. Diabetes 2012;61:2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davoodi-Semiromi A, Hassanzadeh A, Wasserfall CH, Droney A, Atkinson M. Tyrphostin AG490 agent modestly but significantly prevents onset of type 1 in NOD mouse; implication of immunologic and metabolic effects of a Jak-Stat pathway inhibitor. J Clin Immunol 2012;32:1038–1047 [DOI] [PubMed] [Google Scholar]
- 47.Hägerkvist R, Makeeva N, Elliman S, Welsh N. Imatinib mesylate (Gleevec) protects against streptozotocin-induced diabetes and islet cell death in vitro. Cell Biol Int 2006;30:1013–1017 [DOI] [PubMed] [Google Scholar]
- 48.Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. Defective T cell development and function in the absence of Abelson kinases. J Immunol 2007;179:7334–7343 [DOI] [PubMed] [Google Scholar]
- 49.Sherry NA, Chen W, Kushner JA, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology 2007;148:5136–5144 [DOI] [PubMed] [Google Scholar]
- 50.Ablamunits V, Henegariu O, Hansen JB, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes 2012;61:145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]