

Abstract
Humoral immunity to T cell-independent type 2 antigens (TI-2 Ag) is critical for protection against encapsulated bacteria such as Streptococcus pneumoniae. The CD21/35 receptor is thought to promote protective humoral immunity to encapsulated bacteria by enabling complement-decorated capsular polysaccharides to coligate the CD21/35-CD19 signaling complex with the B cell Ag receptor (BCR), thereby enhancing Ag-specific B cell activation. However, antibody responses to S. pneumoniae type 3 capsular polysaccharide (PPS-3) and other strong TI-2 Ags were significantly impaired in CD21/35−/−, but not C3−/− or C4−/− mice. B cells from CD21/35−/− mice expressed significantly higher levels of cell-surface CD19. CD21/35−/− B cells exhibited enhanced BCR-induced calcium responses and significantly higher expression of the inhibitory programmed death-1 (PD-1) receptor following immunization with a TI-2 Ag or BCR crosslinking. Reducing CD19 expression in CD21/35−/− mice normalized BCR-induced calcium responses, PD-1 induction, PPS-3-specific IgG3 responses, and restored protection during S. pneumoniae infection. PD-1 blockade also selectively rescued PPS-3-specific IgG3 responses in CD21/35−/− mice. Thereby, CD21/35 promotes protective humoral immunity to S. pneumoniae and other strong TI-2 Ags through a complement-independent pathway by negatively regulating CD19 expression and PD-1 induction.
Introduction
Humoral immune responses to T cell independent type 2 (TI-2)3 antigens (Ag) are critical for protective immunity to encapsulated bacteria such as Streptococcus pneumoniae, a gram-positive bacterium that is the predominant cause of otitis media, community-acquired pneumonia, septicemia, and meningitis (1). Antibody responses to TI-2 Ags such as pneumococcal polysaccharide (PPS) contribute substantially to protective immunity against infections by S. pneumoniae and other encapsulated bacteria (1). As such, the Pneumovax® vaccine contains 23 types of PPS, including PPS type 3 (PPS-3), a strong TI-2 Ag that is one of the most immunogenic polysaccharides included in the vaccine (2, 3). PPS-3 elicits specific protection against serotype 3 S. pneumoniae, a major cause of fatal invasive pneumococcal disease (1). TI-2 Ags are characterized by high molecular weight, repeating epitopes, slow degradability in vivo, and primarily induce IgM and IgG3 Ab isotypes. TI-2 Ags elicit rapid Ab responses by multivalent crosslinking of the B cell Ag receptor (BCR) in the absence of major histocompatibility complex class II-restricted T cell help (4).
B-1 cells are key players in early humoral responses against pathogens and are the primary source of Abs to TI-2 Ags along with marginal zone (MZ) B cells (5, 6). The B-1b cell subset of B-1 cells is the major B cell population responsible for Ab responses to PPS-3 (7), NP-Ficoll (8) and Borrelia hermsii (9). Humoral responses to TI-2 Ags rely heavily on distinct BCR signaling pathways (10, 11) as well as key regulators of these pathways. Programmed cell death 1 (PD-1), a member of the B7/CD28 family that negatively regulates Ag receptor signaling on both B and T cells (12, 13), is thought to negatively regulate TI-2 Ab responses since PD-1+/− and PD-1−/− mice exhibit significantly enhanced IgG3 production in response to the TI-2 Ag, DNP-Ficoll (13). By contrast, complement receptor 1/2 (CD35/CD21) expression promotes optimal Ab responses to both artificial and pathogen-derived TI-2 Ags (14–17). In particular, CD21/35−/− mice exhibit a striking impairment in IgG3 responses following TI-2 Ag challenge. As a consequence, previous exposure to live or heat-killed S. pneumoniae provides significantly less protection in CD21/35−/− mice compared to wild type littermates (14). CD21/35 binds C3 and C4 cleavage products that become covalently bound to foreign Ags and pathogens following C3 or C4 activation. However, the effects of complement deficiency or depletion on TI-2 Ab responses are variable (16–24).
In mice, CD35 is generated by the addition of six short consensus repeat units to the amino terminal end of the CD21 protein and can therefore bind C4b and C3b in addition to the CD21 ligands, C3d and iC3b (25). CD21/35 is expressed by both follicular dendritic cells and mature B cells where it associates with cell surface CD19, a critical regulator of B cell signaling (25). CD21/35 density varies on mature B cell populations, with B-1a cells expressing the lowest levels and MZ B cells expressing the highest levels. CD21/35 ligation lowers the threshold for Ag-specific B cell activation by binding to C3d-Ag complexes and inducing CD19 signaling (25). The reported effects of CD19-deficiency on TI-2 Ab responses are variable. CD19−/− mice generate normal to augmented Ab responses to TI-2 Ags such as PPS-3, DNP-Ficoll and TNP-Ficoll (7, 26, 27), but impaired Ab responses to TI-2 Ags such as phosphorylcholine (PC) displayed either on intact bacteria or as PC6-Ficoll (7, 28). The reverse is true for mice overexpressing CD19 (7, 26). Similarly, B cells expressing a low affinity BCR require CD19 to respond optimally to NP-Ficoll, whereas B cells expressing a high affinity BCR do not (29). These inconsistencies may be explained by differences in Ag valency or differential requirements for CD19 in promoting the development of B cell subsets involved in Ab responses to distinct TI-2 Ags.
CD21/35 is proposed to promote Ab responses to TI-2 Ags in a manner similar to that described for TD Ags (30). Thus, CD21/35-mediated capture of complement-decorated TI-2 Ags on Ag-specific B cells is predicted to coligate CD19 with the BCR and lower the signaling threshold required for B cell activation (4). However, given the complexities associated with complement- and CD19-mediated regulation of TI-2 Ab responses, the molecular basis for impaired TI-2 Ab responses to encapsulated bacteria in CD21/35−/− mice have remained unclear. Therefore, the molecular mechanisms contributing to impaired Ab responses to TI-2 Ags in CD21/35−/− mice were examined in the current study. Remarkably, CD21/35 regulates protective PPS-specific Ab responses through an unexpected C3-independent, CD19-dependent mechanism that results in upregulation of PD-1 expression by CD21/35−/− B cells.
Materials and Methods
Mice
CD19−/− and CD21/35−/− mice were as described (31–33) and were backcrossed with C57BL/6 mice for 15 generations. C3−/− and C4−/− mice were from Dr. Michael Carroll, Center for Blood Research, Boston, MA (34). CD21/35−/−CD19+/−, CD21/35−/−CD19−/−, and CD21/35−/−C3−/− mice were generated by intercrossing CD19−/−, CD21/35−/−, and C3−/− mice. Wild type C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) were used as controls. Unless indicated otherwise, all experiments were initiated in 2–3 month-old mice housed under specific pathogen free conditions. All studies and procedures were approved by the Duke University Animal Care and Use Committee.
Immunizations and ELISAs
TNP-Ficoll-specific B cell expansion and PD-1 upregulation was performed on mice immunized intraperitoneally (i.p.) with 50 μg of TNP4-Ficoll, TNP52-Ficoll (both from Biosearch Technologies, Novato, CA), TNP30-BSA (Calbiochem-Novabiochem Corp., La Jolla, CA), or PBS only. For serum Ab analyses, mice were immunized i.p. with 0.5 μg PPS-3 (derived from type 3 Streptococcus pneumoniae (Klein) Chester; ATCC (169-X), Manassas, VA), or 25 μg of TNP4-Ficoll, TNP52-Ficoll, DNP40-Ficoll, or 50 μg TNP0.4-LPS (all from Biosearch Technologies) in PBS, or 100 μg of DNP-KLH (Calbiochem-Novabiochem Corp.) precipitated with alum. Mice immunized with DNP-KLH were boosted 21 days later with 100 μg of DNP-KLH in alum. Ags were diluted in sterile PBS and injected in a final volume of 200 μl. Serum was obtained 0, 7, 14, 21, and 28 days post-primary immunization. Mice were administered PD-1 mAb (RMP1-14; eBioscience, San Diego, CA) or control rat IgG2a (Southern Biotechnology Associates, Birmingham, AL) in 200 μl sterile PBS via i.p. injection with 200 μg mAb on d0 and 100 μg on d3 and d6. ELISAs were as described (14). Serum samples were diluted in TBS containing 1% BSA, with the exception of PPS-3 samples, which were diluted in TBS containing 1% BSA and 10 μg/ml cell wall polysaccharide (Statens Serum Institut, Denmark) to adsorb non-capsular polysaccharide Abs. PPS-3-, TNP-, and DNP-specific Ab levels were measured by adding diluted serum samples to plates that had been coated with 10 μg/ml PPS-3 in PBS or 5 μg/ml TNP- or DNP-BSA (Biosearch Technologies) in 0.1 M borate buffered saline overnight at 4°C. AP-conjugated polyclonal goat anti-mouse IgM, IgG1, IgG2a, IgG2b, IgG3, and IgG Abs and pNPP (all from Southern Biotechnology Associates) were used to detect Ag-specific Ab. ELISA values are reported as relative ODs (OD reading for serum samples minus OD reading from wells with serum omitted).
Abs and Immunofluorescence Analysis
Single cell spleen and peritoneal cavity lymphocyte suspensions were incubated with fluorochrome-conjugated mAbs on ice for 25 min. Biotinylated- or fluorochrome-conjugated antibodies and secondary detection reagents used included: anti-mouse IgM (Southern Biotechnology Associates, Inc.); or Abs reactive with mouse CD21/35 (7G6), CD35 (8C12), CD19 (ID3), B220 (RA3-6B2), CD23 (B3B4) and phycoerythrin (PE)- and cychrome-conjugated streptavidin (all from BD-Biosciences, San Jose, CA); CD5 (53-7.3), CD11b (M1/70), CD1d (1B1), CD86 (GL1), and PD-1 (J43) mAb (all from eBioscience). TNP-Ficoll binding cells were detected by incubating Fc-blocked (0.5 μg/ml Fc Block; BD Biosciences) cells (2 × 107/ml) with 40 μg/ml TNP30-Ficoll-FITC (Biosearch Technologies) for 1 hr at room temperature, followed by subsequent staining with fluorochrome-labeled mAbs on ice for 25 min. Cells were analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA), with positive and negative cell populations determined using unreactive isotype-matched Abs (BD Biosciences and eBioscience).
Cell sorting and purification
B cell subsets were purified from peritoneal cavity lavage by staining with B220, CD5, and CD11b mAbs followed by cell-sorting of B220+CD5+CD11b+ (B-1a), B220+CD5−CD11b+ (B-1b), and B220+CD5−CD11b− (B-2) B cells using a FACSVantage SE flow cytometer (Becton Dickinson) with purities of ~85–95% as described (7). Spleen and peritoneal lavage cell suspensions enriched for B cells using were also isolated using B220+ bead selection (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions, with CD19+ B cell purities ≥95%.
Transfections
A full-length mouse CD21 cDNA clone (ID:40054528) was obtained from Open Biosystems (Huntsville, AL) and subcloned into pCDNA3.1(−) (Invitrogen, Carlsbad, CA). CHO cells stably expressing murine CD19 (CHO-CD19) were transfected with either mouse CD21 or mouse CD22 cDNA in pcDNA3.1(−) using Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's instructions. Two days after transfection, the cells were harvested using Versene-EDTA (Gibco, Invitrogen) and stained with PE-conjugated 1D3 (CD19) and FITC-conjugated 7G6 (CD21/35) or MB22-10 (CD22; ref. 35) mAb and analyzed by flow cytometry.
Real time PCR
Total RNA was extracted from B220+ purified spleen cells (B220 selection kit, Miltenyi Biotec) with an RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Random hexamer primers (Promega, Madison, WI) were used to generate cDNA. Forward and reverse primers spanned exons 4 and 7 for CD19 and exons 3 and 5 for CD20. Primer sequences were as follows: CD19 forward, 5'-TTCTG CCCCA AGCCA CAGCT TTAGA-3', CD19 reverse, 5'-CTGGC CAGAG GTAAG ATGTA GGAAGG-3' and CD20 forward, 5'-CCTCG CCATG CAACC TGCTCC-3', and CD20 reverse, 5'-CTGCA GCTGC CAGGA GTGATCC-3'. Melting curve analysis was done to ensure amplification of appropriate PCR products. Relative CD19 and CD20 mRNA expression was determined by real-time PCR on a LightCycler Instrument with analysis software Version 3 and a LightCycler FastStart DNA MasterPLUS SYBR Green I kit (all from Roche). Pairwise comparisons between CD19 cDNA abundance relative to CD20 cDNA abundance were made with the relative expression software tool (REST) program for groupwise comparison and statistical analysis (http://www.bioinformatics.gene-quantification.info/). Results are expressed as the ratio of CD19 and CD20 transcript abundance relative to the ratio calculated for wild type mice.
B cell activation and proliferation assays
For activation assays, purified splenic B cells or FACS-sorted peritoneal B cells (2 × 106/ml) were cultured in complete RPMI 1640 medium containing 10% FCS (Gibco Certified serum, Invitrogen). The cells were cultured in medium alone or in the presence of recombinant murine rBlyS (200 ng/ml; Peprotech, Rocky Hill, NJ), hamster anti-mouse CD40 mAb (1 μg/ml; HM40-3, BD Biosciences), F(ab')2 anti-mouse IgM Ab (40 μg/ml; Cappel, West Chester, PA), or LPS (25 μg/ml, E. coli 0111:B4, Sigma, St. Louis, MO). Due to temporary discontinuation of the Cappel-supplied F(ab')2 anti-mouse IgM Ab, a F(ab')2 anti-mouse IgM Ab from Jackson Immunoresearch (West Grove, PA) was used in some experiments as indicated. The F(ab')2 anti-mouse IgM Ab preparations from Jackson Immunoresearch contained detectable levels of endotoxin (Limulus Amoebocyte Lysate assay; Lonza) and were therefore treated with polymyxin B-agarose (Sigma) prior to use.
[Ca2+]i measurements
Splenocytes (107/ml) and B220+ isolated peritoneal B cells (2 ×106/ml) were loaded with 1 μM indo-1/AM ester (Molecular Probes, Eugene, OR) at 37°C for 30 min in RPMI 1640 medium containing 5% (v/v) FBS and 10 mM HEPES as described (36). In the final 15 min of indo-1 labeling, FITC-conjugated B220 Ab (splenocytes) or FITC-conjugated CD5 and PE-cy5.5-conjugated CD11b mAbs (peritoneal B cells) were added to the cells. The cells were washed and resuspended in media (2×106/ml) and protected from light at room temperature (less than 2 hr) until flow cytometric analysis. Fluorescence ratios (405 nm/525 nm) were measured using a FACStar flow cytometer (BD PharMingen) with 10–40 μg/ml F(ab')2 goat anti-mouse IgM Ab (Cappel) added after 1 min of baseline readings.
S. pneumoniae infections
All infections and whole bacteria immunizations used WU2, a type 3 strain of S. pneumoniae, as previously described (14). For i.p. infections, the bacteria were diluted in 200 μl sterile PBS with the number of CFUs reconfirmed. Mice (2–4 months of age) were immunized i.p. with 0.5 μg of PPS-3 (American Type Culture Collection, Rockville, MD) or heat-killed (105 CFU) S. pneumoniae in PBS (14).
Statistical analysis
Data are shown as means ± SEM. Differences between sample means were assessed using Student's t test. Differences in endpoint survival were assessed using Fisher's exact test. Differences in survival curves were assessed using the Log-rank test.
Results
Impaired humoral immune responses to PPS-3 in CD21/35−/−, but not C3−/− and C4−/− mice
CD21/35−/− mice generate poor humoral immune responses to encapsulated type 3 S. pneumoniae (14). To determine whether CD21/35-deficiency impairs the protective immune response to the pneumoccocal capsule, CD21/35−/−, C3−/− and C4−/− mice were immunized with PPS-3, the major capsule constituent. CD21/35−/− mice generated significantly lower PPS-3-specific IgM and IgG responses compared to wild type mice (Fig. 1A). PPS-3 specific IgG3, the main IgG isotype produced in response to TI-2 Ags, was also significantly lower in CD21/35−/− mice. By contrast, C3−/−, C4−/−, and wild type mice generated similar PPS-3-specific IgM, IgG, and IgG3 Ab responses. IgG and IgG3 responses to a highly-haptenated TI-2 Ag, TNP52-Ficoll, were also impaired in CD21/35−/− mice (Fig. 1B), as also described for DNP-Ficoll (14). By contrast, CD19−/− mice generated normal to augmented Ab responses to TNP52-Ficoll (Fig. 1B), as also described for PPS-3 and DNP-Ficoll (7, 26). Thus, humoral responses to PPS-3 and other model TI-2 Ags were impaired in CD21/35−/− mice, but this was not due to the inability of C3 or C4 to target Ag complexes to the CD21/35 receptor or an absence of CD19 signaling.
Figure 1. Reduced PPS-3-specific Ab responses in CD21/35−/− mice.

A, PPS-3-specific IgM, IgG, and IgG3 serum Ab levels in wild type (WT), CD21/35−/−, C3−/−, and C4−/− mice at 0, 7, and 14 d post i.p. immunization with PPS-3. B, Serum TNP-specific IgM, IgG, and IgG3 Ab levels in WT, CD21/35−/−, and CD19−/− mice post immunization with TNP52-Ficoll. A–B, Results were generated using 5–10 mice/genotype. Mean Ab levels (± SEM) were determined by ELISA. Significant differences between mean Ab levels are indicated by asterisks; *, p<0.05.
CD21/35−/− B cells are hyperresponsive to BCR signaling
Whether CD21/35-deficiency inhibits BCR signaling was examined following BCR crosslinking using anti-IgM Ab. BCR-induced [Ca2+]i responses were increased in CD21/35−/− B cells in response to optimal (40 μg/ml) but not suboptimal (10 μg/ml) anti-IgM Ab relative to wild type splenic B cells (Fig. 2A). Peritoneal B-1b cells from CD21/35−/− mice also generated higher BCR-induced [Ca2+]i responses than wild type B cells, but noticeable differences were not observed between wild type and CD21/35−/− B-1a cell [Ca2+]i responses (Fig. 2A). The proliferation of spleen and peritoneal B cells from CD21/35−/− and wild type mice was also similar in response to anti-IgM Ab (data not shown). Thus, CD21/35-deficiency increased [Ca2+]i responses but did not affect BCR-induced proliferation.
Figure 2. Augmented [Ca2+]i responses and normal in vivo expansion by CD21/35−/− B cells.

A, BCR-induced [Ca2+]i responses by B220+ splenocytes, and peritoneal B-1b and B-1a cells from wild type (WT) and CD21/35−/− mice. F(ab')2 anti-IgM Ab (10 or 40 μg/ml) was added to the cells after 1 min (arrowhead) with relative [Ca2+]i concentrations were assessed by flow cytometry. Results represent those obtained in 2–4 experiments. B–E, In vivo expansion of Ag-specific spleen and peritoneal B cells 3 days following immunization. B, Representative flow cytometry analysis of TNP30-FITC-Ficoll-binding by splenic and peritoneal B220+ B cells from WT mice before and after immunization with TNP52-Ficoll. C, TNP30-FITC-Ficoll-binding B cell frequencies in WT mice immunized with TNP52-Ficoll, TNP4-Ficoll, TNP30-BSA, or PBS. D, Frequencies and numbers of B220+TNP30-FITC-Ficoll+ cells from WT, CD21/35−/−, and CD19−/− mice immunized with TNP52-Ficoll or PBS. C–D, Values represent means (±SEM) generated using 3–5 mice per group. Means significantly different from PBS control means are indicated by asterisks; *, p<0.05.
CD21/35−/− B cells expand normally following TI-2 Ag activation in vivo
To further examine the basis for impaired TI-2 Ab responses in CD21/35−/− mice, B cell clonal expansion was assessed in vivo 3 days following TNP52-Ficoll immunization. The frequency of spleen and peritoneal cavity B cells that bind TNP-Ficoll was significantly expanded (1.6- to 1.9-fold) in wild type mice after immunization (Fig. 2B–C). By contrast, immunization with a weak TI-2 Ag TNP4-Ficoll, a T cell-dependent Ag TNP30-BSA, or PBS alone did not expand the frequency of peritoneal cavity B cells that bind TNP-Ficoll (Fig. 2C). The frequencies and numbers of TNP-Ficoll-specific spleen and peritoneal B cells increased similarly in wild type, CD21/35−/− and CD19−/− mice following TNP52-Ficoll immunization (Fig. 2D). Thus, Ag-specific B cells expanded in response to a strong TI-2 Ag in both CD21/35−/− and CD19−/− mice.
CD21/35−/− B cells express higher levels of PD-1 that inhibits IgG3 Ab responses
Since PD-1 negatively regulates TI-2 Ab responses, the influence of CD21/35 and CD19 expression on PD-1 induction was examined in vitro. The vast majority of freshly isolated peritoneal B-1b cells and spleen B cells from wild type, CD19−/− and CD21/35−/− mice did not express PD-1 (Fig. 3A–B). However, BCR ligation by anti-IgM Ab induced PD-1 expression, which was significantly enhanced on peritoneal B-1b (1.3-fold higher) and splenic B cells (1.6-fold higher) from CD21/35−/− mice when compared with wild type B cells (Fig. 3A–C). Similar results were obtained when B cells were stimulated with anti-IgM Ab combined with either CD40 mAb or recombinant BlyS (rBlyS) (Fig. 3A–C, and data not shown). LPS or CD40 mAb treatments alone did not induce significant PD-1 expression (data not shown), as published (data not shown, and 37, 38). By contrast, PD-1 induction on CD19−/− peritoneal B-1b and splenic B cells was impaired in response to anti-IgM Ab (60–70% reduction) when compared with wild type B cells (Fig. 3A–C). PD-1 induction by CD19−/− B cells was also impaired in response to anti-IgM Ab plus either CD40 mAb or rBlyS. Despite differences in PD-1 upregulation, wild type, CD21/35−/−, and CD19−/− splenic B and B-1b cells strongly upregulated CD86 expression in response to anti-IgM Ab and anti-IgM plus CD40 mAb (data not shown). Thus, CD19 expression inhibited PD-1 upregulation following BCR engagement, while CD21/35 expression augmented PD-1 induction.
Figure 3. Activated CD21/35−/− B cells express elevated PD-1 levels.

A–C, PD-1 expression on purified B220+ spleen cells (A) or peritoneal B-1b (B) cells from wild type (WT), CD19−/−, and CD21/35−/− mice 2 d following culture with either medium alone, F(ab')2 anti-IgM Ab, anti-IgM Ab and anti-CD40 mAb, or anti-IgM Ab and rBlyS. C, Fold induction in PD-1 expression on B cells 2 d following F(ab')2 anti-IgM activation. Fold induction was calculated by dividing the PD-1PE mean fluorescent intensity (MFI) of anti-IgM Ab activated cells by that of control resting cells. Values represent the mean fold-induction in PD-1 expression (±SEM) derived from 3–6 separate experiments. D–E, PD-1 and hamster IgG staining for TNP30-FITC-Ficoll+ spleen or peritoneal B220+cells isolated from WT naïve mice or from mice 3 d after immunization with 50 μg TNP52-Ficoll (D–E) or TNP4-Ficoll, TNP30-BSA, or PBS (E). Significant differences between control (PBS) and immunized mice are indicated by asterisks; *, p<0.05, n=3–5 mice/condition. F–G, PD-1 and hamster IgG staining on TNP30-FITC-Ficoll+ spleen and peritoneal B220+ cells from WT, CD21/35−/− and CD19−/− mice immunized with TNP52-Ficoll or PBS. Fold PD-1 induction values (± SEM) were obtained by dividing PD-1-PE MFI values obtained from TNP52-Ficoll immunized mice by that of PBS-immunized mice. H, PD-1 blockade enhances IgG3 responses in CD21/35−/− mice. WT and CD21/35−/− mice immunized with PPS-3 received PD-1 blocking mAb or control rat IgG2a mAb. Day 7 sera were analyzed by ELISA for PPS-3-specific Abs. Significant differences in mean Ab (±SEM) levels are indicated by asterisks; *, p<0.05. (C, G) Mean fold induction levels significantly different than WT mice are indicated by asterisks; *, p<0.05. A paired Student's t-test was used to analyze data in (C).
TNP-Ficoll-binding B cells isolated from wild type mice immunized with TNP52-Ficoll also expressed significantly higher levels of PD-1 than TNP-Ficoll-binding B cells from naïve mice or mice immunized with TNP4-Ficoll, TNP30-BSA, or PBS alone (Fig. 3D–E, data not shown). No difference was observed in control hamster IgG binding between TNP-Ficoll-binding B cells from TNP52-Ficoll immunized or naïve mice. TNP-Ficoll-specific spleen B cells from CD21/35−/− mice also expressed significantly higher PD-1 densities (13% higher) following TNP52-Ficoll immunization, whereas spleen B cells from CD19−/− mice expressed significantly less PD-1 (14% lower) when compared with wild type mice (Fig. 3F–G). PD-1 levels on Ag-specific peritoneal B cells from CD19−/− mice were similarly reduced. Control hamster IgG binding was absent on TNP-Ficoll-specific spleen B cells from wild type, CD21/35−/−, and CD19−/− mice immunized with TNP52-Ficoll, and no differences in PD-1 staining were detected for TNP-Ficoll-specific spleen B cells from unimmunized mice (Fig. 3F, and data not shown). Thus, Ag-specific CD21/35−/− B cells expressed higher PD-1 levels than wild type B cells following immunization with a strongly haptenated TI-2 Ag, with CD19−/− B cells expressing lower PD-1 levels than wild type B cells.
The effect of PD-1 overexpression on TI-2 Ab responses in CD21/35−/− mice was assessed in mice given a mAb that blocks PD-1 interactions with its ligands, PDL1 and PDL2 (39). After PPS-3 immunizations, Ag-specific IgG3 levels in CD21/35−/− mice were significantly increased (6-fold) by PD-1 mAb treatment (Fig. 3H), while Ag-specific IgM levels were not altered. Thus, PD-1 interactions with its ligands inhibits PPS-3-specific IgG3 responses in CD21/35−/− mice.
CD21/35 expression levels control cell surface CD19 densities
B cells from CD21/35−/− mice express significantly higher levels of cell surface CD19 (14). Whether CD21 expression regulates CD19 cell surface density directly was therefore assessed using CHO cells stably expressing mouse CD19. CD19-CHO cells were transiently transfected with murine CD21 cDNA, with subsequent CD19 and CD21 expression levels analyzed by flow cytometry. Remarkably, inducing cell surface CD21 expression reduced CD19 expression (Fig. 4A). By contrast, CD22 cDNA transfection of CD19-CHO cells as a control did not affect CD19 expression. Thus, CD21 expression directly regulates CD19 cell surface density.
Figure 4. CD21/35 negatively regulates CD19 expression.

A, Co-expression of CD21 reduces CD19 expression in CHO cells stably transfected with mouse CD19 (CD19-CHO). CD19-CHO cells were transiently transfected with either mouse CD21 or CD22 cDNAs. Two days following transfection, the cells were stained and analyzed for CD19, CD21, and CD22 expression by flow cytometry. Results represent 5 independent experiments. B–D, CD19 expression by peritoneal (B–C) and spleen (C–D) B cell subsets in wild type (WT), CD21/35−/−, and CD21/35−/−CD19+/− mice. B, CD19 expression by WT peritoneal B cell subsets as gated in (E). C, Mean CD19 MFI values (± SEM) for WT and CD21/35−/− B cell subsets. D, CD19 mRNA expression in CD21/35−/− and CD21/35−/−CD19+/− spleen B cells. Values represent the mean (±SEM) ratios of CD19/CD20 transcripts in CD21/35−/− and CD21/35−/−CD19+/− mice relative to wild type mice as determined by real-time PCR analysis from 3 independent experiments. F, CD21/35 expression by freshly isolated peritoneal B cell subsets as defined by the gating strategy shown in (E). CD5+CD11b+ (B-1a, thick line), CD5−CD11b+ (B-1b, dotted line), CD5−CD11b− (B-2, thin line) peritoneal B cells from wild type mice were analyzed for CD21 and CD35 expression by flow cytometry with CD21/35 MFI quantified (right panel). CD35 staining of CD21/35−/− B220+ peritoneal B cells is depicted by a dotted line. Asterisks (*) indicate mean MFIs (±SEM) are significantly different, p<0.05. Negative isotype control staining is indicated by a dashed line (B, F).
When B cell subsets from wild type mice were analyzed for relative differences in CD19 expression, peritoneal B-1a cells expressed CD19 at the highest levels, B-1b cells expressed intermediate levels, while B-2 cells expressed the lowest levels (Fig. 4B). Peritoneal B-1b and B-2 cells, and splenic follicular and MZ B cells from CD21/35−/− mice expressed cell-surface CD19 at 1.3-to 2.3-fold higher densities than wild type mice (Fig. 4C). B-1a cells from CD21/35−/− mice expressed wild type levels of cell surface CD19 (Fig. 4C) as described (14), and splenic B cells from CD21/35−/− and wild type mice expressed CD19 transcripts at similar levels (Fig. 4D). CD21/35 expression was inversely correlated with CD19 expression by B cell subsets, with peritoneal B-2 cells expressing the highest levels of CD21/35, B-1b cells expressing intermediate levels, and B-1a cells expressing the lowest levels (Fig. 4F). Thus, the absence of CD21/35 results in augmented cell surface CD19 protein expression on mature B cells including the B-1b, MZ, and B-2 subsets.
CD19 overexpression in CD21/35−/− mice enhances PD-1 upregulation
The contribution of CD19 overexpression in CD21/35−/− mice to augmented PD-1 upregulation was examined by comparing CD21/35−/− mice with CD21/35−/− mice that either lacked CD19 (CD21/35−/− CD19−/−) or expressed ~50% lower CD19 levels (CD21/35−/−CD19+/−). CD19 cell surface expression in CD21/35−/−CD19+/− mice was reduced to levels equal to or lower than those expressed by wild type B cell subsets (Fig. 5A–B). B cell subset development in CD21/35−/−CD19+/−, CD21/35−/−, and wild type mice was similar, whereas CD21/35−/−CD19−/− peritoneal and spleen B cell subset numbers closely resembled those of CD19−/− mice (Table I). B cell [Ca2+]i responses following IgM ligation were similar in CD21/35−/−CD19−/− and CD19−/− B cells, in contrast to the augmented [Ca2+]i responses of CD21/35−/− B cells (Fig. 5C). CD19−/− B cells generated near normal [Ca2+]i responses with a delayed peak during the acute phase and a prolonged late phase response as described (40). BCR-induced PD-1 upregulation in CD21/35−/−CD19−/− B cells was significantly impaired, similar to CD19−/− B cells (Fig. 5D). However, BCR-induced PD-1 levels were similar in CD21/35−/−CD19+/− and wild type B cells, while PD-1 induction on CD21/35−/− B cells was significantly (40%) higher (Fig. 5D). Thus, enhanced CD19 expression and/or function in the absence of CD21/35 expression contributed significantly to increased BCR-induced [Ca2+]i responses and PD-1 expression.
Figure 5. Reducing or eliminating CD19 expression on CD21/35−/− B cells normalizes BCR-induced Ca2+ responses and PD-1 upregulation.

A–B, CD19 expression (A) and mean CD19 MFI values (B) for B cell subsets from wild type (WT), CD21/35−/−, and CD21/35−/−CD19+/− mice. Peritoneal and spleen cells (n≥4 mice/genotype) were stained with B220, CD5, and CD11b mAbs to identify peritoneal B cell subsets or IgM and CD1d to identify follicular (FO) and MZ B cells. C, Relative BCR-induced [Ca2+]i responses in B220+ splenocytes from wild type, CD21/35−/−, CD19−/−, and CD21/35−/−CD19−/− mice. were assessed by flow cytometry. F(ab')2 anti-IgM Ab (40 μg/ml) was added to the cells after 1 min (arrowhead). [Ca2+]i responses are shown for 2 independent experiments. D, PD-1 expression on purified B220+ spleen cells from WT, CD21/35−/−, CD21/35−/−CD19+/−, CD21/35−/−CD19−/−, and CD19−/− mice 2 d following culture with 20 μg/ml F(ab')2 anti-IgM (Jackson Immunoresearch). Dotted lines indicate isotype control staining. Values represent the mean PD-1 fold induction (±SEM) levels derived from 5 separate experiments. Significant differences between means are indicated by asterisks; *, p<0.05.
Table I.
B lymphocyte frequencies and numbers
| Spleen |
Peritoneal cavity |
|||||
|---|---|---|---|---|---|---|
| B cells (x10−7)a | MZ B cell frequency (%)b | B cells (x10−5)a | B cell frequency (%)b |
|||
| B-2 | B-1a | B-1b | ||||
|
|
||||||
| Wild type | 5.1 ± 0.3 | 4.0 ± 0.1 | 7.0 ± 3.0 | 34 ± 7 | 26 ± 4 | 40 ± 3 |
| CD21/35−/− | 6.4 ± 0.9 | 5.2 ± 0.9 | 7.6 ± 0.8 | 32 ± 3 | 19 ± 3 | 49 ± 3 |
| CD21/35−/−CD19+/− | 6.3 ± 0.9 | 4.5 ± 1.0 | 5.2 ± 1.8 | 28 ± 6 | 17 ± 2 | 55 ± 4 |
| CD 19−/− | 1.7 ± 0.2* | 0.4 ± 0.1* | 2.3 ± 1.2 | 47 ± 5 | 6 ± 2* | 44 ± 4 |
| CD21/35−/−CD19−/− | 3.2 ± 0.6* | 0.9 ± 0.1* | 0.8 ± 0.2 | 44 ± 4 | 4 ± 1* | 52 ± 4 |
Mean B cell numbers (± SEM) were calculated based on the total number of B220+ cells harvested from spleen or the peritoneal cavity.
Values represent the mean frequency (± SEM) of lymphocytes expressing the indicated surface markers as determined by immunofluorescence staining with flow cytometry analysis.
The mean percentage or number of cells was significantly different from means of wild type mice, p<0.05. Results were from 4–6 mice of each genotype.
CD19 overexpression inhibits TI-2 Ab responses in CD21/35−/− mice
The effect of CD19 overexpression on impaired TI-2 Ab responses in CD21/35−/− mice was assessed by comparing CD21/35−/−, CD21/35−/−CD19+/−, CD21/35−/−CD19−/−, and CD19−/− mice immunized with PPS-3 or DNP40-Ficoll. PPS-3-specific IgM and IgG3 responses were significantly lower in CD21/35−/− mice compared to wild type mice 14 days following immunization (IgM, 65% of wild type; IgG3, 20% of wild type; Fig. 6A). By contrast, PPS-3-specific Ab responses were normal in CD21/35−/−CD19+/−, CD21/35−/−CD19−/−, and CD19−/− mice. In response to DNP40-Ficoll immunization, DNP-specific IgG3 responses were significantly impaired in CD21/35−/− mice (60% of wild type) in comparison with CD21/35−/−CD19+/− and wild type mice (Fig. 6B). CD21/35−/−CD19−/− and CD19−/− mice generated significantly higher (3-fold) DNP-specific IgG3 responses than wild type mice. DNP-specific IgM responses were impaired at day 7 in CD21/35−/−CD19−/− and CD19−/− mice, but were comparable to wild type levels by day 14. This is likely due to significantly lower natural DNP-reactive IgM Ab levels in CD21/35−/−CD19−/− and CD19−/− mice, which were undetectable at day 0. The TD Ag, DNP-KLH in alum, induced remarkably different Ab responses with modest responses in CD21/35−/−CD19−/− and CD19−/− mice (Fig. 6C). DNP-specific IgG3 responses to DNP-KLH were significantly impaired in both CD21/35−/− (35% of wild type) and CD21/35−/−CD19+/− mice (30% of wild type). Moreover, impaired IgG3 responses in CD21/35−/− mice to the TI-1 Ag, TNP-LPS, were not rescued by reducing CD19 expression since responses were similarly impaired in CD21/35−/− CD19+/− mice (Fig. 6D). Thus, reducing or eliminating CD19 expression specifically rescued TI-2, but not TI-1 or TD Ab responses in CD21/35−/− mice.
Figure 6. TI-2 Ab responses are selectively rescued by decreasing or eliminating CD19 expression in CD21/35−/− mice.

A–D, Ag-specific Ab responses of wild type (WT), CD21/35−/−, CD19−/−, CD21/35−/−CD19−/− and CD21/35−/−CD19+/− mice following immunization with (A) PPS-3, (B) DNP40-Ficoll, (C) DNP-KLH, or (D) TNP-LPS (CD19−/− and CD21/35−/−CD19−/− mice were omitted). Sera were collected at the indicated timepoints and analyzed by ELISA for PPS-3- (A) DNP- (B, C), or TNP-specific (D) Abs (3–8 mice/genotype). E, Ab responses to TNP52-Ficoll (upper panel) and TNP4-Ficoll (lower panel) in WT, CD21/35−/−, C3−/−, CD21/35−/−C3−/−, and CD21/35−/−CD19−/− mice. Sera were analyzed for TNP-specific Abs 14 d post-immunization by ELISA. Data points for individual mice are shown (n=4–5 mice/group). A–E, Mean Ab (±SEM) levels significantly different than WT mice are indicated by asterisks; *, p<0.05.
Humoral responses to weak TI-2 Ags in CD21/35−/− mice are complement-dependent
Whether the current conflicting reports regarding the relative importance of CD21/35, C3, and CD19 in generating optimal TI-2 Ab responses is due to differences in Ag strength was examined. Wild type, C3−/−, CD21/35−/−, CD21/35−/−C3−/−, and CD21/35−/−CD19−/− mice were immunized with TNP52-Ficoll, a strong TI-2 Ag, or TNP4-Ficoll, a weak TI-2 Ag. C3−/− mice generated normal Ag-specific IgM and IgG3 responses 14 days following TNP52-Ficoll immunization, whereas IgG3 responses were significantly impaired in CD21/35−/− mice (28% of wild type) and CD21/35−/−C3−/− mice (39% of wild type; Fig. 6E). By contrast, CD21/35−/−CD19−/− mice generated normal IgG3 responses to TNP52-Ficoll. Humoral responses to TNP4-Ficoll were substantially different. Fourteen days following immunization, TNP-specific IgM levels were significantly lower in CD21/35−/−CD19−/− mice (20% of wild type) and TNP-specific IgG3 levels were significantly impaired in C3−/−, CD21/35−/−, CD21/35−/−C3−/−, and CD21/35−/−CD19−/− mice. Therefore, the degree of TI-2 haptenation dictates the importance that C3, CD21/35, and CD19 play in humoral immune responses to TI-2 Ags. Optimal Ab responses to strongly haptenated TI-2 Ags occurred in the absence of CD19 and C3 expression, while Ab responses to weakly haptenated TI-2 Ags required CD19 and C3. Thus, CD21/35 expression normally promotes Ab responses to strong TI-2 Ags via a complement-independent and CD19 density-dependent pathway and promotes responses to weak TI-2 Ags through a complement-dependent pathway.
CD19 overexpression in CD21/35−/− mice inhibits protective immunity to S. pneumoniae
Immunization of CD21/35−/− mice with heat-killed S. pneumoniae was not protective during subsequent challenges with 104 CFU type 3 S. pneumoniae, but generated significant protective responses in wild type mice (p=0.03, Fisher's exact test; data not shown) as described (14). Therefore, the effect of CD19 overexpression on immune responses in CD21/35−/− mice was assessed using wild type, CD21/35−/−, CD21/35−/−CD19+/−, and CD21/35−/−CD19−/− mice. Immunization of mice with PPS-3 protected the majority of these mice during subsequent challenge with 104 CFU, although CD21/35−/− mice (75% survival) were less protected than wild type (91%), CD21/35−/−CD19+/− (100%), and CD21/35−/−CD19−/− (100%) mice (Fig. 7A). PPS-3 immunization did not protect CD21/35−/− mice (0% survival; p=0.02 vs. wild type) against challenge with 105 CFU S. pneumonia, while protection was observed in wild type (66% survival), CD21/35−/−CD19+/− (100%), and CD21/35−/−CD19−/− (100%) mice (Fig. 7A). Thus, CD21/35−/− mice generated protective immune responses following PPS-3 immunizations when CD19 expression was reduced by half or eliminated.
Figure 7. Protective immunity to S. pneumoniae is restored in CD21/35−/− mice by decreasing or eliminating CD19 expression.
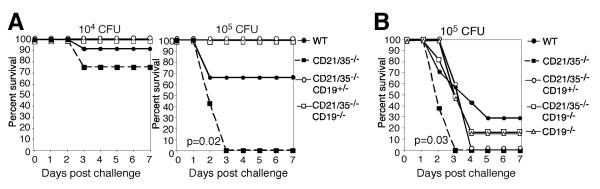
A, WT, CD21/35−/−, CD21/35−/−CD19−/− and CD21/35−/−CD19+/− mice were immunized with 0.5 μg PPS-3. Fourteen to eighteen days later, mice were challenged with 104 or 105 CFU type 3 S. pneumoniae and monitored for survival. B, Wild type (WT), CD21/35−/−, CD19−/−, CD21/35−/−CD19−/− and CD21/35−/−CD19+/− mice were immunized with 105 heat-killed type 3 S. pneumoniae. Ten days later, mice were challenged with 104 or 105 CFU type 3 S. pneumoniae and monitored for survival. Each experiment used 6–12 mice per group.
CD21/35−/− mice immunized with heat-killed (105 CFU) type 3 S. pneumoniae were significantly less protected than wild type mice during challenge with 105 CFU S. pneumoniae, as all mice died rapidly during infection with significantly shorter mean survival times (p=0.03, Log-rank test; Fig. 7B). However, survival was increased significantly in CD21/35−/−CD19+/− (p=0.01), CD21/35−/−CD19−/− (p=0.02), and CD19−/− (p=0.02) mice in comparison with CD21/35−/− mice. Survival in CD21/35−/−CD19+/−, CD21/35−/−CD19−/−, and CD19−/− mice was not significantly different from that of wild type mice. Thus, reducing or eliminating CD19 expression in CD21/35−/− mice significantly enhanced protective immunity after both PPS-3 and S. pneumoniae immunizations.
Discussion
This study reveals an unanticipated equilibrium between CD21/35 and CD19 cell surface densities that balances PD-1 expression and humoral immune responses to TI-2 Ags. CD21/35 deficiency resulted in impaired Ab production to strong TI-2 Ags, TNP52-Ficoll, DNP40-Ficoll, PPS-3, as well as intact type 3 S. pneumoniae (14). By contrast, CD19 deficiency results in normal to augmented Ab responses to strong TI-2 Ags (7, 26, 27). Remarkably, impaired TI-2 Ab responses in CD21/35−/− mice were due to elevated CD19 expression, which enhanced PD-1 induction. Thereby, reducing or eliminating CD19 expression in CD21/35−/− mice normalized PD-1 expression in response to Ag receptor crosslinking and selectively rescued Ab responses to strong TI-2 Ags, including the protective immune response to PPS-3 and intact S. pneumoniae. C3- and C4-deficiencies had no effect on Ab responses to PPS-3, indicating that CD21/35 function is complement-independent during these responses. By contrast, optimal Ab responses to weak or poorly haptenated TI-2 Ags required C3 as well as CD21/35 and CD19 expression. Thus, Ab responses to TI-2 Ags that efficiently crosslink the BCR are balanced by CD19 regulation of PD-1 expression, whereas weaker Ags are likely to require iC3b/C3d-Ag complex binding and CD21/35 ligation to initiate CD19 signaling function and lower the threshold for BCR-induced B cell activation (30).
B cell PD-1 induction was significantly influenced by CD21/35 and CD19 expression. BCR crosslinking in vitro induced significantly more cell surface PD-1 on CD21/35−/− B cells than wild type B cells, whereas PD-1 induction was significantly impaired in CD19−/− B cells. Similarly, Ag-specific B cells from CD21/35−/− mice expressed higher PD-1 levels following immunization with a strong TI-2 Ag, whereas Ag-specific B cells from CD19−/− mice expressed less PD-1. Decreasing CD19 expression on CD21/35−/− B cells restored PD-1 upregulation to wild type levels, indicating that CD19 overexpression in CD21/35−/− mice enhanced PD-1 induction. Independently, PD-1 has significant effects on TI-2 Ab responses, since PD-1−/− mice generate significantly higher Ab responses to DNP-Ficoll, with striking increases in IgG3 levels, despite normal TD Ab responses (13). PD-1+/− mice also generate augmented TI-2 Ab responses, clearly indicating that the relative dosage of cell surface PD-1 is functionally important in vivo. Inhibiting PD-1 binding to its ligands also significantly increased PPS-3-specific IgG3 responses in CD21/35−/− mice. Although PD-1 negatively regulates BCR signaling (12, 13), the mechanisms by which PD-1 regulates B cell activity in vivo are largely unexplored. Whether PD-1+ B cells are maintained in a state of exhaustion similar to what has been described for Ag-experienced T cells (41) is also unknown, although PD-1 expression by memory B cells may suppress protective anti-Env Ab responses during SIV infection (42). It is likely that PD-1 expression by autoreactive B cells similarly suppresses the development of autoimmune disease. PD-1 expression by B cells must therefore be delicately balanced to allow for protective humoral immunity during infections while preventing autoimmunity. Thus, CD21/35 and CD19 represent critical regulatory proteins that influence protective immunity and autoimmunity through multiple pathways, including the regulation of PD-1 expression by B cells.
A striking negative correlation exists between cell surface CD21/35 and CD19 expression. CD19 expression was significantly increased on most B cell populations in CD21/35−/− mice, including B-1b and marginal zone B cell subsets that are responsible for TI-2 Ab production. Differences in CD19 mRNA levels between CD21/35−/− and wild type spleen B cells were not detected. Moreover, differences in CD19 transcript levels between B-1a and B-2 subsets were not detected by microarray analysis (unpublished observations) even though B-1a cells express nearly 2-fold more cell surface CD19 than follicular B cells (32). Thus, differences in CD19 expression are controlled at the protein level. CD21/35 may control CD19 cell surface density by inducing CD19 internalization and turnover, although the mechanisms responsible for this remain to be fully elucidated. Regardless, CD21/35 plays a critical function by directly controlling cell surface CD19 density on B cells, in addition to serving as a receptor for C3 and C4 cleavage products.
Alterations in CD19 expression have profound effects on B cell development, subset distributions, activation, proliferation, and humoral immunity (7, 26, 27, 31, 33, 43). While CD19−/− mice are generally hyporesponsive to transmembrane signals, they generated normal to augmented IgG responses to PPS-3, TNP52-Ficoll, and DNP40-Ficoll. By contrast, B cells from transgenic mice that overexpress CD19 (CD19Tg) are hyperresponsive to transmembrane signals, but have severely impaired Ab responses to PPS-3 as well as DNP40-Ficoll (7, 26). B cells from CD19Tg mice also have constitutively increased levels of PD-1 expression (data not shown). Thereby, decreased PD-1 expression by CD19−/− B cells and increased PD-1 expression by B cells with elevated CD19 expression parallels the TI-2 Ab responses observed in these mice. Reciprocal TI-2 Ab responses in CD19−/− and CD19Tg mice are explained in part by their increase or lack of B-1b cells, respectively (7). CD19 expression also has intrinsic functional effects on B cells (7, 26, 27, 31, 33, 43) and CD19 signaling is differentially regulated in B-1 subsets as compared to conventional B cells. B-1a cells flux intracellular calcium poorly following CD19 ligation induced through either CD19 mAbs or C3dg-complexes in comparison to conventional B-2 cells (44–46). The levels of key cytoplasmic molecules involved in CD19 signaling such as Vav-1 and Vav-2 are also significantly diminished in B-1 cells compared to B-2 cells, as is the association of PI3-K with CD19 before and after BCR crosslinking (45). Thus, in addition to its role in regulating PD-1 expression, CD19 may regulate unique functions of B-1a and B-1b cells due to its altered signaling activities and expression.
CD21/35 regulates CD19 activity beyond regulating its expression levels. In addition to engaging CD19 signaling function following ligand binding, CD21/35 has been proposed to physically sequester CD19 away from the BCR, thereby dampening BCR-mediated signals (47). Recent evidence suggests a portion of cell-surface CD19 may physically associate with the BCR to regulate signaling intensity independent of its association with CD21/35 (48). Consistent with this, CD21/35−/− B cells were hyperresponsive to membrane bound Ag (48), similar to the augmented intracellular calcium responses we observe for CD21/35−/− B cells following strong anti-IgM crosslinking. By contrast, extensive independent ligation of CD19 or CD21/35 via mAb or C3dg-complexes dampens BCR signaling following anti-IgM crosslinking (36, 49). Whether hyperresponsiveness in CD21/35−/− B cells is due to increased CD19 associations with the BCR or solely due to increased CD19 expression levels has not been determined. Regardless, it is clear that Ab responses to TI-2 Ags which extensively crosslink the BCR did not require CD19 for optimal signaling, but were inhibited by CD19-facilitated PD-1 induction.
This study demonstrates the complex roles that CD21/35, CD19, and C3 play in regulating humoral responses to weak and strong TI-2 Ags, as well as TI-1 and TD Ags. While CD19 expression is not required for responses to strong TI-2 Ags, CD19 is required for optimal responses to TI-1, TD, and weak TI-2 Ags. Thus, deficient Ab responses to TI-1, TD, and weak TI-2 Ags are not rescued by reducing CD19 expression levels in CD21/35−/− mice. C3 is required for optimal Ab responses to weak TI-2 Ags and primary responses to TD Ags. Reduced TI-1 IgG3 responses in CD21/35−/− mice are reportedly due to excessive C3 activation (50). Our results agree with this, since reduced TNP-LPS Ab responses in CD21/35−/− mice were restored in CD21/35−/−C3−/− mice (data not shown), but not in CD21/35−/−CD19+/− mice. Thus, B cell responses to strong TI-2 Ags are regulated by C3, CD19, and CD21/35 differently than responses to TI-1, T-D, and weak TI-2 Ags. The explanation for this is undoubtedly complex as differential pathways of B cell activation, differential CD19 regulatory activity, and distinct B cell subsets may be involved in each of these responses.
In summary, the current study demonstrates a novel C3-independent mechanism by which CD21/35 and CD19 regulate humoral responses to strong TI-2 Ags. CD19 promotes PD-1 expression following Ag receptor crosslinking and this results in decreased TI-2 Ab responses. Thus, by negatively regulating CD19 expression, CD21/35 dampens the level of PD-1 induced following B cell activation and promotes optimal TI-2 Ab responses. Intact responses to TI-2 Ags are critical for the generation of protective immunity to encapsulated extracellular bacteria including S. pneumoniae, Haemophilus influenzae, and Neisseria meningitidis (51). Indeed, a significant number of pathogens express surface polysaccharides that may function as immunogenic TI-2 Ags (51). The finding that CD19 and CD21/35 play unique roles in regulating B-1b cell responses to these types of Ags uncovers new pathways by which TI-2 Ab responses are regulated. As such, further elucidation of the factors involved in regulating these responses may provide novel opportunities to develop new vaccines and improve existing vaccine strategies.
Acknowledgements
The authors thank Dr. Suman Sen for performing real-time PCR studies and transfection experiments.
Footnotes
This project was supported by NIH grants CA96547, CA105001, and AI056363. KMH was supported by a Special Fellow Award from the Leukemia and Lymphoma Society and by a Small Project grant from the Duke Center for Translational Research.
Abbreviations used Ag, antigen; TI-2, T cell-independent type 2; TI-1, T cell-independent type 1 antigen; TD, T cell-dependent; PPS-3, Streptococcus pneumoniae type 3 capsular polysaccharide; MZ, marginal zone; PD-1, programmed cell death 1; PC, phosphorylcholine.
References
- 1.Wuorimaa T, Kayhty H. Current state of pneumococcal vaccines. Scand. J. Immunol. 2002;56:111–129. doi: 10.1046/j.1365-3083.2002.01124.x. [DOI] [PubMed] [Google Scholar]
- 2.Russell ND, Corvalan JR, Gallo ML, Davis CG, Pirofski L. Production of protective human antipneumococcal antibodies by transgenic mice with human immunoglobulin loci. Infect. Immun. 2000;68:1820–1826. doi: 10.1128/iai.68.4.1820-1826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett DJ, Lee CG, Ammann AJ, Ayoub EM. IgG and IgM pneumococcal polysaccharide antibody responses in infants. Pediatr. Res. 1984;18:1067–1071. doi: 10.1203/00006450-198411000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol. Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 5.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 6.Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a “natural immune memory”. Immunol. Rev. 2000;175:70–79. [PubMed] [Google Scholar]
- 7.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Hsu MC, Toellner KM, Vinuesa CG, Maclennan IC. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. USA. 2006;103:5905–5910. doi: 10.1073/pnas.0601502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 10.Patterson HC, Kraus M, Kim YM, Ploegh H, Rajewsky K. The B cell receptor promotes B cell activation and proliferation through a non-ITAM tyrosine in the Igα cytoplasmic domain. Immunity. 2006;25:55–65. doi: 10.1016/j.immuni.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Fruman DA, Satterthwaite AB, Witte ON. Xid-like phenotypes: a B cell signalsome takes shape. Immunity. 2000;13:1–3. doi: 10.1016/s1074-7613(00)00002-9. [DOI] [PubMed] [Google Scholar]
- 12.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 13.Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998;10:1563–1572. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- 14.Haas KM, Hasegawa M, Steeber DA, Poe JC, Zabel MD, Bock CB, Karp DR, Briles DE, Weis JH, Tedder TF. Complement receptors CD21/35 link innate and protective immunity during Streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity. 2002;17:713–723. doi: 10.1016/s1074-7613(02)00483-1. [DOI] [PubMed] [Google Scholar]
- 15.Szomolanyi-Tsuda E, Seedhom MO, Carroll MC, Garcea RL. T cell-independent and T cell-dependent immunoglobulin G responses to polyomavirus infection are impaired in complement receptor 2-deficient mice. Virology. 2006;352:52–60. doi: 10.1016/j.virol.2006.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pozdnyakova O, Guttormsen HK, Lalani F, Carroll MC, Kasper DL. Impaired antibody response to group B streptococcal type III capsular polysaccharide in C3- and complement receptor 2-deficient mice. J. Immunol. 2003;170:84–90. doi: 10.4049/jimmunol.170.1.84. [DOI] [PubMed] [Google Scholar]
- 17.Ochsenbein AF, Pinschewer DD, Odermatt B, Carroll MC, Hengartner H, Zinkernagel RM. Protective T cell-independent antiviral antibody responses are dependent on complement. J. Exp. Med. 1999;190:1165–1174. doi: 10.1084/jem.190.8.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Neil KM, Ochs HD, Heller SR, Cork LC, Morris JM, Winkelstein JA. Role of C3 in humoral immunity. Defective antibody production in C3-deficient dogs. J. Immunol. 1988;140:19391945. [PubMed] [Google Scholar]
- 19.Matsuda T, Martinelli GP, Osler AG. Studies on immunosuppression by cobra venom factor. II. On responses to DNP-ficoll and DNP-polyacrylamide. J. Immunol. 1978;121:2048–2051. [PubMed] [Google Scholar]
- 20.Pepys MB. Role of complement in induction of antibody production in vivo. Effect of cobra venom factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J. Exp. Med. 1974;140:126–145. doi: 10.1084/jem.140.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markham RB, Nicholson-Weller A, Schiffman G, Kasper DL. The presence of sialic acid on two related bacterial polysaccharides determines the site of the primary immune response and the effect of complement depletion on the response in mice. J. Immunol. 1982;128:2731–2733. [PubMed] [Google Scholar]
- 22.Test ST, Mitsuyoshi JK, Hu Y. Depletion of complement has distinct effects on the primary and secondary antibody responses to a conjugate of pneumococcal serotype 14 capsular polysaccharide and a T-cell-dependent protein carrier. Infect. Immun. 2005;73:277–286. doi: 10.1128/IAI.73.1.277-286.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pryjma J, Humphrey JH. Prolonged C3 depletion by cobra venom factor in thymus-deprived mice and its implication for the role of C3 as an essential second signal for B-cell triggering. Immunology. 1975;28:569–576. [PMC free article] [PubMed] [Google Scholar]
- 24.Guinamard R, Okigaki M, Schlessinger J, Ravetch JV. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral responses. Nat. Immunol. 2000;1:31–36. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- 25.Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv. Exp. Med. Bio. 2005;560:125–139. doi: 10.1007/0-387-24180-9_16. [DOI] [PubMed] [Google Scholar]
- 26.Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc. Natl. Acad. Sci. USA. 1995;92:11558–11562. doi: 10.1073/pnas.92.25.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH. The physiologic role of CD19 cytoplasmic tyrosines. Immunity. 2002;17:501–514. doi: 10.1016/s1074-7613(02)00426-0. [DOI] [PubMed] [Google Scholar]
- 29.Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 2002;3:399–406. doi: 10.1038/ni776. [DOI] [PubMed] [Google Scholar]
- 30.Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 31.Engel P, Zhou L-J, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 32.Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J. Immunol. 1996;157:4371–4378. [PubMed] [Google Scholar]
- 33.Sato S, Steeber DA, Jansen PJ, Tedder TF. CD19 expression levels regulate B lymphocyte development: human CD19 restores normal function in mice lacking endogenous CD19. J. Immunol. 1997;158:4662–4669. [PubMed] [Google Scholar]
- 34.Wessels MR, Butko P, Ma M, Warren HB, Lage A, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haas KM, Sen S, Sanford IG, Miller AS, Poe JC, Tedder TF. CD22 ligand binding regulates normal and malignant B lymphocyte survival in vivo. J. Immunol. 2006;177:3063–3073. doi: 10.4049/jimmunol.177.5.3063. [DOI] [PubMed] [Google Scholar]
- 36.Lee Y, Haas KM, Gor DO, Ding X, Karp DR, Greenspan NS, Poe JC, Tedder TF. Complement component C3d-Ag complexes can either augment or inhibit B lymphocyte activation and humoral immunity in mice depending on the degree of CD21/CD19 complex engagement. J. Immunol. 2005;175:8011–8023. doi: 10.4049/jimmunol.175.12.8011. [DOI] [PubMed] [Google Scholar]
- 37.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 38.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 39.Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K. Blockade of B7-H1 on macrophages suppresses CD4+ T cell proliferation by augmenting IFN-γ-induced nitric oxide production. J. Immunol. 2005;175:1586–1592. doi: 10.4049/jimmunol.175.3.1586. [DOI] [PubMed] [Google Scholar]
- 40.Fujimoto M, Bradney AP, Poe JC, Steeber DA, Tedder TF. Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity. 1999;11:191–200. doi: 10.1016/s1074-7613(00)80094-1. [DOI] [PubMed] [Google Scholar]
- 41.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 42.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2008;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J. Immunol. 2000;165:6635–6643. doi: 10.4049/jimmunol.165.11.6635. [DOI] [PubMed] [Google Scholar]
- 44.Lyubchenko T, dal Porto J, Cambier JC, Holers VM. Coligation of the B cell receptor with complement receptor type 2 (CR2/CD21) using its natural ligand C3dg: activation without engagement of an inhibitory signaling pathway. J. Immunol. 2005;174:3264–3272. doi: 10.4049/jimmunol.174.6.3264. [DOI] [PubMed] [Google Scholar]
- 45.Sen G, Wu HJ, Bikah G, Venkataraman C, Robertson DA, Snow EC, Bondada S. Defective CD19-dependent signaling in B-1a and B-1b B lymphocyte subpopulations. Mol. Immunol. 2002;39:57–68. doi: 10.1016/s0161-5890(02)00047-0. [DOI] [PubMed] [Google Scholar]
- 46.Krop I, Shaffer AL, Fearon DT, Schlissel MS. The signaling activity of murine CD19 is regulated during B cell development. J. Immunol. 1996;157:48–56. [PubMed] [Google Scholar]
- 47.Rickert RC. Regulation of B lymphocyte activation by complement C3 and the B cell coreceptor complex. Curr. Opin. Immunol. 2005;17:237–243. doi: 10.1016/j.coi.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, Batista FD. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat. Immunol. 2008;9:63–72. doi: 10.1038/ni1547. [DOI] [PubMed] [Google Scholar]
- 49.Chakravarty L, Zabel MD, Weis JJ, Weis JH. Depletion of Lyn kinase from the BCR complex and inhibition of B cell activation by excess CD21 ligation. Int. Immunol. 2002;14:139–146. doi: 10.1093/intimm/14.2.139. [DOI] [PubMed] [Google Scholar]
- 50.Jacobson AC, Weis JJ, Weis JH. Complement receptors 1 and 2 influence the immune environment in a B cell receptor-independent manner. J. Immunol. 2008;180:5057–5066. doi: 10.4049/jimmunol.180.7.5057. [DOI] [PubMed] [Google Scholar]
- 51.Weintraub A. Immunology of bacterial polysaccharide antigens. Carbohydrate Res. 2003;338:2539–2547. doi: 10.1016/j.carres.2003.07.008. [DOI] [PubMed] [Google Scholar]