

Abstract
Background
Decades-old animal experiments suggested dietary long-chain monounsaturated fatty acids (LCMUFA) caused cardiotoxicity, leading, for example, Canada to develop Canadian-oil-low-in-erucic-acid (Canola) from rapeseed. However, potential cardiotoxicity in humans and contemporary dietary sources of LCMUFA are unknown.
Methods and Results
We prospectively investigated associations of plasma phospholipid LCMUFA (20:1, 22:1, and 24:1), objective biomarkers of exposure, with incidence congestive heart failure (CHF) in two independent cohorts: 3,694 older adults (mean age=75.2±5.2 years) in the Cardiovascular Health Study (CHS, 1992–2006), and 3,577 middle-aged adults (mean age=54.1±5.8 years) in the Atherosclerosis Risk in Communities Study Minnesota subcohort (ARIC, 1987–2008). We further examined dietary correlates of circulating LCMUFA in CHS and ARIC, and US dietary sources of LCMUFA in the 2003–2010 National Health and Nutrition Examination Survey (NHANES). In CHS, 997 CHF events occurred during 39,238 person-years; and in ARIC, 330 events during 64,438 person-years. After multivariable-adjustment, higher levels of 22:1 and 24:1 were positively associated with greater incident CHF in both CHS and ARIC: hazard ratios (95% confidence interval)=1.34 (1.02–1.76) and 1.57 (1.11–2.23) for highest vs. lowest quintiles of 22:1, respectively; and 1.75 (1.23–2.50) and 1.92 (1.22–3.03) for 24:1, respectively (P-trend≤0.03 each). A variety of foods related to circulating LCMUFA in CHS and ARIC, consistent with food sources of LCMUFA in NHANES, including fish, poultry, meats, whole grains, and mustard.
Conclusions
Higher circulating levels of 22:1 and 24:1, with apparently diverse dietary sources, were associated with incident CHF in two independent cohorts, suggesting possible cardiotoxicity of LCMUFA in humans.
Keywords: congestive heart failure, fatty acids, diet, epidemiology
In the 1960s–1980s, feeding experiments in rodents, pigs, and non-human primates suggested that consumption of erucic acid (22:1n9) and cetoleic acid (22:1n11) caused cardiac steatosis.1–4 Although potential effects in humans were never studied, mechanistic studies suggest that exposure to long-chain monounsaturated fatty acids (LCMUFA. 20:1, 22:1 and 24:1 fatty acids) might impair myocardium. The heart preferentially uses fatty acids as fuel.5–7 Long-chain fatty acids including LCMUFA are predominantly oxidized in peroxisomes, rather than mitochondria, which lack membrane-transporting enzymes for long-chain fatty acids.3, 8 Peroxisomal fatty acid oxidation produces reactive oxygen species and various cytosolic lipid metabolites8 that stimulate several signaling pathways, thereby inhibiting mitochondrial fatty acid oxidation, synthesizing cardiac lipid droplets, inhibiting glycolysis, and inducing apoptosis.5–7 In sum, such effects can cause cardiotoxicity.5–7
This decades-old evidence led Canadian farmers to modify rapeseed oil, a major source of erucic acid (~30–60% of fatty acids), and market it as CANadian Oil Low in Erucic Acid (Canola) oil9, as well as some governments to limit content of erucic acid in rapeseed oil.9, 10 Thereafter, the potential cardiotoxicity and dietary sources of LCMUFA have been largely forgotten and, to our knowledge, never studied in humans. However, food composition data indicate LCMUFA remain present in mustard oils (20–50%) and related products, some fish species (5–30%), and meat and poultry products (0–5%).2, 11 Given experimental induction of cardiac fibrosis and steatosis, key risk factors for CHF12–14, we hypothesized that LCMUFA exposure may increase incidence of CHF. To address this hypothesis, we investigated prospective associations between LCMUFA exposure, assessed as objective biomarkers in plasma phospholipids, and incident CHF in two independent US cohorts: the Cardiovascular Health Study (CHS) and the Atherosclerosis Risk in Communities Study (ARIC). We further characterized dietary factors related to LCMUFA biomarker levels and evaluated potential dietary sources of LCMUFA consumption in based on the U.S. National Health and Nutrition Examination Survey (NHANES) and the USDA food composition database.11
METHODS
Design and Population
In 1989–90, CHS recruited 5,201 ambulatory, noninstitutionalized adults age 65 years or older who were randomly selected from Medicare lists in four US communities; an additional 687 black participants were similarly recruited in 1992–93.15, 16 ARIC recruited 15,792 adults aged 45–64 years in four US communities in 1987–89 based on multiple databases including driver's license listings, community lists, local health census lists, or area sampling.17 In CHS and ARIC, 57% and 60% among eligible adults agreed to enroll and gave informed consent, respectively.
Circulating fatty acid concentrations were measured in 3,941 CHS participants using 1992–93 blood samples; and in 3,705 ARIC participants in the Minneapolis subcohort using 1987–89 blood samples. These years of fatty acid measurement were considered the baseline for all analyses. Sociodemographic characteristics, lifestyle behaviors, dietary consumption, and laboratory measures were assessed as previously described15–17 (see Supplemental Materials for the details). After excluding participants with prevalent CHF in CHS (n=247) and ARIC (n=128) and without circulating fatty acid measures, the present analyses included 3,694 CHS and 3,577 ARIC participants.
Phospholipid Fatty Acids
Methods for assessments of plasma phospholipid fatty acids in CHS and ARIC slightly differed, as described in Supplemental Material. In CHS, 42 known individual fatty acids were quantified; and in ARIC, 29 fatty acids. In this study, we evaluated LCMUFA as the main exposure variables: gadoleic acid (20:1; the first number referring to the number of carbons, and the second to the number of double bonds, in the fatty acid), erucic acid (22:1) and nervonic acid (24:1). Intra-assay CVs were <5% for 20:1 and 24:1, and 15% for 22:1. We assessed reproducibility of LCMUFA levels, potentially affected by both measurement error and biologic variation over time that would attenuate findings toward the null, evaluating serial measures from blood samples drawn in 1992–93, 1998–99, and 2005–06 in a subset (n=100) of CHS. Within-individual correlations were highest for 24:1 (r=0.66 at 6 years and 0.43 at 13 years) and lower for 22:1 (r=0.26 and 0.18) and 20:1 (r=0.26 and 0.26). This reproducibility for 24:1 was comparable or superior to reproducibility for major cardiovascular risk factors as blood pressure.18
Cardiovascular Risk Factors and Incident Congestive Heart Failure
In each cohort, we evaluated cross-sectional associations of LCMUFA levels with cardiovascular risk factors (see Supplemental Material for the assessments) and longitudinal associations of LCMUFA with incident CHF. In CHS, potential CHF events were identified from annual examinations, interim 6-month phone contacts, and hospital discharge records, with review and classification by a centralized committee of physicians.16, 19 CHF event was confirmed when all three criteria were met: 1) CHF symptoms (shortness of breath, fatigue, orthopnea, paroxysmal nocturnal dyspnea) and signs (edema, rales, tachycardia, gallop, displaced apical impulse), or clinical findings from echocardiography, ventriculography, or chest radiography; 2) diagnosis of CHF by a treating physician; and 3) CHF medical therapy (a diuretic plus either digitalis or a vasodilator). In ARIC, potential CHF events were first identified by annual phone contacts, review of hospitalization discharge codes, and death certificates, and then ascertained by either a hospitalization including a CHF discharge diagnosis (ICD9 code=428) or a death certification listing CHF.20 In CHS, CHF events with sufficient information were further subclassified as primarily due to ischemic or nonischemic etiologies and due to valvular or nonvalvular etiologies.19
To determine whether LCMUFA-CHF associations were myocardium-specific, we analyzed incident stroke as a prespecified negative-control outcome.21 We selected stroke because it shares many risk factors with CHF (e.g., hypertension) but should be unaffected by any causal processes specific to cardiac steatosis, i.e. the hypothesized cardiotoxicity of LCMUFA. Each cohort defined stroke as a neurological deficit of rapid onset lasting >24 hours, or as a subarchnoid hemorrhage confirmed by CT or MRI when available.22, 23
Dietary Correlates of Circulating LCMUFA and Estimated US LCMUFA Consumption
To examine independent dietary correlates to circulating LCMUFA levels, we assessed cross-sectional associations of habitual food consumption with phospholipid LCMUFA levels. We evaluated 43 food groups in CHS and 41 food groups in ARIC derived from interviewer-administered validated food-frequency questionnaires (mustard consumption and fried fish were only available in CHS). In CHS, we related averages dietary intakes over the two questionnaires in 1989–90 and 1996–97 to LCMUFA levels in 1992–93, as previously performed.24 In ARIC, dietary intakes were related to LCMUFA levels at the same data-collection cycle in 1987–89.
To evaluate the validity of dietary correlates to LCMUFA biomarkers in CHS and ARIC, as well as consider current consumption levels in the US, we assessed major food sources of LCMUFA evaluating food-composition databases and food intakes in the 2003–2010 NHANES (n adults=20,150) that implemented two 24-hour dietary recalls per person (see Supplemental Material for the analytical methods). Food consumption data were available for 20:1 and 22:1, but not 24:1.
Statistical Analyses
We assessed independent dietary correlates to circulating LCMUFA levels by multivariable-adjusted stepwise linear regression (p<0.05 to retain and p>0.1 to remove) as performed previously24, relating food groups (serving/week) to LCMUFA levels that were standardized to a standard deviation after log-transformation to improve normality.
To assess cross-sectional associations of LCMUFA with cardiovascular risk factors, we evaluated LCMUFA levels as independent variables and cardiovascular risk factors as dependent variables by multivariable-adjusted linear regression. Prospective relationships of phospholipid LCMUFA with incident CHF were examined by multivariable-adjusted Cox proportional hazards in each cohort separately and then estimates for quintile category scores were pooled by random-effects meta-analysis. The proportionality assumption was not rejected by examining cross-product terms of follow-up time by exposure (p>0.3). Time at risk was from time of blood draw to the first CHF (or stroke) diagnosis, death, loss of follow up, or administrative censoring (2006 in CHS; 2008 in ARIC). Loss of follow-up was ≤2% of person-times in both CHS and ARIC.
To minimize confounding, we adjusted for covariates based on previously published associations or clinical relevance, including demographics, clinical histories, and lifestyle factors. We recognized that numerous dietary factors and circulating fatty acids could be confounders. Consumption of both generally healthy foods such as plant oils and fish could influence circulating levels of LCMUFA and other fatty acids, and incident CHF. Importantly, fish consumption has been associated with lower CHF incidence in CHS and ARIC.20, 25 To select and control for potential confounders of diet and circulating fatty acids, we adopted a confounder-selection strategy developed previously.26 Briefly, we selected covariates when their removal caused >5% change in the measure of association of LCMUFA levels with CHF.
To assess whether the associations between LCMUFA and incident CHF was independent of traditional cardiovascular risk factors or potential mediators, we further adjusted for body-mass index, waist circumference, blood lipids, inflammatory markers, and incident CHD during follow-up as a time-varying covariate. We tested multiplicative interactions by prespecified factors of age, gender, race, and prevalent CHD by evaluating the Wald test for a cross-product term of exposure and covariate in the model. We also evaluated interaction by prevalent diabetes post hoc.
In longitudinal analyses, time-dependent misclassification in both exposures and covariates causes regression dilution bias and residual confounding. We performed sensitivity analyses to correct for this bias by means of multivariate regression calibration, based on within-individual correlations of serial measures of physical activity, dietary habits, and phospholipid FA (in a subset) in CHS.18 Because comparable serial measures were unavailable in ARIC, we extended regression dilution ratios in CHS to analysis in ARIC, recognizing the limited generalizability and thus considering these corrected risk values in ARIC as only estimates. We examined non-linear associations in each cohort using 4-knot restricted cubic splines. Statistical analyses were performed using STATA 10.0, two-tailed α=0.05.
RESULTS
Participants in CHS were older at baseline (age=75.2±5.2y) than in ARIC (54.1±5.8y), with concomitantly higher prevalence of chronic diseases (Supplemental Table 1). Majority were white, 88% in CHS and 100% in ARIC. Both cohorts included broad mixtures of gender, education, and smoking. Lifestyle habits were relatively similar in both cohorts.
Mean±SD levels of 24:1 were 1.96±0.44 and 0.57±0.17 percent of total fatty acids in CHS and ARIC, respectively. The interdecile ranges (10th and 90th percentiles) of 24:1 were 1.21% in CHS and 0.41% in ARIC. Levels of 20:1 and 22:1 were much lower than 24:1 levels; in ARIC, 43% of adults exhibited 22:1 levels lower than the detection limit (0.01%). Levels of the three LCMUFA were interrelated moderately in CHS (Pearson correlation coefficients, r= 0.25 to 0.63) and weakly in ARIC (r=0.10 to 0.27). In both cohorts, LCMUFA levels were also weakly or moderately correlated with levels of other phospholipid fatty acids: 20:0, 22:0, and 24:0 (r=0.16 to 0.67); 20:3n3 and 22:6n3 (r=0.21 to 0.49); 18:3n6 and trans-18:2n6 (r=−0.17 to −0.36); 16:1n7 and 18:1n9 (r=−0.13 to −0.15), and most strongly 20:0 (with 24:1) (r=0.54 in CHS and r=0.67 in ARIC).
Cross-sectional Associations of Circulating LCMUFA with Cardiovascular Risk Factors
Higher levels of 24:1 were independently associated with several cardiovascular risk factors, but with associations that would predict different directions of risk (Table 1). These included greater adiposity (BMI and waist circumference), higher levels of CRP, fibrinogen, and leukocyte count, but also trend toward lower diastolic blood pressure, higher HDL-C, and substantially lower triglycerides. Results were concordant across the two cohorts. Levels of 20:1 were associated with generally healthier profiles of adiposity and physiologic measures, and 22:1 showed either weaker or null results (Supplemental Table 2).
Table 1.
Multivariable-adjusted associations between plasma phosphohpid 24:1 fatty acid and cardiovascular risk factors
| Multivariable-adjusted difference (95% confidence interval)* | ||
|---|---|---|
| Cardiovascular Health Study (n=3,694) | Atherosclerosis Risk in Communities Study (n=3,577) | |
| Body-mass index, kg/m2 | 1.1 (0.4, 1.7)† | 0.7 (0.2, 1.2)‡ |
| Waist circumference, cm | 3.5 (1.8, 5.3)† | 2.1 (0.8, 3.5)† |
| Systolic blood pressure, mm Hg | −2.5 (−5.4, 0.3) | −0.7 (−2.3, 0.9) |
| Diastolic blood pressure, mm Hg | −0.8 (−2.5, 0.9) | −0.9 (−1.8, 0.1) |
| LDL cholesterol, mmol/L | 0.44 (0.29, 0.58)† | −0.03 (−0.11, 0.06) |
| HDL cholesterol, mmol/L | 0.05 (0.00, 0.10) | 0.06 (0.02, 0.1)‡ |
| Triglyceride, mmol/L | −0.72 (−0.83, −0.61)† | −0.37 (−0.45, −0.30)† |
| Glucose, mmol/L | −0.09 (−0.23, 0.06) | −0.05 (−0.17, 0.07) |
| Insulin, pmol/L | −6.4 (−14.9, 2.1) | 3.2 (−7.8, 14.2) |
| Fibrinogen, μmol/L | 0.73 (0.47, 0.99)† | 1.05 (0.83, 1.28)† |
| Leukocyte count, 103/mm3 | 1.1 (0.8, 1.3)† | 0.3 (0.1, 0.5)‡ |
| C-reactive protein, nmol/L | 0.3 (0.0, 0.6)‡ | not available |
| Left-ventricular hypertrophy, yes or no§ | 0.80 (0.40, 1.50) | 1.87 (0.91, 3.83) |
Values according to the interdecile range (90th vs. 10th percentile) of plasma phospholipid 24:1 fatty acids (1.21% of total fatty acids in Cardiovascular Health Study [CHS] and 0.42% of total fatty acids in Atherosclerosis Risk in Communities Study [ARIC]), adjusted for age (years), sex, enrollment sites (in CHS), employment status (in ARIC), education (<high school, high school or vocational school, or ≥college), smoking (current, former, never), physical activity (kcal/week in CHS; five-point score in ARIC), alcohol consumption (servings/week), hypertension medication (yes/no), prevalent coronary heart disease (yes/no), prevalent diabetes (yes/no), total caloric intake (kcal/week), and dietary factors (serving/week) (eggs, salad dressing, and mustard use in CHS, and eggs, whole grains, margarines, dairy products in ARIC). Individual phospholipid fatty acids were further adjusted for, after backward selection (see text in detail), including cis-trans-18:2n6, 22:2n6, 17:1n9, 20:0, and 18:1n7 in CHS, and 17:0, 20:2n6, 16:0, 18:2n6, 20:3n6, and 23:0 in ARIC. Covariates also included body-mass index and waist circumference, except when these were outcomes.
p<0.001
p<0.05
Odds ratio (95% CI) evaluated by multivariable-adjusted logistic regression.
Prospective Associations of Circulating LCMUFA with Incidence of Congestive Heart Failure
In CHS, 997 CHF events were documented during 39,238 person-years; and in ARIC, 330 events during 64,438 person-years. In multivariable-adjusted analyses, higher levels of both 22:1 and 24:1, but not 20:1, were significantly associated with higher CHF incidence (ptrend=0.01 and 0.004, respectively) (Table 2). As expected, adjustment for other individual phospholipid FA strengthened the associations. In CHS, individuals in the highest quintile of 22:1 had 34% higher CHF incidence (HR=1.34 [95%CI=1.02–1.74], ptrend=0.01) compared to those in the lowest quintile; in ARIC, 57% higher incidence (1.57 [1.11–2.23], ptrend=0.03); when pooled together, 42% higher incidence (1.42 [1.15–1.76], ptrend=0.001). Results were more robust for 24:1, with 75% higher CHF incidence comparing top to bottom quintiles in CHS (1.75 [1.23–2.50], ptrend<0.001); 92% higher incidence in ARIC (1.92, [1.22–3.03], ptrend=0.002); and 82% higher incidence when pooled (1.82 [1.37, 2.40], ptrend<0.001).
Table 2.
Incidence of congestive heart failure according to plasma phospholipid long-chain monounsaturated fatty acids
| Quintiles of long-chain monounsaturated fatty acids | ||||||
|---|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 4th | 5th | p trend | |
| 20:1 fatty acid | ||||||
| Cardiovascular Health Study | ||||||
| Median percent of total fatty acids | 0.09 | 0.11 | 0.12 | 0.13 | 0.16 | |
| Cases (rate/1,000 person-years) | 209 (25.8) | 190 (22.9) | 211 (26.2) | 189 (25.3) | 198 (27.1) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.81 (0.64–1.03) | 0.98 (0.77–1.25) | 1.00 (0.78–1.29) | 0.88 (0.68–1.14) | 0.69 |
| + Further adjusted for fatty acids† | 1.0 | 0.82 (0.65–1.04) | 1.02 (0.80–1.30) | 1.09 (0.84–1.42) | 0.95 (0.72–1.26) | 0.81 |
| Atherosclerosis Risk in Communities Study | ||||||
| Median percent of total fatty acids | 0.10 | 0.11 | 0.12 | 0.14 | 0.16 | |
| Cases (rate/1,000 person-years) | 91 (6.3) | 51 (4.4) | 101 (4.7) | 39 (5.9) | 48 (4.7) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.76 (0.54–1.08) | 0.78 (0.58–1.04) | 0.99 (0.67–1.45) | 0.84 (0.59–1.21) | 0.74 |
| + Further adjusted for fatty acids† | 1.0 | 0.78 (0.55–1.11) | 0.85 (0.62–1.16) | 1.13 (0.74–1.72) | 1.05 (0.69–1.59) | 0.37 |
| Pooled estimates ‡ | ||||||
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.79 (0.65–0.96) | 0.89 (0.71–1.10) | 1.00 (0.81–1.23) | 0.87 (0.70–1.07) | 0.89 |
| + Further adjusted for fatty acids† | 1.0 | 0.81 (0.66–0.98) | 0.95 (0.78–1.15) | 1.10 (0.88–1.38) | 0.98 (0.78–1.23) | 0.77 |
| 22:1 fatty acid | ||||||
| Cardiovascular Health Study | ||||||
| Median percent of total fatty acids | 0.02 | 0.02 | 0.03 | 0.04 | 0.05 | |
| Cases (rate/1,000 person-years) | 239 (24.9) | 154 (21.5) | 189 (23.0) | 181 (24.8) | 234 (33.8) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.97 (0.75–1.25) | 0.88 (0.69–1.12) | 0.99 (0.78–1.25) | 1.20 (0.93–1.53) | 0.08 |
| + Further adjusted for fatty acids† | 1.0 | 0.97 (0.75–1.26) | 0.89 (0.70–1.14) | 1.04 (0.81–1.34) | 1.34 (1.02–1.76) | 0.01 |
| Atherosclerosis Risk in Communities Study | ||||||
| Median percent of total fatty acids | 0.00 | 0.01 | 0.02 | 0.03 | 0.06 | |
| Cases (rate/1,000 person-years) | 187 (5.0) | 45 (5.5) | 37 (4.2) | 20 (5.5) | 41 (6.6) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 1.25 (0.90–1.74) | 0.79 (0.55–1.13) | 1.23 (0.77–1.96) | 1.48 (1.05–2.09) | 0.06 |
| + Further adjusted for fatty acids† | 1.0 | 1.23 (0.88–1.72) | 0.79 (0.55–1.13) | 1.32 (0.82–2.11) | 1.57 (1.11–2.23) | 0.03 |
| Pooled estimates ‡ | ||||||
| Multivariable-adjusted HR (95% CI)* | 1.0 | 1.07 (0.84–1.36) | 0.85 (0.70–1.04) | 1.03 (0.84–1.27) | 1.29 (1.05–1.58) | 0.01 |
| + Further adjusted for fatty acids† | 1.0 | 1.07 (0.86–1.32) | 0.86 (0.70–1.05) | 1.10 (0.88–1.37) | 1.42 (1.15–1.76) | 0.001 |
| 24:1 fatty acid | ||||||
| Cardiovascular Health Study | ||||||
| Median percent of total fatty acids§ | 1.40 | 1.71 | 1.93 | 2.16 | 2.61 | |
| Cases (rate/1,000 person-years) | 200 (24.9) | 185 (22.5) | 187 (23.4) | 219 (28.9) | 206 (27.9) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 1.00 (0.77–1.32) | 1.10 (0.85–1.44) | 1.31 (1.02–1.68) | 1.27 (0.97–1.66) | 0.019 |
| + Further adjusted for fatty acids* | 1.0 | 1.10 (0.84–1.44) | 1.26 (0.95–1.69) | 1.62 (1.22–2.16) | 1.75 (1.23–2.50) | <0.001 |
| Atherosclerosis Risk in Communities Study | ||||||
| Median percent of total fatty acids§ | 0.39 | 0.49 | 0.56 | 0.65 | 0.80 | |
| Cases (rate/1,000 person-years) | 75 (5.1) | 50 (4.0) | 67 (5.3) | 61 (5.0) | 77 (6.2) | |
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.84 (0.58–1.20) | 1.11 (0.79–1.56) | 1.01 (0.72–1.43) | 1.27 (0.92–1.76) | 0.09 |
| + Further adjusted for fatty acids† | 1.0 | 1.01 (0.68–1.48) | 1.41 (0.97–2.05) | 1.43 (0.95–2.16) | 1.92 (1.22–3.03) | 0.002 |
| Pooled estimates ‡ | ||||||
| Multivariable-adjusted HR (95% CI)* | 1.0 | 0.94 (0.76–1.17) | 1.11 (0.90–1.36) | 1.19 (0.93–1.51) | 1.27 (1.03–1.57) | 0.004 |
| + Further adjusted for fatty acids† | 1.0 | 1.07 (0.85–1.33) | 1.32 (1.05–1.65) | 1.56 (1.23–1.97) | 1.82 (1.37–2.40) | <0.001 |
Hazard ratio (HR) and 95% confidence interval (CI), with the first quintile category as the reference, adjusted for the same covariates as those in the Table 1, except for phospholipid fatty acids not incorporated in this first model.
Further adjusted for phospholipid fatty acids, after backward selection (see text in detail and the footnote of Table 1).
CHS and ARIC estimates were pooled using random-effects meta-analysis: little evidence for heterogeneity across estimates for each fatty acid (I2<3%).
Due to unclear reasons, 24:1 fatty acid levels were significantly higher in CHS than ARIC. When evaluated continuously in each cohort: in CHS, fully-adjusted HR (95% CI)=1.82 (1.33–2.49) per interdecile range (1.21%); in ARIC, fully-adjusted HR (95% CI)=1.74 (1.19–2.54) per interdecile range (0.41%); when pooled, fully-adjusted HR (95% CI)=1.78 (1.40–2.27).
Further adjustment for potential mediators, including BMI, waist circumference, HDL-C, fibrinogen, CRP (CHS only), and leukocyte count partly attenuated the associations: in CHS, extreme-quintile HRs (95% CI)=1.27 (0.96–1.67; ptrend=0.04) for 22:1 and 1.62 (1.12–2.34; ptrend=0.003) for 24:1 and in ARIC, 1.55 (1.09–2.20; ptrend=0.06) for 22:1 and 1.53 (0.96–2.42; ptrend=0.04) for 24:1. We examined incident CHD (n=471 in CHS, n=349 in ARIC) as a potential mediator. Adjusted for the incident events as time-varying variables, results were generally similar: in CHS, the extreme-quintile HR (95%CI) was 1.39 (1.11–1.74) for 22:1 and 1.44 (1.06–1.94) for 24:1 (ptrend<0.002 each); and in ARIC, these HRs were 1.72 (1.21–2.45) and 1.43 (0.89–2.28) (ptrend=0.01 and 0.11), respectively. We assessed multiplicative interaction for association of circulating LCMUFA with incident CHF in each cohort according to age, gender, race, prevalent CHD, and prevalent diabetes. None of these factors appeared significantly modified the associations (pinteraction>0.05 each).
Subtypes of Congestive Heart Failure
We separately evaluated CHF subtypes centrally adjudicated in 857 cases (86%) in CHS, including ischemic CHF, valvular CHF, and non-ischemic non-valvular CHF. There was no evidence of substantial differences in associations of LCMUFA with CHF overall and CHF subtypes (Supplemental Table 3). For example, the multivariable-adjusted HR (95% CI) across the interdecile range (90th to 10th percentile) of 24:1 was 1.65 (1.19–2.29) for overall CHF, 1.70 (1.06–2.71) for ischemic CHF, 1.83 (0.93–3.60) for valvular CHF, and 1.85 (1.04–3.30) for non-ischemic non-valvular CHF.
Regression Dilution Bias
In sensitivity analyses, we evaluated the associations of 22:1 and 24:1 with incident CHF after correcting for regression dilution bias (measurement error over time) in both LCMUFA levels and time-varying covariates. In multivariable-adjusted models (as in Table 2, fully adjusted model) further corrected for the bias, the observed associations were estimated to be 4.0-fold stronger for 20:1, 4.2-fold stronger for 22:1, and 3.0-fold stronger for 24:1. For example, the multivariable-adjusted HR (95%CI) comparing the top to the bottom quintile of 22:1 was 3.37 (1.19–9.52) in CHS and 6.58 (1.74–24.8) in ARIC; and of 24:1, 5.44 (1.81–16.3) in CHS and 7.14 (1.75–29.1) in ARIC.
Dose-Response Relationships
Dose-response relationships between circulating LCMUFA levels and incident CHF were evaluated by restricted cubic splines. While non-linear associations of 22:1 levels was suggested in CHS (pcurve=0.01), higher levels of LCMUFA, in particular 24:1 levels, appeared monotonically associated with higher incidence CHF in both cohorts (plinear<0.001 in CHS and plinear=0.003 in ARIC) (Figure 1).
Figure 1.
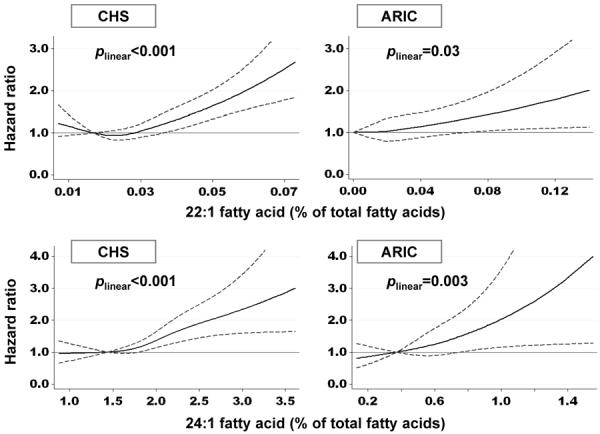
Multivariable-adjusted associations of plasma phospholipid 22:1 and 24:1 fatty acids with incidence of congestive heart failure – the Cardiovascular Health Study (CHS) and the Atherosclerosis Risk in Communities Study (ARIC, Minneapolis subcohort). Solid and dashed lines represent hazard ratios and 95% confidence intervals, respectively. The reference value was 10th percentile. Levels below 0.1st and above 99.9th percentile values were truncated for presentation purpose. P for linear associations are presented. Non-linear association was suggested for 22:1 in CHS (pcurve=0.01). Estimates were adjusted for potential confounders (see Table 2 footnote).
Incident Stroke
Given many shared risk factors for CHF and stroke, we evaluated whether LCMUFA were associated with stroke as a prespecified negative control, i.e., to evaluate the specificity for CHF. None of the LCMUFA were associated with incident stroke when separately evaluated in both cohorts and when pooled by meta-analysis (Supplemental Table 3). The multivariable-adjusted pooled HR (95% CI) per an interdecile rage were 1.13 (0.94–1.36) for 20:1, 0.96 (0.75–1.25) for 22:1, and 1.12 (0.78–1.63) for 24:1.
Dietary Correlates of Circulating LCMUFA and Estimated US LCMUFA Consumption
A variety of foods were positively associated with LCMUFA levels consistently between CHS and ARIC (Figure 2, Supplemental Figure 1). These included both generally more healthful foods such as seafood and whole grains; and generally less healthful foods such as meat products.
Figure 2.

Dietary factors independently associated with plasma phospholipid long-chain monounsaturated fatty acid (LCMUFA) concentrations – the Cardiovascular Health Study (CHS) and the Atherosclerosis Risk in Communities Study (ARIC, Minneapolis subcohort). Values represent each standard deviation (SD) difference of LCMUFA levels according to 1 servings/week of each food group; mean±SD of 20:1, 22:1, and 24:1 levels (% fatty acids) were 0.12±0.04, 0.03±0.01, and 1.96±0.44 in CHS and 0.12±0.03, 0.01±0.03, and 0.57±0.17 in ARIC, respectively. Food groups were selected by backward stepwise approach (pretain<0.05 and premove >0.1). Covariates were the same as those in the Table 1, except that the other phospholipid fatty acids were not adjusted for, which were potential mediators in this analysis. Negative correlates of LCMUFA included margarine, butter, and other sugary foods (Supplemental Figure 1), that may have reflected high levels of major shorter chain fatty acids. *Vegetables were first grouped into 7 categories based on 30; vegetables herein included celery, beet, zucchini, garlic, vegetable sauces, and mixed vegetables. Mixed meals included pasta, lasagna, pizza and other miscellaneous meals.
In NHANES 2003–2010, mean±SD intakes of 20:1 and 22:1 were 239±23 and 34±15 mg/d, respectively. Many of the food sources of LCMUFA consumption in US were similar to those seen associated with circulating phospholipid LCMUFA levels in CHS and ARIC, including seafood, poultry, meats, and mustard (Figure 3). Because 24:1 consumption was not reported in NHANES, we assessed the USDA nutrient database to identify possible sources of LCMUFA.11 Reported sources included mustard, seafood, and edible oil products (Supplemental Table 4 and 5), although only 6% of all items indexed 24:1 food contents.
Figure 3.
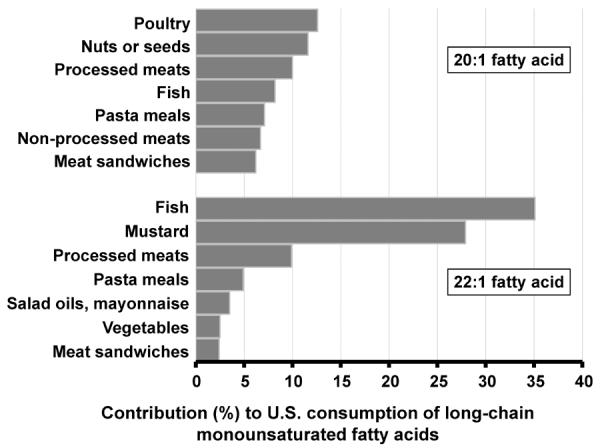
Contribution of food groups to consumption of 20:1 and 22:1 fatty acids in US adults (age≥20 years) – the US National Health and Nutrition Examination Survey, 2003–2010 (n=20,150). The food groups presented here contributed to 62.4% of total 20:1 fatty acid consumption and 86.2% of total 22:1 fatty acid consumption. No data on 24:1 fatty acid consumption are available in the survey.
DISCUSSION
In two independent community-based prospective cohorts, we found higher circulating levels of 22:1 and 24:1 to be associated with higher incidence of CHF. Each plasma phospholipid LCMUFA, in particular 24:1, was also associated with specific physiologic risk factors. In both cohorts, we identified diverse dietary correlates of circulating LCMUFA concentrations, including fish, poultry, nuts, mustard, and meat products. The latter findings were broadly consistent with the current major food sources of estimated LCMUFA consumption in the US.
Existing evidence from animal experiments of LCMUFA-rich oil feeding and human studies on cardiac steatosis both supports our observations, providing plausible biologic pathways whereby LCMUFA exposure increases CHF risk, including CHF with both preserved and reduced ejection fraction. In studies in rodents, pigs, and non-human primates, experimental feeding of LCMUFA caused cardiac steatosis and fibrosis1–4, which relate to both systolic and diastolic dysfunction in humans.12–14 For example, among patients with aortic valve disease and healthy volunteers without known cardiac diseases, presence of greater cardiac fibrosis and greater myocardial lipid content, as quantified by cardiac biopsy or MRI, are independently associated with lower ejection fraction, greater passive ventricular stiffness, and other indices of impaired early and late diastolic function.27–29
The potential cardiotoxicity of LCMUFA may relate to their oxidative metabolism, to which the heart is likely to be especially susceptible due to its preferential use of fatty acids for energy.5–7 Experimental studies suggest that, in contrast to most other fatty acids, long-chain fatty acids including LCMUFA are poorly oxidized in mitochondria, due to lack of mitochondrial membrane-transporting enzymes.3, 8 Consequently, long-chain fatty acids predominantly undergo peroxisomal oxidation, which generates reactive oxygen species including H2O2 and releases various lipid metabolites into the cytosol.8 Accumulated lipids include malonyl-CoA that could inhibit mitochondrial fatty acid transport mediated by carnitine palmitoyltransferase-1 (CPT-1); other acyl-CoAs that could stimulate lipogenic signals suppressing glycolysis and forming lipid droplets linked to cardiac steatosis; ceramides that serve as a second messenger for apoptotic signaling; and phospholipids and diacylglycerols that alter membrane lipid composition and electrophysiological homeostasis, enhancing calcium overload linked to apoptosis as well as contractile dysfunction and arrhythmia.5–7 These exacerbations have been partly demonstrated by research in transgenic animals.6, 7 For example, mice with myocardium-specific overexpression of peroxisome proliferator-activated receptor-α (PPARα) exhibited interrelated phenotypes30, including greater peroxisomal fatty acid oxidation; greater cytosolic lipid accumulation; inhibition of pyruvate dehydrogenase, a rate-limiting enzyme of glycolysis; as well as greater left-ventricular size and wall thickness and reduced systolic function. Other evidence supports the notion of links between mitochondrial dysfunction and cardiac steotosis and heart failure: in cardiomyocytes biopsied from patients with aotic regurgitation,28 cardiac steatosis was associated with greater systolic dysfunction and overexpression of lipogenic enzymes including sterol-regulatory element binding protein (SREBP)-1c.
As described above, one potential mechanism of cardiotoxicity is malonyl-CoA accumulation resulting in inhibition of CPT-1. However, in animal studies and small clinical studies, anti-anginal drugs include CPT-1 inhibitors which block mitochondrial oxidation, activating glycolysis and resulting in lower myocardial oxygen consumption.6, 31 Thus, our findings highlight the need for better understanding of the potential mechanisms that might underlie the observed toxicity of LCMUFA, including investigation of pathways related to mitochondrial function and energetic changes induced by fatty acids of all types.
When we evaluated subtypes of incident CHF in CHS, 24:1 appeared similarly associated with all subtypes. Conversely, 22:1 appeared most strongly related to ischemic CHF; however, we cannot rule out possibility that the associations for CHF subtypes were similar, as the 95% confidence intervals were broad and associations for different CHF subtypes were not significantly different. Potential harms of LCMUFA for diverse CHF etiologies are consistent with the multiple pathophysiologic pathways implicated in experimental studies, which together could lead to both systolic and diastolic dysfunction, both in the setting of ischemia and in the absence of ischemia. Interestingly, we also found that higher LCMUFA levels were associated with substantially lower triglyceride levels and higher inflammatory biomarkers, consistent with hepatic mitochondrial toxicity exhibiting hepatic inflammation and impaired hepatic lipid secretion.32 Our findings demonstrate the need for future studies to better characterize associations of LCMUFA exposure with differing etiologies of CHF as well as with measures of steatosis, fibrosis, and function of both cardiac and non-cardiac tissues, for example as assessed by cardiac and hepatic MRI.
Circulating levels of 22:1 and 24:1 were robustly and consistently associated with CHF in both cohorts. Findings for 22:1 are consistent with animal-experimental evidence of cardiotoxicity due to consumption of oil rich in 22:1. In addition, dietary 22:1 appears to be elongated to 24:1 in humans,33 which together with our findings suggests that experimentally observed cardiotoxicity of dietary 22:1 could be partly attributable to effects of its metabolite 24:1. In contrast to 22:1 and 24:1, we did not find significant associations of 20:1 with incident CHF. Evidence from cellular studies suggests that 20:1 undergoes mitochondrial oxidation to a greater extent than 22:13, which could limit its cardiotoxicity and explain our findings. Additionally, why potential cardiotoxicity of 22:1 and 24:1 may be greater than for other long-chain fatty acids is unknown. One study suggested that peroxisomal oxidation of 24:1 is faster than that of 24:0,34 supporting greater toxicity of the former.
Animal-experimental evidence of cardiotoxicity due to LCMUFA consumption stimulated Canadian farmers to develop Canola oil which became commercially available during the 1960s.9 Thereafter, governmental regulations in Canada (1975), the UK (1977), and the US (1985), as well as recommendations by FAO/WHO (1982), were instituted to lower the content of 22:1n9 in rapeseed-derived oils.9, 10 For instance, the US Food and Drug Administration mandated producers to reduce the 22:1n9 content in rapeseed oil from the original 30–60% to less than 2% by weight.10 Although these steps might reduce population exposure to LCMUFA in these countries, our findings indicate remaining dietary exposure to LCMUFA in US. Additionally, in other countries, LCMUFA exposure could be even higher due to habitual consumption of unaltered rapeseed oil as well as mustard oil.35
We found that many different foods could influence LCMUFA exposure, including generally more healthful foods (fish, mustard seeds and oil, salad oils, poultry) and less healthful foods (processed meats and mixed meals, for example, pizza and meat sandwiches). This indicates that potential cardiotoxicity of LCMUFA cannot be attributed to any single dietary source and depends on overall exposure to LCMUFA. As corollary of this, the net effects of any food would depend not just on its LCMUFA content, but also on other constituents in the food. For example, in the case of fish which has been inversely associated with incident CHF in these cohorts,20, 25 potential benefits of long-chain omega-3 polyunsaturated fatty acids (PUFA) in fish36 would plausibly outweigh any harmful effects of LCMUFA content. This does not obviate the suggestion of our findings that LCMUFA exposure itself increases the risk of CHF, compared to absence of such exposure. Our results support the need for interventional studies to determine whether lowering LCMUFA in foods could reduce their harms or, in the case of beneficial foods such as fish, further increase their benefits. While diet clearly influences circulating LCMUFA levels, contribution of metabolic pathways remains uncharacterized. Few studies suggest that both diet and metabolism may influence circulating LCMUFA levels. For example, in a small (n=29) 2-year intervention among patients with inherited peroxisomal disorder (X-linked adorenoleukodystrophy), to reduce circulating levels of long-chain saturated fatty acids, 22:1 feeding (0.3 g/kg/day) elevated levels of both circulating 22:1 and 24:1 by 3-fold.33 These findings suggest that dietary 22:1 directly influences circulating 22:1 levels and is also elongated to 24:1.
To the best of our knowledge, this is the first investigation to evaluate LCMUFA and any health outcome in humans. Despite our findings and the prior animal-experimental evidence, any single study should not alter dietary guidelines or clinical practice. Policy makers and clinicians should recognize a potential cardiotoxicity of LCMUFA exposure, and be supportive of and watchful for further investigations. Our findings highlight the clear need to understand how LCMUFA may influence health. This includes better characterization of the dietary sources of LCMUFA in various populations, as well as intervention studies to explore possible benefits of lowering LCMUFA exposure in populations having or at risk for CHF, examining endpoints including cardiac imaging metrics, left ventricular systolic and diastolic function, symptoms and exercise tolerance, and clinical CHF.12–14, 27–29
Potential limitations deserve consideration. Methods for assessment of study variables were somewhat different in each cohort. Methodologic differences in fatty acid measurements could partly explain the observed differences in absolute levels of 24:1 in the two cohorts. Despite these differences, consistency of our findings between the two cohorts increases confidence in validity and generalizability of our findings. Our findings should be interpreted cautiously, because causality is unknown due to confounding and exposure misclassification. Lesser reproducibility over time could partly explain weaker associations for 20:1 and 22:1, compared with 24:1. Although we performed multivariate adjustment and regression dilution correction, such methods are imperfect and remaining residual errors could bias our results toward or away from the null. When examining potential confounding, circulating LCMUFA were not associated with incident stroke, which shares many CHF risk factors. Moreover, both generally healthful and unhealthful foods were associated with LCMUFA exposure. These results, together with the magnitude and consistency of our findings, suggest that residual confounding could not entirely explain the observed associations. Our analysis was based on cohorts comprising generally white Americans, stimulating future research on LCMUFA exposure and CHF events and etiologies in other races/ethnicities.
In summary, our observations indicate that higher circulating LCMUFA levels are associated with greater incidence of CHF. Our findings in two cohorts, together with prior animal experiments, support the need to better characterize dietary sources of LCMUFA, further elucidate molecular mechanisms of effects, and design and implement interventions to test the effects of reducing LCMUFA exposure.
Supplementary Material
Acknowledgment
We thank the participants and staff of CHS and ARIC (See http://www.chs-nhlbi.org/ and http://www.cscc.unc.edu/aric/).
Funding sources: The National Heart, Lung, and Blood Institute (NHLBI) with co-funding from the Office of Dietary Supplements (R01-HL085710), and Administrative Supplement Award (3R01-HL085710-03S1). CHS was supported by NHLBI (N01-HC-85239, N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01-HC-15103, N01-HC-55222, N01-HC-75150, N01-HC-45133, and HL080295), with additional contributions from the National Institute of Neurological Disorders and Stroke and from the National Institute of Aging (AG-023629, AG-15928, AG-20098, and AG-027058). A Searle Scholar Award additionally supported CHS fatty acid assessment. ARIC was supported by NHLBI (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C).
Footnotes
Conflict of Interest Disclosures: DM reported support from GlaxoSmithKline, Sigma Tau, and Pronova for a trial of fish oil and post-surgical complications; ad hoc travel reimbursement and/or honoraria for presentations from Aramark, Unilever, SPRIM, and Nutrition Impact; ad hoc consulting fees from Foodminds and McKinsey Health Systems Institute; and royalties from UpToDate. None from the other authors.
References
- 1.Roine P, Uksila E, Teir H, Rapola J. Histopathological changes in rats and pigs fed rapeseed oil. Zeitschrift fur Ernahrungswissenschaft. 1960;1:118–124. [Google Scholar]
- 2.Schiefer B, Loew FM, Laxdal V, Prasad K, Forsyth G, Ackman RG, Olfert ED. Morphologic effects of dietary plant and animal lipids rich in docosenoic acids on heart and skeletal muscle of cynomolgus monkeys. Am. J. Pathol. 1978;90:551–564. [PMC free article] [PubMed] [Google Scholar]
- 3.Bremer J, Norum KR. Metabolism of very long-chain monounsaturated fatty acids (22:1) and the adaptation to their presence in the diet. J. Lipid Res. 1982;23:243–256. [PubMed] [Google Scholar]
- 4.van Vleet JF, Ferrans VJ. Myocardial diseases of animals. Am J Pathol. 1986;124:98–178. [PMC free article] [PubMed] [Google Scholar]
- 5.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: Ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 6.Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg Ira J, Trent Chad M, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metabolism. 2012;15:805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy JK, Hashimoto T. Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: An adaptive metabolic system. Ann Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 9.Daun J. Erucic acid levels in western canadian canola and rapeseed. J. Am. Oil Chem. Soc. 1986;63:321–324. [Google Scholar]
- 10.United States Food and Drug Administration Federal Register. 1985;50:3746. [Google Scholar]
- 11.U.S. Department of Agriculture Agricultural Research Service Usda national nutrient database for standard reference, release 22. 2009
- 12.McGavock JM, Victor RG, Unger RH, Szczepaniak LS. Adiposity of the heart*, revisited. Ann Int Med. 2006;144:517–524. doi: 10.7326/0003-4819-144-7-200604040-00011. [DOI] [PubMed] [Google Scholar]
- 13.Lindsey JB, Marso SP. Steatosis and diastolic dysfunction: The skinny on myocardial fat. J Am Coll Cardiol. 2008;52:1800–1802. doi: 10.1016/j.jacc.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 14.Dhoble A, Patel MB. Cardiac steatosis and myocardial dysfunction. J Am Coll Cardiol. 2009;53:636. doi: 10.1016/j.jacc.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 15.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A. The cardiovascular health study: Design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 16.Ives DG, Fitzpatrick AL, Bild DE, Psaty BM, Kuller LH, Crowley PM, Cruise RG, Theroux S. Surveillance and ascertainment of cardiovascular events. The cardiovascular health study. Ann Epidemiol. 1995;5:278–285. doi: 10.1016/1047-2797(94)00093-9. [DOI] [PubMed] [Google Scholar]
- 17.The ARIC Investigators The atherosclerosis risk in communities (aric) study: Design and objectives. Am. J. Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 18.Clarke R, Shipley M, Lewington S, Youngman L, Collins R, Marmot M, Peto R. Underestimation of risk associations due to regression dilution in long-term follow-up of prospective studies. Am. J. Epidemiol. 1999;150:341–353. doi: 10.1093/oxfordjournals.aje.a010013. [DOI] [PubMed] [Google Scholar]
- 19.Gottdiener JS, Kitzman DW, Aurigemma GP, Arnold AM, Manolio TA. Left atrial volume, geometry, and function in systolic and diastolic heart failure of persons >=65 years of age (the cardiovascular health study) Am. J. Cardiol. 2006;97:83–89. doi: 10.1016/j.amjcard.2005.07.126. [DOI] [PubMed] [Google Scholar]
- 20.Nettleton JA, Steffen LM, Loehr LR, Rosamond WD, Folsom AR. Incident heart failure is associated with lower whole-grain intake and greater high-fat dairy and egg intake in the atherosclerosis risk in communities (aric) study. J. Am. Diet. Assoc. 2008;108:1881–1887. doi: 10.1016/j.jada.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipsitch M, Tchetgen Tchetgen E, Cohen T. Negative controls: A tool for detecting confounding and bias in observational studies. Epidemiology. 2010;21:383–388. doi: 10.1097/EDE.0b013e3181d61eeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Longstreth WT, Jr., Bernick C, Fitzpatrick A, Cushman M, Knepper L, Lima J, Furberg CD. Frequency and predictors of stroke death in 5,888 participants in the cardiovascular health study. Neurology. 2001;56:368–375. doi: 10.1212/wnl.56.3.368. [DOI] [PubMed] [Google Scholar]
- 23.Rosamond WD, Folsom AR, Chambless LE, Wang C-H, McGovern PG, Howard G, Copper LS, Shahar E. Stroke incidence and survival among middle-aged adults. Stroke. 1999;30:736–743. doi: 10.1161/01.str.30.4.736. [DOI] [PubMed] [Google Scholar]
- 24.Micha R, King IB, Lemaitre RN, Rimm EB, Sacks F, Song X, Siscovick DS, Mozaffarian D. Food sources of individual plasma phospholipid trans fatty acid isomers: The cardiovascular health study. Am J Clin Nutr. 2010;91:883–893. doi: 10.3945/ajcn.2009.28877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mozaffarian D, Bryson CL, Lemaitre RN, Burke GL, Siscovick DS. Fish intake and risk of incident heart failure. J Am Coll Cardiol. 2005;45:2015–2021. doi: 10.1016/j.jacc.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 26.Maldonado G, Greenland S. Simulation study of confounder-selection strategies. Am J Epidemiol. 1993;138:923–936. doi: 10.1093/oxfordjournals.aje.a116813. [DOI] [PubMed] [Google Scholar]
- 27.Villari B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, Krayenbuehl HP. Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol. 1993;22:1477–1484. doi: 10.1016/0735-1097(93)90560-n. [DOI] [PubMed] [Google Scholar]
- 28.Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F, D'Amico M, Paolisso G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009;50:2314–2323. doi: 10.1194/jlr.P900032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rijzewijk LJ, van der Meer RW, Smit JWA, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52:1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 30.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by pparα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters AM, Porter GA, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circulation Research. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser HW, Borel J. Dietary management of x-linked adrenoleukodystrophy. Ann Rev Nutr. 1995;15:379–397. doi: 10.1146/annurev.nu.15.070195.002115. [DOI] [PubMed] [Google Scholar]
- 34.Sandhir R, Khan M, Chahal A, Singh I. Localization of nervonic acid β-oxidation in human and rodent peroxisomes: Impaired oxidation in zellweger syndrome and x-linked adrenoleukodystrophy. J Lipid Res. 1998;39:2161–2171. [PubMed] [Google Scholar]
- 35.Wallingford JC, Yuhas R, Du S, Zhai F, Popkin BM. Fatty acids in chinese edible oils: Value of direct analysis as a basis for labeling. Food Nutr Bull. 2004;25:330–336. doi: 10.1177/156482650402500402. [DOI] [PubMed] [Google Scholar]
- 36.Stanley WC, Dabkowski ER, Ribeiro RF, Jr, O'Connell KA. Dietary fat and heart failure: Moving from lipotoxicity to lipoprotection. Circulation Res. 2012;110:764–776. doi: 10.1161/CIRCRESAHA.111.253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.