

SUMMARY
Monitoring cancer and aging in vivo remains experimentally challenging. Here, we describe a luciferase knockin mouse (p16LUC), which faithfully reports expression of p16INK4a, a tumor suppressor and aging biomarker. Lifelong assessment of luminescence in p16+/LUC mice revealed an exponential increase with aging, which was highly variable in a cohort of contemporaneously housed, syngeneic mice. Expression of p16INK4a with aging did not predict cancer development, suggesting that the accumulation of senescent cells is not a principal determinant of cancer-related death. In 14 of 14 tested tumor models, expression of p16LUC was focally activated by early neoplastic events, enabling visualization of tumors with sensitivity exceeding other imaging modalities. Activation of p16INK4a was noted in the emerging neoplasm and surrounding stromal cells. This work suggests that p16INK4a activation is a characteristic of all emerging cancers, making the p16LUC allele a sensitive, unbiased reporter of neoplastic transformation.
INTRODUCTION
Although murine models have provided insight into the mechanisms governing senescence and tumorigenesis, the dynamics of these processes in vivo remain elusive. Genetic engineering has produced murine systems that faithfully mimic important aspects of human cancer. However, monitoring disease in these systems is challenging, particularly in the setting of early neoplasia. Imaging techniques akin to those used in the clinic (e.g., MRI, FDG-PET, and ultrasound) are now being applied to rodent models; however, the specificity, cost, and technical challenges of these approaches have limited their utility, particularly for early detection and serial imaging. In an attempt to address these challenges, lineage-specific luminescent and fluorescent reporters have been employed to dynamically track tumor growth (reviewed in O’Neill et al., 2010). Although these alleles have provided insight into the development, progression, and therapeutic response of de novo tumors, their expression is not limited to would-be cancer cells but is instead limited to the lineage from which the tumors originate. In contrast, several “tumor-specific” reporter alleles have been reported in the literature (e.g., E2f-LUC [Uhrbom et al., 2004], ARF-GFP [Zindy et al., 2003], ODD-Luc [Goldman et al., 2011; Safran et al., 2006], and p21-p-luc [Ohtani et al., 2007]), but these models are not ideal as they are activated at late stages during tumorigenesis, are limited for in vivo and serial imaging, are specific to the underlying tumor genetics, and/or demonstrate high background with a small dynamic range of expression. Similarly, long-term monitoring is challenging in the case of murine aging, in which no validated in vivo biomarkers exist. As a result, the discovery of genetic modifications and therapeutic interventions that influence aging currently relies upon costly 2 to 3 year rodent life span studies. To address these needs, we sought to exploit the unusual properties of the endogenous p16INK4a promoter.
Expression of p16INK4a functions to limit cell-cycle progression and to promote cellular senescence in response to multiple stressors, including oncogene activation, telomere erosion, reactive oxygen species, and stalled replication forks (Collado et al., 2007; Sharpless and DePinho, 2007). Expression of p16INK4a in healthy cells is low, but once induced, p16INK4a binds and inhibits cyclin-dependent kinase 4/6 (CDK4/6) activity, thereby promoting a retinoblastoma (RB)-dependent cell-cycle arrest. This tumor-suppressive mechanism is believed to limit the growth of would-be neoplasms, and accordingly, the p16INK4a-CDK4/6-RB axis is disrupted in most, if not all, human cancers, with inactivation of p16INK4a being the most common lesion of this pathway (see COSMIC [Forbes et al., 2006]). Although induction of p16INK4a in response to oncogenic stimuli results in a beneficial, anticancer outcome, expression of this tumor suppressor also contributes to aspects of mammalian aging. Both senescent cells and levels of p16INK4a progressively accumulate with age (Krishnamurthy et al., 2004; Nielsen et al., 1999; Zindy et al., 1997) and are associated with a decline in the replicative capacity of many tissue types (Chen et al., 2011; Janzen et al., 2006; Krishnamurthy et al., 2006; Liu et al., 2011; Molofsky et al., 2006). As such, human polymorphisms affecting p16INK4a expression influence susceptibility to multiple age-associated diseases (e.g., cancer, type 2 diabetes, and atherosclerosis) (Jeck et al., 2012). Further supporting a causal role for p16INK4a in aging, measures to reduce the formation of or to destroy p16INK4a-expressing cells in mice have been shown to ameliorate several age-associated phenotypes (e.g., cataracts, sarcopenia, and pancreatic β cell dysfunction) (Baker et al., 2011; Berent-Maoz et al., 2012; Carlson et al., 2008; Chen et al., 2011).
Exploiting the highly dynamic induction of p16INK4a observed in response to oncogenic insult and senescence, we generated an in vivo reporter system, p16LUC. In contrast to previous transgenic reporter systems driven by fragments of the p16INK4a promoter (Baker et al., 2011; Yamakoshi et al., 2009), we elected to employ a targeted “knockin” strategy that preserves known cis-regulatory elements centromeric to the p16INK4a open reading frame (Visel et al., 2010; Witcher and Emerson, 2009). We employed this p16LUC allele to monitor senescence and the earliest steps of tumorigenesis in well-defined murine systems.
RESULTS
Generation and Characterization of the p16LUC Allele
To generate the p16LUC allele, the firefly luciferase complementary DNA (cDNA), followed by an SV40 polyadenylation signal, was targeted into the translational start site of the endogenous p16INK4a locus (Figure 1A). The resulting knockin allele was expected to be null for p16INK4a expression yet to retain intronic structures surrounding exon 1α. After validation of targeting by Southern analysis and PCR (Figures S1A and S1B available online), we tested the functionality of the allele by culturing p16+/+, p16+/LUC, and p16LUC/LUC murine embryo fibroblasts (MEFs) on a 3T3 schedule with serial assessment of growth, luminescence, and Ink4/Arf gene expression. In accord with prior results (Krimpenfort et al., 2001; Sharpless et al., 2001), MEFs of each genotype displayed similar growth kinetics at early passage (Figure S1C). Moreover, transcription of p19ARF and p15INK4b was not altered by luciferase insertion into exon 1α of the Ink4a/Arf locus (Figures S1D and S1E). As expected, no endogenous p16INK4a mRNA or protein was detected in p16LUC/LUC cells, and expression was reduced by 50% in p16+/LUC cells (Figures 1B–1D). Cultured p16+/LUC and p16LUC/LUC MEFs showed rapid increases in luciferase activity with passage, exploiting the full dynamic range of this assay (Figure 1E). For example, luminescence was initially low in p16LUC/LUC cells (~300 light units/105 cells) but increased more than 150-fold (>50,000 light units/105 cells) after 2 weeks in culture (Figure 1E). Induction of p16LUC closely paralleled transcription of the wild-type allele in p16+/LUC MEFs (Figure 1F, r2 = 0.81, p < 0.0001), and the absolute level of luciferase transcript was nearly identical to that of the native p16INK4a transcript (data not shown).
Figure 1. Design and Validation of the p16LUC Allele.

(A) Schematic of the p16LUC knockin targeting strategy. “+ Flp” denotes the targeted allele after Flp-recombinase-mediated excision of the neomycin selection cassette.
(B) Induction of p16INK4a mRNA is shown in MEFs of indicated genotypes passaged on a 3T3 schedule. Fold induction was calculated with respect to p16INK4a transcript levels at day 3. Data shown correspond to three biological replicates performed in triplicate. Error bars represent SEM.
(C) p19ARF and p16INK4a western blots for littermate MEFs cultured on a 3T3 schedule and harvested at the indicated time points.
(D) Fold p16INK4a induction is shown for the western blots represented in (C). Bands were quantified by using a LICOR Odyssey system, normalized to total protein, and analyzed as in (B).
(E) Luciferase activity in MEFs of the indicated genotypes, with results calculated and presented as in (B). Error bars are ±SEM.
(F) Correlation of luciferase activity and endogenous p16INK4a expression in p16+/LUC MEFs passaged on a 3T3 schedule. Data represent ≥3 biological replicates at multiple in vitro time points with best-fit line and 95% confidence intervals (dashed lines) shown. Linear regression was used to calculate the p value and correlation coefficient (r).
(G) Representative, 2 min luminescent images of a lactating p16+/LUC mouse at the indicated time points relative to the weaning date (WD).
(H) Representative, 2 min photographic and luminescent images of a p16+/LUC mouse with healing wounds.
See Figure S1 for additional related data.
Next, we sought to validate the proper expression of p16LUC in vivo through examination of two physiological processes previously associated with p16INK4a activation: wound healing and mammary involution. To achieve optimal sensitivity during serial assessments, the p16LUC allele was backcrossed onto a hairless, immunocompetent strain, SKH1-E. As has been reported for endogenous p16INK4a expression (Gadd et al., 2001; Jun and Lau, 2010; Natarajan et al., 2003), luminescent signal increased focally during both wound healing and mammary involution (Figures 1G and 1H) and resolved upon completion of these processes. Together, these results show that the p16LUC targeting strategy produced an allele null for p16INK4a expression without affecting the production of p15INK4b and p19ARF. Moreover, induction of p16LUC both in vitro and in vivo faithfully and robustly reported endogenous p16INK4a expression.
p16LUC Activation Correlates with Chronological Aging but Fails to Predict Cancer Death
Increases in p16INK4a are associated with cellular senescence and correlate with chronological age in mammals (Krishnamurthy et al., 2004; Nielsen et al., 1999; Zindy et al., 1997). To determine more precisely the relationship between p16INK4a expression and age, we serially analyzed luminescence in p16+/LUC mice during physiologic aging. Total body luciferase (TBL) activity was monitored every 2 months beginning at 16 weeks of age (young adulthood) in a large cohort of p16+/LUC animals (n = 32). In this cohort, but not in a contemporaneously analyzed group of p16+/+ animals, TBL activity increased during a period of 80 weeks (Figures 2A, 2B, and S2A; average 6.9-fold increase from 16 to 80 weeks of age). Likewise, luciferase mRNA levels, detected by using single-molecule in situ hybridization, increased in the epidermal keratinocytes and dermis of aged murine skin in comparison to young controls (Figure 2C). These data confirm previous findings regarding p16INK4a expression in aging tissues (Dimri et al., 1995; Jeyapalan et al., 2007; Krishnamurthy et al., 2004; Ressler et al., 2006; Waaijer et al., 2012; Zindy et al., 1997), showing that p16LUC reports faithful, age-related increases in gene expression.
Figure 2. Induction of p16LUC Correlates with Age, but Not with Life Span.

(A) Total body luciferase activity in hairless (SKH1-E) p16+/LUC mice quantified using a 2 min, ventral image and plotted versus age (n = 32). The blue line represents the median value for each time point. Luciferase activity in 80-week-old p16+/+ mice is shown for comparison. For statistical analyses, at each time point, the ratio of total body luminescence relative to week 16 was calculated. Wilcoxon signed-rank tests were used to test whether each ratio was significantly different than 1. p < 0.0001 for all comparisons except week 24 (p = 0.018).
(B) Representative serial luciferase images of mice from the study shown in (A). Each 2 min image shows the same group of mice, arranged identically. Mouse 2 is p16+/+, whereas all other mice are p16+/LUC.
(C) Skin from the hind quarter of old and young p16+/LUC animals was fixed in the same paraffin block and subjected to single-molecule luciferase RNA in situ hybridization. Shown are representative images of tissues from the same paraffin block with luciferase message appearing as brown dots. Original magnification = 40×.
(D) The average total body luciferase activity for the cohort shown in (A) is plotted in blue with 95% confidence intervals depicted by dotted lines. Plotted in black are points representing total body luciferase activity in the image preceding the life span event (death of tumor formation) for each mouse (n = 14).
(E) Total body luciferase activity was calculated for each living mouse at 56 weeks of age. Using the median luciferase activity at this time point, mice were divided into high and low groups. Kaplan Meier curves for each group are shown with p values calculated by using a log rank test.
(F) By using linear regression, the rate of luciferase increase was calculated for each mouse alive at 56 weeks. By using the median rate of increase at this time point, mice were divided into high and low groups. Kaplan Meier curves for each group are shown, and p values were calculated by using a log rank test.
See Figure S2 for additional related data.
Given the ability of p16LUC to serially assess aging in vivo, we next compared the dynamics of p16INK4a induction with chronological aging. Analysis of aggregate data collected during 80 weeks demonstrated a strong, linear correlation (r2 = 0.963) between log2-transformed luminescence and chronological age, indicating exponential growth with a “p16INK4a-doubling time” of 0.45 years (Figure S2A). By using combined data of p16INK4a expression in T cells from two cohorts of healthy human donors aged 18–91 (n = 208) (Liu et al., 2009; H.B. Muss et al., 2011, J. Clin. Oncol., abstract), we estimated the average human p16INK4a-doubling time to be 17.8 years (Figure S2B). These data suggest that the ratio of human/mouse p16INK4a-doubling time (40-fold) is similar to the human/mouse ratio of median and maximal life span (Austad, 1997). Moreover, the rate and expression of p16LUC did not differ significantly between genders, although females appeared to have slightly higher p16LUC levels early in life (p = 0.094; Figure S2C). Interestingly, even in this cohort of contemporaneously housed, syngeneic mice, we observed significant interindividual variability in the rate of change in luminescence with aging (Figures 2A and S2D), demonstrating a considerable nongenetic contributor to the rate of molecular aging.
Based upon extensive work from our group and others suggesting that age-related phenotypes, including cancer, are promoted by the accumulation senescent cells (Baker et al., 2008, 2011; Bavik et al., 2006; Berent-Maoz et al., 2012; Campisi et al., 2011; Chen et al., 2011; Coppé et al., 2008; Kang et al., 2011; Krishnamurthy et al., 2006; Krtolica et al., 2001; Laberge et al., 2012; Liu and Hornsby, 2007; Liu et al., 2011; Molofsky et al., 2006; Parrinello et al., 2005), we hypothesized that p16LUC expression with aging would predict mortality. If correct, p16+/LUC animals with the highest total body luminescence should be more likely to develop age-related phenotypes than age-matched mice with lower p16LUC expression. Exploiting the large heterogeneity in luciferase activity at any given time point in our aging cohort, we were able to directly test this assumption. However, in contrast to expectation, total body p16LUC expression failed to predict two age-related phenotypes: overall mortality and development of spontaneous malignancy. Imaging 4–6 weeks prior to an animal’s terminal event (death or euthanasia for overt tumor formation) showed no increase in p16LUC activity compared to surviving age-matched contemporaries (Figure 2D). Additionally, after excluding animals with wounds or cancer and stratifying our cohort into groups with higher- or lower-than-median luminescence, Kaplan Meier analysis showed that animals with higher total body luminescence at any age exhibited the same overall mortality rates as those with lower total body luminescence (Figures 2E and S2E–S2G). Likewise, the rate of change in luminescence activity at any age was not predictive of death (Figures 2F and S2H–S2J). Consistent with previous assessments of mortality in several strains of inbred as well as outbred mice (Chrisp et al., 1996; Miller et al., 2011; Wilkinson et al., 2012), all autopsied animals from the study showed signs of advanced cancer. Moreover, no significant decrease in median survival was observed between p16+/+ and p16+/LUC animals (Figure S2K). Therefore, these findings demonstrate that total body expression of p16INK4a, an accepted in vivo marker of cellular senescence, does not, at any age, predict the risk of death from spontaneous malignancy during normal murine aging.
p16LUC Signals Cancer
We next turned to a study of p16INK4a induction in early neoplasia by crossing the p16LUC allele to well-characterized, genetically engineered mouse models of autochthonous cancer: C3(1)TAg (basal-like breast cancer driven by SV40 large T-antigen [Tag]) (Herschkowitz et al., 2007; Maroulakou et al., 1994) and TRIA (Tyr-RasG12V, Ink4a/Arf−/− melanoma) (Chin et al., 1997). Notably, TRIA mice harboring the p16LUC knockin allele are null for functional p16INK4a (p16−/LUC) but retain a single copy of p19ARF in cis to the targeted luciferase allele. However, we have labeled TyrRasG12V p16−/LUC Arf−/+ mice TRIA for the sake of brevity. Of note, C3(1)Tag mice also develop mixed eccrine sweat gland tumors of the paw in addition to mammary tumors (Maroulakou et al., 1999) (Figure S3A). The resulting cohorts were monitored biweekly, starting at young adulthood. Luminescent activity was not apparent in young p16+/LUC or p16−/LUC mice harboring these oncogenic transgenes; but with aging, intense, highly focal regions of luminescence developed within the mammary glands and paws of C3(1)TAg mice or the skin of TRIA mice (Figures 3A–3D and S3A). These regions showed focal luminescent intensities >10 times higher than that observed with aging, clearly distinguishing them from age-related p16INK4a activation (Figure S3B). On average, 62 days later (range = 0–408 days), tumors developed in these precise locations, providing an earlier and more dynamic means of identifying neoplastic growth than traditional methods (Figures 3E–3G and S3C). No difference in luminescence activity was observed between age-matched p16+/LUC and tumor-free p16−/LUC TRIA mice (Figure S3D), suggesting that oncogene activation in melanocytes alone was not sufficient to induce marked p16INK4a expression. Instead, subsequent events associated with neoplastic conversion appeared to trigger p16LUC transcription.
Figure 3. p16LUC Marks Early, De Novo Tumorigenesis.

(A) Representative serial images showing p16LUC accumulation at p16+/LUC, C3(1)TAg+ tumor foci during a period of 8 weeks. Several focal, mammary, and paw tumors are visible. A palpable tumor, smaller than could be measured by calipers, is highlighted in the red box.
(B) Representative serial images showing p16LUC accumulation at a p16−/LUC TRIA ear tumor during a period of 6 weeks.
(C) Haematoxylin and eosin staining showing the morphology of the TRIA ear tumor shown in (B).
(D) Haematoxylin and eosin staining showing the morphology of the barely palpable C3(1)TAg tumor highlighted in (A).
(E) The number of days prior to palpation or visualization where luciferase activity was observed in tumors of p16+/LUC C3(1)TAg mice. Data are shown for individual tumors with the median detection advantage indicated by a line.
(F) The number of days prior to palpation or visualization where luciferase activity was observed in tumors from p16−/LUC TRIA mice. Data are depicted as in (E).
(G) Combined Kaplan-Meier analysis of tumor-free survival in p16+/LUC C3(1)TAg and p16−/LUC TRIA mice by using tumor detection by either luminescence or palpation. Significance was determined by using a log rank test.
(H) Luciferase images of a p16+/LUC C3(1)TAg mouse with early mammary tumors (2 min ventral image, 1 min side image).
(I) Photographic images of mammary tumors dissected from the mouse in (H) and (J). Ruler hash marks represent 1 mm units.
(J) FDG-PET images of the same p16+/LUC C3(1)TAg mouse shown in (H). Tumor 1 is not visualized by FDG-PET.
See additional supporting data in Figure S3.
When initially noted, luminescent foci of the skin (TRIA) or breast (C3(1)TAg) appeared normal visually and by palpation. Therefore, we further explored the tumor detection advantage provided by the p16LUC allele. On average, luminescent foci were discovered 41 (C3(1)TAg; range: 14–73) or 87 (TRIA; range: 0–408) days before the tumors became palpable or visible (Figures 3E–3G). Representative tumors visualized by luminescence, but not yet palpable, were excised from p16+/LUC C3(1) TAg mice and measured. An unequivocal luminescence signal was detectable in mice with tumors as small as 1 mm3 (Figures 3H and 3I). Moreover, comparison of fluorodeoxyglucose positron emission tomography (FDG-PET) with luminescence detection revealed increased specificity and sensitivity by using p16LUC as a tumor detection modality (Figure 3J). Signal intensity was not uniform in tumors of the same size from different animals, owing to interanimal differences in tumor biology and/or technical features related to imaging (e.g., tumor location) (Figure S3E). Subsequent analysis of de novo C3(1)TAg tumors by using luciferase RNA in situ hybridization confirmed activation of the p16INK4a promoter in the early stages of neoplasia, prior to detectability by total body luminescent imaging (Figure S5B). Therefore, use of the p16LUC allele provides a detection advantage over conventional modalities, allowing for the visualization, excision, and analysis of de novo tumors at considerably earlier stages.
Unbiased Tumor Detection with p16LUC
Use of p16LUC to detect de novo tumor formation was not limited to genetically engineered mouse models harboring defects in the RB tumor suppressor pathway. Melanomas driven by oncogenic K-RasG12D (Liu et al., 2012) or B-RafV600E (Dankort et al., 2009), as well as hematological malignancies caused by Myc overexpression (Harris et al., 1988), displayed intense, focal luminescent signals (Figures S4A–S4C). In addition, spontaneous tumors arising in aged p16+/LUC mice were readily detected (Figures 4A, 4B, and S4D). Not only were primary tumors easily detectable in these mice, but spontaneous hematologic malignancies disseminated to the brain, bone marrow, spleen, and liver were also routinely observed (Figures 4A and S4D). In fact, testing of 14 different tumors models failed to identify a tumor type wherein p16LUC is not focally induced in emerging tumors (Table S1). Taken together, these data suggest that activation of p16LUC marks the vast majority of, if not all, malignancies, independent of cell type or driver mutation.
Figure 4. p16LUC Activity Detects Spontaneous Cancers In Vivo.
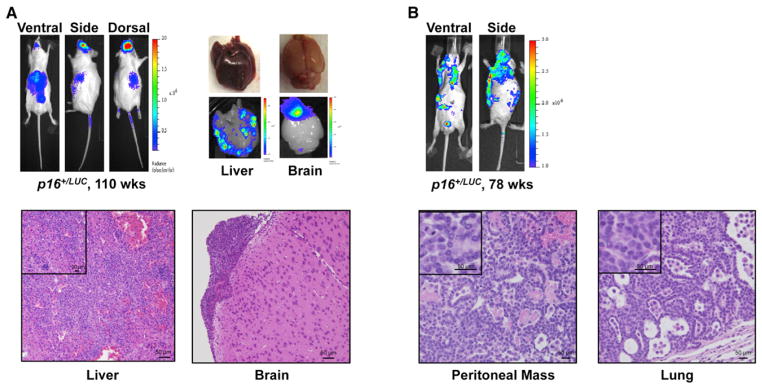
(A) (Top) Detection of a spontaneous histiocytic malignancy in a 110-week-old p16+/LUC mouse. All luminescent images shown were 1 min in length except the ventral image, which was 2 min long. Organ images were taken immediately following imaging. (Bottom) Haematoxylin and eosin stained fixed tissues confirm the presence of malignancy within luciferase positive regions of the brain and liver.
(B) (Top) Luminescent detection of a spontaneous, disseminated lung adenocarcinoma in a 78-week-old p16+/LUC mouse. Images were generated as in (A) with confirmatory histology shown below.
Non-Cell-Autonomous Activation of p16LUC
To address the contribution of p16LUC expression in tumor versus tumor-associated stroma, we performed orthotopic transplantation of C3(1)TAg tumor cells without the p16LUC allele into syngeneic p16+/LUC mice (Figure 5A) and then serially imaged these animals at early stages of tumor formation. Strikingly, induction of p16LUC was observed in the area of tumor formation, but not in the contralateral, matrigel-injected mammary gland. Moreover, injection with MEFs failed to induce p16LUC (Figure S5A), demonstrating that stromal p16INK4a activation is triggered by local malignant growth. To assess the universality of stromal p16LUC induction, we transplanted five different, syngeneic nonluminescent tumor cell lines (two breast, one pancreatic, one endometrial, and one lung) harboring diverse oncogenic driver mutations (Lkb1 loss, p53 loss, K-RasG12D, and Her2-Neu overexpression) into p16+/LUC recipients (Figures 5B and S5A). In all cases, luciferase activity localized to the site of tumorigenesis but was not observed in the vehicle-injected control. Furthermore, examination of de novo C3(1)TAg tumors by using luciferase RNA-ISH showed activation of p16LUC in the stroma surrounding early neoplastic lesions (Figure S5B). Together, these results provide evidence that extrinsic signals present in an emerging tumor induce local, non-cell-autonomous p16INK4a expression.
Figure 5. p16LUC Activity Signals Cancer via Non-Cell-Autonomous Mechanisms.

(A) Immune competent, FVB/N p16+/LUC mice were injected orthotopically as depicted with syngeneic p16+/+ C3(1)TAg cells in matrigel or matrigel vehicle alone and imaged upon tumor establishment. A representative 2 min image is shown.
(B) Syngeneic p16+/+ breast (MMTV-HER2/Neu and K14-CRE p53Lox/Lox), pancreatic (Pdx-CRE LSL-K-rasG12D p53Lox/Lox), and endometrial (Sprr2f-CRE Lkb1Lox/Lox) cancer cell lines were injected subcutaneously into p16+/LUC mice and imaged upon tumor establishment. Representative 2 min luminescent images are shown for each model.
(C) Syngeneic transplantations of p16+/LUC bone marrow into p16+/+ recipients were conducted as depicted. Following immune reconstitution, syngeneic p16+/+ C3(1)TAg cells were orthotopically transplanted into the mice and imaged as in (A). Tumor size at the time of imaging is noted. Robust luciferase activity was seen in all transplanted tumors, but not in contralateral sites injected with matrigel only.
See also Figure S5.
To establish whether the host hematopoietic system contributes to the p16LUC signal in transplanted tumors, FVB/n mice underwent lethal irradiation and bone marrow transplantation with total marrow from syngeneic p16+/LUC donors (Figure 5C). Following engraftment, mice were orthotopically injected with syngeneic C3(1)TAg tumor cells lacking the p16LUC allele. Two months after bone marrow transplantation, luciferase imaging revealed strong induction of p16LUC, specifically in the area of tumor formation (Figure 5C). These results indicate that neoplasia either induces p16INK4a expression in infiltrating hematopoietic cells or recruits p16INK4a-expressing bone-marrow-derived cells to the developing tumor.
DISCUSSION
This work establishes the p16LUC knockin allele as a faithful reporter of p16INK4a transcriptional activation in vivo. We have used this system to serially assess the activation of p16INK4a with aging, tissue wounding, breast involution, and neoplastic progression. Surprising findings from these analyses are the lack of association between organismal expression of a cellular senescence marker and cancer-related mortality and the potent, nonautonomous induction of p16INK4a in the benign stroma of a nascent neoplasm.
Expression of p16INK4a has been considered one of the best in vivo markers of cellular senescence. Although activation of p16LUC directly correlated with chronological age in our cohort (Figures 2A and S2A), we observed a surprising variability in total body p16INK4a expression. For example, in 80-week-old mice, total body luminescence differed by a factor of 10 between the highest- and lowest-expressing mice. We used this heterogeneity in expression to directly test whether a common age-associated phenotype, risk of spontaneous malignancy, was predicted by the in vivo activation of p16INK4a, a classical indicator of cellular senescence. In contrast to our expectations, total body luminescence and the rate of change in luminescence were not predictive of mortality (Figures 2A, 2B, 2D–2F, S2A, and S2E–S2J). In accord with prior work (Chrisp et al., 1996; Miller et al., 2011; Wilkinson et al., 2012), all deaths in our cohort appeared to result from malignancy. Therefore, this analysis indicates that total body expression of a marker of cellular senescence is not associated with an important age-associated phenotype: risk of developing cancer.
This finding is not the predicted result. Work from several groups, including our own, has shown that the accumulation of p16INK4a-positive and/or senescent cells in vivo promotes numerous age-related phenotypes (Baker et al., 2008, 2011; Berent-Maoz et al., 2012; Chen et al., 2011; Krishnamurthy et al., 2006; Liu et al., 2011; Molofsky et al., 2006), including tumorigenesis (Bavik et al., 2006; Campisi et al., 2011; Coppé et al., 2008; Kang et al., 2011; Krtolica et al., 2001; Laberge et al., 2012; Liu and Hornsby, 2007; Parrinello et al., 2005). We believe there are two ways to reconcile these prior results with the present work. First, we show that p16INK4a expression reports tissue insults that may not represent cellular senescence (e.g., tissue wounding). It is therefore formally possible that the massive, stereotypical increase in p16INK4a expression noted with aging in all mammals does not reflect the accumulation of senescent cells but rather indicates cellular stresses independent of senescence. This would be surprising to the field given the extensive body of work suggesting that p16INK4a expression is one of the most reliable and robust markers of senescence in vivo in both murine and human tissues (Baker et al., 2011; Edwards et al., 2007; Koppelstaetter et al., 2008; Krishnamurthy et al., 2004; Lemster et al., 2008; Liu et al., 2009; Melk et al., 2004; Nielsen et al., 1999; Signer et al., 2008; Waaijer et al., 2012; Zindy et al., 1997). The finding that p16INK4a does not faithfully report senescence in vivo would undermine the majority of prior work describing senescence in intact organisms, particularly studies in mice. More likely, we believe these data indicate that spontaneous cancer, which was the cause of death in our colony, is not caused by the accumulation of senescent cells and the expression of cytokines related to the senescence-associated secretory phenotype (SASP). A caveat to this analysis is that spontaneous human malignancies differ from cancers of experimentally housed mice in important ways such as time to progression and degree of aneuploidy, and it is possible that SASP cytokines play a more critical role in human tumor progression.
We did not examine age-related phenotypes other than cancer susceptibility and mortality. Therefore, our results do not contradict the view that some age-related phenotypes (e.g., decreased T cell replication [Liu et al., 2011] and sarcopenia [Baker et al., 2011]) are associated with p16INK4a activation, senescence, and the elaboration of SASP factors. For example, Van Deursen and colleagues recently demonstrated the amelioration of several age-associated phenotypes in progeroid mice through the in vivo clearance of senescent cells (Baker et al., 2011), but these measures were not able to extend life span, in accord with our present findings. We continue to believe that the measurement of p16INK4a expression in human tissues will be of clinical value to predict regenerative decline (Koppelstaetter et al., 2008; Liu et al., 2009; McGlynn et al., 2009; Nelson et al., 2012), but our findings represent an important step in reconciling which age-associated phenotypes result from the in vivo accumulation of senescent cells and which (such as development of murine cancers) do not.
We demonstrate that the p16LUC allele is an unexpectedly robust detector of nascent tumorigenesis. Although cancer reporter systems have been published that are tissue specific or require ex vivo transduction (reviewed in O’Neill et al., 2010), the apparent universality and early detection capabilities of this system provide significant advantages. Tumor detection by p16LUC is independent of CRE recombinase expression, allowing this system to be used to monitor carcinogen-induced and spontaneously arising cancers (Figures 3, 4, S3A, and S4A–S4D; data not shown). Unlike previous cancer reporters, p16LUC appears to function robustly in tumors of any genetic background or tissue type. To wit, we have observed activation of the p16INK4a promoter in 14 distinct cancer models, including transgenic autochthonous, transplanted, and spontaneous tumors (see Table S1 and Figures 3, 4, 5, S3, S4, and S5), and we have yet to identify a neoplasm that is not associated with p16INK4a induction. Even using furred, heterozygous p16LUC mice, we noted a distinct spatial and temporal advantage over standard approaches (i.e., FDG-PET and palpation) (Figures 3H–3J), with better results noted in hairless, p16LUC/LUC mice. Preliminary results using tail vein injection of syngeneic cancer cells show that internal tumors can be readily detected by p16LUC (data not shown), suggesting that the reporter will also be of value to studies of metastatic spread. However, we have yet to test the allele in a setting of true de novo metastasis. Given the versatility of p16LUC, we believe this reporter will be generally useful for early cancer detection, serial assessment of tumor progression, and anticancer compound testing.
We believe two features of this system account for its utility in the detection of early cancers. First, prior work has shown that the p16INK4a promoter is unusually dynamic in vivo (Krishnamurthy et al., 2004), and accordingly, we note a >150-fold change in luminescence during the serial passaging of p16+/LUC MEFs (Figure 1E). We believe this massive increase in expression on a per cell basis underlies the allele’s sensitivity, allowing for in vivo detection of extremely small tumors (Figures 3H–3J and S5B). Second, robust induction of p16INK4a in the stroma of developing neoplasia amplifies the tumor-specific signal and occurs regardless of a given tumor’s driver genetics (Figures 5 and S5). The resulting focal, sustained, and intense signal allows emerging neoplasms to be readily distinguished from p16INK4a increases associated with aging and other nonmalignant states (e.g., wound healing) (Figure S3B).
Since the original observation that oncogenes induce senescence in part through upregulation of p16INK4a (Serrano et al., 1997), research has largely focused on cell-intrinsic activators of p16INK4a expression such as DNA damage (Pavey et al., 1999), telomere shortening (Bartkova et al., 2006; Jacobs and de Lange, 2004), production of reactive oxygen species (Ito et al., 2004), and stalled replication forks (Ressler et al., 2006). These results indicate that p16INK4a also responds to extrinsic “field effects” present within every type of emerging neoplasm thus far examined. Additionally, this work speaks to the cell-intrinsic/extrinsic debate that pervades mammalian gerontology as we show that an emerging neoplasm triggers local, non-cell-autonomous expression of a validated effector of cellular senescence. Indeed, non-cell-autonomous factors (e.g., Wnt/β-catenin, Hh/Gli, PDGF, and IL-7) have been reported to regulate p16INK4a expression as a potential means of developmental control (Berent-Maoz et al., 2012; Bishop et al., 2010; Chen et al., 2011). Whether such extrinsic signals also serve to induce p16INK4a expression and cellular senescence in the context of normal aging remains to be established.
In summary, the p16LUC system represents a substantial advance for cancer and aging studies that use the mouse as a model system. The system also allows for simple, serial, and noninvasive assessment of expression of p16INK4a in vivo, which is a critical step in the activation of cellular senescence. The allele’s utility reflects the highly dynamic range of p16INK4a expression in vivo as well as the unexpected finding of p16INK4a activation in tumor-associated, nonmalignant stroma, thereby allowing the allele to report emergence of every tumor type tested, regardless of the underlying tumor genetics. As cellular senescence is a principal barrier to tumorigenesis and contributes to mammalian aging, the p16LUC system will be a valuable asset for the serial assessment of cancer and aging, as well for the testing of pharmacological interventions intent on altering these processes in vivo.
EXPERIMENTAL PROCEDURES
Husbandry and Generation of the p16LUC Allele
Animal work was conducted in accordance with protocols approved by the institutional care and use committee for animal research at the University of North Carolina. Standard homologous recombination procedures were used to target firefly luciferase, followed by an SV40 polyadenylation site to exon 1α of the endogenous p16INK4a gene. The primers and conditions used for p16LUC genotyping were as follows: p16-LUC-F 5′-CTATGGCGGGCTGTG GAG-3′ (0.15 μM); p16-LUC-R 5′-CACGGTAGGCTGCGAAATG-3′ (0.15 μM), p16-LUCR3 5′ 3′ (0.2 μM); 95°C for 15 min, 36× [94°C for 30 s, 58°C for 30 s, and 72°C for 45 s], 72°C for 2 min. The resulting PCR products are 312 (wild-type) and 543 (p16LUC) base pairs long.
Cell Isolation, Culture, and Detection of Endogenous Ink4a/Arf Transcripts
MEFs were generated from p16LUC heterozygous parents by using a Gentle-MACS dissociator (Miltenyi Biotec, Germany). Briefly, embryos were harvested, chopped coarsely in 0.5% trypsin-EDTA, and then allowed to digest for 5–10 min. Cells were then placed into a GentleMACS C-tube, and program “A” was used twice in succession to generate single-cell suspensions. A similar protocol was used to generate tumor cell lines from the C3(1)TAg and TRIA models.
MEFs, C3-TAg, PDAC1 (Bardeesy et al., 2006), and Kp53 (murine lung adenocarcinoma cells, gift of W.Y. Kim, UNC) cells were cultured in DMEM supplemented with 4.5g/l glucose, 10% fetal bovine serum (FBS), 1% penicillin streptomycin, and 2 mM L-glutamine. Her2/Neu-overexpressing NT2 cells were cultured in RPMI-1640 supplemented with 20% FBS, 2 mM L-glutamine, 12 mM HEPES, 0.1 mM NEAA, 1 mM sodium pyruvate, 1% penicillin streptomycin, 50 μM β-mercaptoethanol, and 0.2 units/ml Novolin R-insulin. Endometrial Lkb1 null tumor lines derived from metastatic tumors in Sprr2f-CRE Lkb1Lox/Lox females (Contreras et al., 2010) were cultured in DMEM with low glucose supplemented with 110 mg/l pyruvate, 10% FBS, 2 mM L-glutamine, and 1× antibiotic antimycotic (15240, Invitrogen).
RT-PCR strategies for the detection of Ink4a/Arf transcripts were previously described (Burd et al., 2010; Krishnamurthy et al., 2004). Western blots employed the following antibodies and were quantified on a LICOR Odyssey system: p16INK4a (Santa Cruz M126; 1:500), p19ARF (Abcam AB80; 1:1,000).
In Vivo and In Vitro Luciferase Detection
Isoflurane-anesthetized mice were injected intraperitoneally with D-luciferin substrate (Caliper Life Sciences; 15 mg/ml in PBS) and were imaged by using an IVIS Lumina or IVIS Kinetic system (Caliper Life Sciences). Peak luminescence was determined to begin 6–8 min after substrate administration. To ensure that analysis was performed at the same time point, a set sequence of sequential images between 10 s and 2 min in length was begun immediately after intraperitoneal injection of D-luciferin. A wide-angle lens was used to simultaneously capture images from >3 animals. Images were taken by using a binning of 4 (medium). For tumor studies in which mice were approximately the same size, 300 μl of substrate was administered. In aging studies, in which animal size varied, mice were injected with 10 μl/g body weight of substrate. Living Image Software (Caliper Life Sciences) was used to compare multiple images taken at the same exposure and time point following substrate administration. For all analyses, an additional ROI was employed to normalize for background luminescence on each image. In vitro luciferase assays were conducted by using a standard approach and were normalized for cell number. RNA-ISH was conducted by using RNAscope 2.0 technology as described by the manufacturer (Advanced Cell Diagnostics).
Analysis of Luminescence with Aging
Beginning at 16 weeks of age (adulthood), mice were imaged bimonthly in groups of three to five, as above. Analysis was conducted on 2 min ventral images taken 6 min after D-luciferin administration. The contour of each mouse was traced, and the flux/pixel was determined by using Living Image Software (Caliper Life Sciences). Flux/pixel was also calculated for an unoccupied region of the image and subtracted from the traced area as background. Images were excluded from the analysis if a mouse had visible wounds. Animals were only allowed to re-enter the study upon complete wound resolution. Focal luminescent intensity in the mesenteric and cervical lymph nodes was common in healthy mice of all ages. However, intense luminescent signal in other areas of the body was often indicative of tumor formation; therefore, these images were excluded from analysis (see example in Figure 2B). Never did these “tumor regions” resolve. As such, these mice never re-entered the study. Of the 32 p16+/LUC mice in this study, the number excluded or dead at each time point was the following: 16 weeks, 0; 24 weeks, 1; 32 weeks, 1; 40 weeks, 2; 48 weeks, 2; 56 weeks, 2; 64 wks, 3; 72 weeks, 7; and 80 weeks, 12. Kaplan-Meier analysis showed no significant difference between the life span of p16+/+ and p16+/LUC mice in our cohort (Figure S2K), suggesting that p16INK4a heterozygous animals were as susceptible to cancer-related death as their wild-type counterparts in this study.
Genetically Engineered Tumor Models
Female C3(1)TAg animals express SV40 large T-antigen (TAg) from the rat C3 promoter and develop mammary tumors with transcriptional profiles resembling human basal-like breast cancers (Herschkowitz et al., 2007; Maroulakou et al., 1994) as well as salivary and mixed eccrine sweat gland tumors of the paws (Maroulakou et al., 1999). TRIA mice are germline null for the Ink4/Arf locus and express oncogenic H-RasG12V from the melanocyte-specific tyrosinase promoter (Chin et al., 1997) (NCI Mouse Repository strain 01XB1). TRIA mice analyzed in this work were Arf+/− and were therefore not fully deficient for Arf function (see Results). The Eμ-Myc model was obtained from Jackson Laboratories (Stock 002728) (Harris et al., 1988). The Lkb1−/−p53−/− LSL-K-RasG12D melanoma model has been previously described (Liu et al., 2012). The B-RafV600E Pten−/− melanoma model was obtained from Jackson Laboratories (Stock 013590) (Dankort et al., 2009). Following crosses with p16LUC, the C3(1)TAg and TRIA models were 50% C57Bl/6 and 50% FVB/n. Eμ-Myc p16LUC mice were fully backcrossed to albino C57Bl/6. The Lkb1−/− p53−/− LSL-K-RasG12D and B-RafV600E Pten−/− melanoma models were analyzed on a mixed background.
Syngeneic Tumor-Grafting Experiments
Cultured tumor cells were harvested, washed in Hank’s Balanced Salt Solution (HBSS), and resuspended at the following concentrations in either matrigel (M; BD Biosciences) or HBSS (H): 8.0 × 105 C3(1)Tag cells/100 μl injection (M); 5.0 × 105 NT2 cells/100 μl (M); 5.0 × 105 PDAC1 cells/100 μl injection (H); 5.0 × 105 endometrial cancer cells/100 μL injection (M); 5.0 × 105 KP53 cells/100 μl (H); 8.0 × 105 MEFs cells/100 μL (H). T11 cells were maintained through serial passaging in wild-type BALB/c mice. For each passage, ~5 × 105 cells resuspended in 100 μl of a 1:1 Matrigel:HBSS solution were injected subcutaneously.
Bone Marrow Transplantation
Fifteen 10- to 12-week-old wild-type FVB/n mice were lethally irradiated (8 Gy) by using a cesium source. The same day, bone marrow from a p16LUC heterozygous FVB/n mouse was isolated, washed, and resuspended in HBSS. Cells were injected intravenously at a concentration of 2 × 106 cells/100 μl injection. Two to three weeks after transplant, mice were orthotopically injected with tumor cells as described above. Prior to sacrifice due to advancing tumor burden, mice were imaged to detect luciferase activity, and blood was harvested. RT-PCR genotyping of blood DNA was compared to a standard curve of mixed wild-type and p16LUC heterozygous blood to determine chimerism following transplantation. Blood chimerism was found to be >89% p16+/LUC positive at the time of imaging.
PET Imaging
FDG-PET imaging was performed as previously described (Liu et al., 2011).
Supplementary Material
Acknowledgments
We thank H. Yuan, K. Guley, and the UNC BRIC Small Animal Imaging Facility for imaging assistance; A. Moisan (MGH Cancer) for technical assistance; C.G. Peña (UT Southwestern), E.A. Akbay (UT Southwestern), D.T. Gammons (UNC), W.Y. Kim (UNC), and W. Liu (UNC) for providing reagents; the UNC Animal Clinical Chemistry, Animal Histopathology, and Gene Expression Laboratories, as well as G. Aloisio (UT Southwestern), for tissue processing and routine genotyping; the UNC Microscopy Services Laboratory and UNC Animal Studies facility for technical assistance; and the UNC Mouse Phase I unit for providing animals and expertise. This work was supported by the NIH (AG024379; CA137181; CA163896; AG036817), the Paul Glenn Foundation, and the Burroughs Wellcome Fund.
Footnotes
Supplemental Information includes five figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2012.12.010.
References
- Austad SN. Comparative aging and life histories in mammals. Exp Gerontol. 1997;32:23–38. doi: 10.1016/s0531-5565(96)00059-9. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- Berent-Maoz B, Montecino-Rodriguez E, Signer RA, Dorshkind K. Fibroblast growth factor-7 partially reverses murine thymocyte progenitor aging by repression of Ink4a. Blood. 2012;119:5715–5721. doi: 10.1182/blood-2011-12-400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CL, Bergin AM, Fessart D, Borgdorff V, Hatzimasoura E, Garbe JC, Stampfer MR, Koh J, Beach DH. Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A) Mol Cell. 2010;40:533–547. doi: 10.1016/j.molcel.2010.10.027. [DOI] [PubMed] [Google Scholar]
- Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011;21:354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, Horner JW, II, DePinho RA. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11:2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisp CE, Turke P, Luciano A, Swalwell S, Peterson J, Miller RA. Lifespan and lesions in genetically heterogeneous (four-way cross) mice: a new model for aging research. Vet Pathol. 1996;33:735–743. doi: 10.1177/030098589603300620. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, Bardeesy N, Takahashi M, Settleman J, Wong KK, et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech. 2010;3:181–193. doi: 10.1242/dmm.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, Prolla TA. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007;8:80. doi: 10.1186/1471-2164-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR. Cosmic 2005. Br J Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M, Pisc C, Branda J, Ionescu-Tiba V, Nikolic Z, Yang C, Wang T, Shackleford GM, Cardiff RD, Schmidt EV. Regulation of cyclin D1 and p16(INK4A) is critical for growth arrest during mammary involution. Cancer Res. 2001;61:8811–8819. [PubMed] [Google Scholar]
- Goldman SJ, Chen E, Taylor R, Zhang S, Petrosky W, Reiss M, Jin S. Use of the ODD-luciferase transgene for the non-invasive imaging of spontaneous tumors in mice. PLoS ONE. 2011;6:e18269. doi: 10.1371/journal.pone.0018269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol. 2004;14:2302–2308. doi: 10.1016/j.cub.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell. 2012;11:727–731. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Koppelstaetter C, Schratzberger G, Perco P, Hofer J, Mark W, Ollinger R, Oberbauer R, Schwarz C, Mitterbauer C, Kainz A, et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell. 2008;7:491–497. doi: 10.1111/j.1474-9726.2008.00398.x. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemster BH, Michel JJ, Montag DT, Paat JJ, Studenski SA, Newman AB, Vallejo AN. Induction of CD56 and TCR-independent activation of T cells with aging. J Immunol. 2008;180:1979–1990. doi: 10.4049/jimmunol.180.3.1979. [DOI] [PubMed] [Google Scholar]
- Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, Thomas NE, Sharpless NE. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8:439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, Sharpless NE. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117:3257–3267. doi: 10.1182/blood-2010-09-304402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Monahan KB, Pfefferle AD, Shimamura T, Sorrentino J, Chan KT, Roadcap DW, Ollila DW, Thomas NE, Castrillon DH, et al. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell. 2012;21:751–764. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci USA. 1994;91:11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Shibata MA, Anver M, Jorcyk CL, Liu Ml, Roche N, Roberts AB, Tsarfaty I, Reseau J, Ward J, Green JE. Heterotopic endochondrial ossification with mixed tumor formation in C3(1)/Tag transgenic mice is associated with elevated TGF-beta1 and BMP-2 expression. Oncogene. 1999;18:5435–5447. doi: 10.1038/sj.onc.1202926. [DOI] [PubMed] [Google Scholar]
- McGlynn LM, Stevenson K, Lamb K, Zino S, Brown M, Prina A, Kingsmore D, Shiels PG. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging Cell. 2009;8:45–51. doi: 10.1111/j.1474-9726.2008.00447.x. [DOI] [PubMed] [Google Scholar]
- Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004;65:510–520. doi: 10.1111/j.1523-1755.2004.00438.x. [DOI] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. 2003;163:477–491. doi: 10.1016/s0002-9440(10)63677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JA, Krishnamurthy J, Menezes P, Liu Y, Hudgens MG, Sharpless NE, Eron JJ., Jr Expression of p16(INK4a) as a biomarker of T-cell aging in HIV-infected patients prior to and during antiretroviral therapy. Aging Cell. 2012;11:916–918. doi: 10.1111/j.1474-9726.2012.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen GP, Stemmer-Rachamimov AO, Shaw J, Roy JE, Koh J, Louis DN. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest. 1999;79:1137–1143. [PubMed] [Google Scholar]
- O’Neill K, Lyons SK, Gallagher WM, Curran KM, Byrne AT. Bioluminescent imaging: a critical tool in pre-clinical oncology research. J Pathol. 2010;220:317–327. doi: 10.1002/path.2656. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Imamura Y, Yamakoshi K, Hirota F, Nakayama R, Kubo Y, Ishimaru N, Takahashi A, Hirao A, Shimizu T, et al. Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc Natl Acad Sci USA. 2007;104:15034–15039. doi: 10.1073/pnas.0706949104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey S, Conroy S, Russell T, Gabrielli B. Ultraviolet radiation induces p16CDKN2A expression in human skin. Cancer Res. 1999;59:4185–4189. [PubMed] [Google Scholar]
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Dürr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- Safran M, Kim WY, O’Connell F, Flippin L, Günzler V, Horner JW, Depinho RA, Kaelin WG., Jr Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA. 2006;103:105–110. doi: 10.1073/pnas.0509459103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- Signer RA, Montecino-Rodriguez E, Witte ON, Dorshkind K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes Dev. 2008;22:3115–3120. doi: 10.1101/gad.1715808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Nerio E, Holland EC. Dissecting tumor maintenance requirements using bioluminescence imaging of cell proliferation in a mouse glioma model. Nat Med. 2004;10:1257–1260. doi: 10.1038/nm1120. [DOI] [PubMed] [Google Scholar]
- Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, Blow MJ, Cohen JC, Rubin EM, Pennacchio LA. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijer ME, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, de Craen AJ, Sedivy JM, Westendorp RG, Gunn DA, Maier AB. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell. 2012;11:722–725. doi: 10.1111/j.1474-9726.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell. 2009;34:271–284. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya H, et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol. 2009;186:393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, Roussel MF, Sherr CJ. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci USA. 2003;100:15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.