

Abstract
The year 2011 marks the centenary of Francis Peyton Rous’s landmark experiments on an avian cancer virus. Since then, seven human viruses have been found to cause 10–15% of human cancers worldwide. Viruses have been central to modern cancer research and provide profound insights into both infectious and non-infectious cancer causes. This diverse group of viruses reveals unexpected connections between innate immunity, immune sensors and tumour suppressor signalling that control both viral infection and cancer. This Timeline article describes common features of human tumour viruses and discusses how new technologies can be used to identify infectious causes of cancer.
The burden of viral infections in cancer is high but underappreciated by much of the cancer research community. The International Agency for Research on Cancer estimates that one in five cancer cases worldwide are caused by infection, with most caused by viruses1,2. These cancers are particular public health problems for the developing world, as well as for underserved and immunosuppressed populations in developed countries. Most importantly, these cancers have readily identifiable targets for diagnosis, prevention and therapy. Vaccination programmes against two human tumour viruses, hepatitis B virus (HBV) and human papillomavirus (HPV), have already begun to alter age-old cancer patterns on an international scale3-5. In this Timeline article, we provide an overview of the major milestones in research on viruses and human cancer (TIMELINE) and highlight common features among the human cancer viruses (TABLE 1). Non-viral infectious causes for human cancer are reviewed elsewhere6,7.
Timeline.

Some major discoveries and events during the first century of tumour virology
Table 1.
The human cancer viruses
| Virus | Genome | Notable cancers | Year first described |
Refs |
|---|---|---|---|---|
| Epstein–Barr virus (EBV; also known as human herpesvirus 4 (HHV4)) |
Double-stranded DNA herpesvirus | Most Burkitt’s lymphoma and nasopharyngeal carcinoma, most lymphoproliferative disorders, some Hodgkin’s disease, some non-Hodgkin’s lymphoma and some gastrointestinal lymphoma |
1964 | 15 |
| Hepatitis B virus (HBV) | Single-stranded and double-stranded DNA hepadenovirus |
Some hepatocellular carcinoma | 1965 | 25 |
| Human T-lymphotropic virus-I (HTLV-I) |
Positive-strand, single-stranded RNA retrovirus |
Adult T cell leukaemia | 1980 | 20 |
| High-risk human papillomaviruses (HPV) 16 and HPV 18 (some other α-HPV types are also carcinogens) |
Double-stranded DNA papillomavirus |
Most cervical cancer and penile cancers and some other anogenital and head and neck cancers |
1983–1984 | 29, 30 |
| Hepatitis C virus (HCV) | Positive-strand, single-stranded RNA flavivirus |
Some hepatocellular carcinoma and some lymphomas |
1989 | 31 |
| Kaposi’s sarcoma herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) |
Double-stranded DNA herpesvirus | Kaposi’s sarcoma, primary effusion lymphoma and some multicentric Castleman’s disease |
1994 | 33 |
| Merkel cell polyomavirus (MCV) | Double-stranded DNA polyomavirus | Most Merkel cell carcinoma | 2008 | 34 |
Discovery of tumour viruses
On 1 October 1909, Francis Peyton Rous began his famous cancer virus transmission experiments at the Rockefeller Institute, USA, on a 15-month-old barred Plymouth Rock hen that had been brought to him by a farmer from Long Island, New York, USA. The chicken had a sarcomatous chest tumour that Rous successfully transplanted into other chickens that were related to the same brood8,9. By 1911, he had shown that the cancer could be transmitted through cell-free tumour extracts and thus must be caused by a small transmissible agent, probably a virus. These experiments knowingly built on the pioneering work of two Danish scientists, Vilhelm Ellerman and Oluf Bang (FIG. 1), who published similar results in 1908 on the viral transmission of avian erythroblastosis10. As cancers are not contagious, viral causes for chicken cancer were shortly thereafter relegated to being scientific curiosities11. Rous gave up his studies on viral cancers 4 years later and little further progress was made in tumour virology until the 1930s when mammalian tumour viruses began to be described12,13. Rous eventually returned to viral tumour biology through his studies with Richard Shope on the cottontail rabbit papillomavirus in 1934. These studies led to investigations of the co-carcinogenic effects of coal tars on virus-induced tumours. Interest in viral causes for cancer redoubled in the early 1950s following Ludwik Gross’s discovery of an acutely transforming murine retrovirus (BOX 1) and a polyomavirus that caused murine tumours14. The culmination of this first century of tumour virology would be celebrated with Nobel Prizes awarded in 2008 for the discovery by Harald zur Hausen of high-risk HPV strains that cause cervical cancer and the discovery of HIV, an agent that does not initiate cancer but indirectly ‘sets the stage’ for malignancy through immuno suppression, by François Barré-Sinoussi and Luc Montagnier.
Figure 1. Historical figures.

a | The chicken tumour that started it all. This photograph from 1909 shows a sarcoma on the external chest wall of a chicken that was used by Francis Peyton Rous to discover the Rous sarcoma retrovirus8. b | Vilhelm Ellerman (1871–1924), shown on the left, and his assistant, Oluf Bang (1881–1937), shown on the right. These Danish scientists first succeeded in transmitting a leukaemia-inducing avian retrovirus in 1908. These experiments formed a basis for Rous’s subsequent experiments showing a viral cause of a solid cancer. Images courtesy of Medical Museion, Copenhagen University, Denmark.
Box 1. The long, strange trip of retroviruses in cancer.
“One can scarcely suppose that a horde of viruses, each with its more or less limited potentialities, are passed along together in the ovum or sperm, as generation succeeds generation, or that they reach the young organism by way of the uterus or milk.” Francis Peyton Rous, 1960 (REF. 171)
Tumorigenic retroviruses have been central to cancer biology, leading to the development of focus formation assays172, discovery of reverse transcription173,174, identification of more than 20 cellular oncogenes175-177, and ultimately Nobel Prize recognition for Rous 57 years after his initial experiments. The Ellerman and Bang erythroblastosis virus and the Rous sarcoma virus are independently derived from avian leukosis virus (ALV), a simple retrovirus11,178. Simple retroviruses can become carcinogenic by recombination with cell-derived oncogenes (SRC in the case of Rous sarcoma virus and probably ERBB2 in the case of the Ellerman and Bang virus), which disrupt the viral genome, usually rendering it non-infectious or by insertional mutagenesis. ALV is endemic among chickens, but Rous sarcoma virus is not and most strains require co-infection with a helper retrovirus to be transmitted11. Discovery of endogenous retroviral sequences by Robin Weiss and colleagues179 suggested that simple retroviruses (endogenous retroviruses (ERV)) could re-emerge from the host genome, raising a perplexing possibility that some oncogenic retroviruses arose during serial cancer transplantation experiments with the unwitting aid of early experimentalists180,181. Ironically, tumour viruses helped to identify cellular oncogenes177 and tumour suppressor genes182,183, giving rise to the successful somatic mutation theory of cancer that no longer required the confusing biology of tumour viruses. By the 1970s, as Anders Valhne recalls, “...the notion of human cancer viruses became in ill repute and rather than talking of ‘human tumour viruses’ people in science talked of ‘human rumor viruses’.” (REF. 24) Discovery of a complex cancer-causing human retrovirus (human T-lymphotropic virus-I (HTLV-I)) in 1980 (REFS 20,24) came too late to fully rehabilitate their reputation.
But the possibility for simple retroviral involvement in human cancer persists. The exogenous ovine Jaagsietke retrovirus has evolved over the past several hundred years from an ERV to cause a transmissible cancer of in-bred sheep — most famously in the cloned sheep Dolly184. Spontaneously arising human ERV-derived retroviruses might similarly contribute to sporadic human cancers but would be difficult to detect and non-transmissible185. Further, a recent candidate human cancer virus is a simple retrovirus that is controversially associated with prostate cancer and chronic fatigue syndrome186-188. Time, careful epidemiology and careful experimentation will determine the role of these viruses in human cancer.
Human cancer viruses
The first human tumour virus, Epstein–Barr virus (EBV; also known as human herpesvirus 4 (HHV4)), was not described until 53 years after Rous’s initial experiments. Anthony Epstein, Bert Achong and Yvonne Barr used electron microscopy in 1964 to identify EBV particles in cell lines from African patients with Burkitt’s lymphoma15. The unusual geographic distribution of Burkitt’s lymphoma had suggested a novel environmental cause, such as a viral infection, which was confirmed by these early virological studies. This spurred other electron microscopy-based searches for human cancer viruses that largely turned out to be fruitless, as cancer viruses generally do not replicate to form virions in tumours (discussed below). The discovery of EBV also sparked contention among cancer biologists because of the difficulty in reconciling the near ubiquitous infection of adults with EBV and the fact that EBV-associated cancers are uncommon16. Furthermore, although Burkitt’s lymphoma tumours from African and New Guinean patients are almost always positive for EBV, sporadic Burkitt’s lymphomas in developed countries frequently lack EBV but retain signature MYC–IgH or MYC–IgL translocations17. This would require a rethinking of disease causality, as EBV does not follow the Galilean principles of causality 18, which require that a virus must be both necessary and sufficient to be the cause of cancer (BOX 2).
Box 2. The EBV-cancer paradox.
Resolving the Epstein–Barr virus (EBV)–cancer paradox — that a common infection can cause a rare cancer — required the recognition that chronic viral infection functions together with multifactorial non-viral risks to contribute to cancer. The well-known Koch’s postulates are not applicable to viruses such as EBV, which generally cannot be isolated as pure cultures in vitro or used to re-infect susceptible laboratory animals189,190. Hill’s criteria191, used to determine the relationship between cigarette smoking and lung cancer, work well for uncommon agents, such as Kaposi’s sarcoma herpesvirus (KSHV) that causes Kaposi’s sarcoma108, but also have unstated pathobiological biases that limit their use in tumour virology. Subsequent to its discovery, Werner and Gertrude Henle and colleagues showed that EBV infection can immortalize primary B cells192, a property that is unique to EBV; its oncogenes were characterized193; tumour-viral clonality was established94; and additional examples of EBV-associated cancers and lymphoproliferative diseases were described194. From the perspective of Bayesian reasoning, the posterior probability that EBV causes cancer was strengthened by the accumulating clinical and basic data from various sources that ultimately left little doubt that EBV has a causal role in specific tumours. Nonetheless, more than 30 years passed from its discovery until it was officially declared a human carcinogen by an international cancer agency195.
Six more cancer viruses have been discovered, in addition to EBV (TABLE 1), which are now widely accepted as causes for invasive human tumours. Additional candidates are continuously proposed but their roles in human cancer remain controversial and unclear. This list is small but we can still draw at least one startling conclusion from it: human cancer viruses do not fall into a single viral class. Much of the past century was devoted to the search for simple human cancer retroviruses similar to the Rous sarcoma virus (BOX 1), but the only retroviruses associated with human cancer are the complex retroviruses: human T-lymphotropic virus-I (HTLV-I) and HIV-1 and HIV-2. HIV does not directly cause cancer but it is frequently included as a cancer-causing agent by virtue of its induction of immunodeficiency, which promotes the development of cancers caused by other viruses2.
A second surprise is that human cancer viruses span the entire range of virology and include complex exogenous retroviruses (such as HTLV-I), positive-stranded RNA viruses (such as hepatitis C virus (HCV)), DNA viruses with retroviral features (such as HBV) and both large double-stranded DNA viruses (such as EBV and Kaposi’s sarcoma herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) and small double-stranded DNA viruses (such as HPV and Merkel cell polyomavirus (MCV)). There is no obvious molecular rule that either firmly establishes or eliminates an agent as a potential human tumour virus a priori. Also, almost all of the tumour viruses have close relatives that do not cause human cancer. This leads to the conclusion that almost every virus has the potential to cause cancer but only a very small proportion actually do so.
As suggested above, traditional virological techniques have had limited success in identifying human cancer viruses. EBV virions were identified by cell culture and electron microscopy19 but this was followed by only one other human tumour virus, HTLV-I20. HTLV-I was discovered by Bernard Poiesz, Robert Gallo and colleagues in 1980 (and shortly after confirmed by Yorio Hinuma, Isao Miyoshi and collaborators21) from cell lines established from a case of the newly described adult T cell leukaemia/lymphoma (ATLL) syndrome that was previously diagnosed as mycosis fungoides20,22-24. This virus was sought by searching for reverse transcriptase activity in a survey of T cell lines, which was partly initiated owing to the earlier discovery that a gibbon retrovirus causes T cell leukaemia. By contrast, HBV, discovered shortly after EBV in the mid-1960s and leading to a Nobel Prize for Baruch Blumberg in 1976, has only recently been successfully propagated in culture and was first linked by serology to acute hepatitis rather than to cancer25,26. The role of HBV in hepatocellular carcinoma was established more than a decade later by Beasley et al.27 through longitudinal studies of Taiwanese insurance company cohorts.
The remaining four viruses were discovered as genetic elements using molecular biology, rather than virology, techniques. Since the 1840s, sexual activity had been suspected to be a risk factor for cervical cancer, and Harald zur Hausen reasoned that papillomaviruses might contribute to this cancer owing to their role in sexually transmitted genital warts28. He and his colleagues cross-hybridized known papillomavirus DNA to cervical cancer DNA, discovering two novel high-risk papillomaviruses genotypes (HPV-16 and HPV-18) in the early 1980s29,30 that were subsequently confirmed to be present in most cervical cancers. Similar to HBV, HPV also propagates poorly in culture in most cell types although it is maintained as an integrated, non-productive virus in HeLa cells29,30. In the late 1980s, Qui-Lim Choo, Michael Houghton, Daniel Bradley and colleagues sought additional causes for transfusion-transmitted hepatitis (known as the nonA-nonB hepatitis virus) by antibody panning of a randomly primed cDNA library made from the sera of experimentally infected chimpanzees. This led, in 1989, to the isolation of genome fragments from the flavivirus HCV31, which was then immediately exploited by multiple groups to show that this new virus, like HBV, is associated with hepatocellular carcinoma. HCV was only recently propagated in cell lines32.
We specifically sought out viral causes for Kaposi’s sarcoma and Merkel cell carcinoma (MCC), discovering KSHV in 1994 (REF. 33) and MCV (together with Huicheng Feng from our group) in 2008 (REF. 34). These discoveries were both based on nucleic acid subtraction, although the actual experimental approaches were very different. KSHV was identified by physical DNA subtraction (representational difference analysis)35 using Kaposi’s sarcoma and healthy tissue genomes from the same patient. By contrast, MCV sequences were identified by computational subtraction of cDNA sequence data using digital transcriptome subtraction (DTS)36, a technique developed by us over a 10-year period and independently developed by others37. Applying DTS to MCC revealed viral sequences belonging to a new human polyomavirus. Like most other human cancer viruses, MCV and KSHV are not typically transmissible from the cancers that they help to induce.
These examples show how traditional approaches that are used by virologists, such as virus culture and electron microscopy, often fail in tumour virology. Another difficulty to understanding viruses in human cancer has been the slow realization that virus infection alone is never sufficient for tumorigenesis, an unsurprising fact that is also true for non-neoplastic viral diseases. Only in a few specific cases, such as KSHV in Kaposi’s sarcoma and HPV in cervical cancer, can particular viruses be assumed to be necessary, as they are universally present in these tumours. HBV, HCV and chemical carcinogens each contribute to the total attributable liver cancer risk, but none of these factors alone is required for liver cancer38. Thus, the concept of a virus being both necessary and sufficient as the cause of a cancer is too simplistic to be useful in modern cancer research. As cancer is a complex multistep process, it is now obvious that many molecular events, including virus infection, function together to generate the transformed cellular phenotype39-41.
Immunity is an external factor that has particular importance in determining whether a cancer occurs after exposure to a potential tumour virus. This can be seen with signalling lymphocytic activation molecule-associated protein (SAP) mutations in males that cause immunodeficiency and X-linked lymphoproliferative syndrome after EBV infection42. Kaposi’s sarcoma, first described in 1872 (REF. 43), also illustrates the importance of immunity to control the expression of a viral cancer. KSHV co-speciated with humans 80 million years ago44 but infects only ~3% of healthy North Americans45-50. Before the AIDS epidemic, KSHV caused less than three Kaposi’s sarcoma cases per year per million people in the United States, but rates of Kaposi’s sarcoma soared tens of thousands-fold among people with AIDS after the emergence of immune suppression owing to the HIV pandemic51.
Indirect versus direct carcinogenesis
Infectious cancer agents (including, viruses, bacteria and parasites) have been divided into two broad categories: direct carcinogens, which express viral oncogenes that directly contribute to cancer cell transformation, and indirect carcinogens that presumably cause cancer through chronic infection and inflammation, which eventually leads to carcinogenic mutations in host cells52,53. This is a useful description for infectious cancer causes that will undoubtedly change as knowledge accumulates. By definition, a direct viral carcinogen is present in each cancer cell and expresses at least one transcript to maintain the transformed tumour cell phenotype, as occurs with HPV-, MCV-, EBV- and KSHV-related cancers. Evidence supporting this comes from knockdown studies in which the loss of viral proteins results in the loss of host cancer viability54-60. Indirect carcinogens (most notably, the Helicobacter pylori bacterium) could potentially also include ‘hit-and-run’ viruses in which the viral genes are lost as the tumour begins to mature, although good examples of this process have not been documented to date.
Several agents (such as HBV, HCV and HTLV-I), however, do not fit neatly into either the indirect or the direct carcinogen categories. Hepatocellular carcinoma (HCC) generally arises after prolonged liver cirrhosis from chronic virus-induced cell death and regeneration61-63. HBV is clonally integrated into the genomes of tumour cells in almost all HBV-related cancers, but it is not clear whether persistent HBV (or HCV) gene expression is required for HCC cell proliferation61. HTLV-I, like most direct carcinogens, is present as a clonal infection of ATLL, but expression of its putative oncogene v-tax is frequently absent in the mature leukaemia or lymphoma cells64. Transgenic models reveal that various proteins from these viruses, including the HBX protein from HBV, NS5 protein from HCV and TAX from HTLV-I can initiate oncogenic transformation62,65. Thus, for these viruses it remains unclear whether specific viral products maintain mature tumour cells, promote a precancerous cell phenotype or contribute to cancer solely through prolonged infection and chronic inflammation62,65. Bacterial carcinogens can also have features that are reminiscent of direct carcinogens66, showing that the simple dichotomization of direct and indirect carcinogens is probably inadequate.
The indirect versus direct paradigm is nonetheless very useful as it guides our thinking about which cancers are most likely to harbour a new human cancer virus. Cancers that are related to immunosuppression are candidates for being caused by tumour viruses67. Loss of surveillance for specific viral cytotoxic T cell epitopes without generalized immunosuppression, as might occur during ageing, is also likely to promote cancers that are caused by viruses68,69. This makes intuitive sense, particularly for direct carcinogens, as they must express at least one foreign protein in each cancer cell, but even cancers caused by indirect infectious carcinogens have an increased occurrence in immunosuppressed populations67. In a classic epidemiological study, Beral et al.70 used this knowledge of direct and indirect cancer causation on registry data of patients with AIDS to correctly predict most of the major epidemiological features for the virus (KSHV) that caused Kaposi’s sarcoma before its actual discovery. Similarly, analyses by Engels et al.71 of data of patients with AIDS focused our attention on MCC as possessing a potentially infectious origin. Other immunosuppression-related tumours, including non-melanotic skin tumours, EBV-negative Hodgkin’s disease and non-Hodgkin’s lymphomas, are promising candidates for future cancer virus discovery72,73.
This paradigm not only suggests where to look for human cancer viruses but also how to look for them. A cell possesses approximately 200,000 mRNA transcripts, and methods to sequence cDNA substantially beyond this level are readily available. If a direct carcinogen is present and expresses a foreign oncoprotein, carrying out DTS on cDNA from a monomorphous tumour specimen is likely to sequence a viral gene. There are technical difficulties with this approach (particularly in recognizing that a transcript belongs to a novel virus rather than being a missequenced or unannotated human transcript, as outlined elsewhere36) that place constraints on sequencing-based virus discovery. As genomic databases improve, molecular distinctions between self and non-self genomes will become more precise and easier to detect. Equally importantly, sequencing technologies can help to exclude a direct carcinogen if it is not present in a cancer. Deep sequencing of four human mesothelioma tumour cDNAs failed to identify SV40 viral transcripts74, providing evidence against a long-standing hypothesis that SV40, a rhesus polyomavirus, is directly involved in the development of mesothelioma75. Although this does not exclude SV40 as a cause of human cancer, this virus would have to do so in mesotheliomas through a new and undescribed mechanism. Tumour trasnscriptome sequencing can also be paired with sequencing of the appropriate control tissues to determine cancer cell gene expression. Therefore, if properly carried out, even negative searches for viral sequences can provide useful clues about the origins of human cancer.
Latency and pseudo-latency
A common feature for human tumour viruses is that they are persistent latent or pseudo-latent infections that generally do not replicate to form infectious virus particles in tumours. All of the viruses in TABLE 1 have the capacity to form virions and become transmissible at some point in their natural lifecycles, but within tumours these infections are generally latent so that productive virus replication (also known as lytic replication) is either diminished or absent76. Viral latency serves as an immune evasion strategy allowing the virus to hide from the immune system by turning off unnecessary viral proteins that might be sensed by cell-mediated immune recognition. The virus persists as a naked nucleic acid, often as a plasmid or episome, which relies on host cell machinery to replicate whenever the cell divides. Viral latency should not be confused with clinical latency, which means asymptomatic infection. Latent viral infections can be symptomatic, as in viral cancers, and active lytic viral replication can be relatively asymptomatic, as occurs during the prodromal phases of HIV or HCV infection. As early as the 1970s, investigators recognized an inverse relationship between virus replication, or permissiveness, and cell transformation for tumour viruses. SV40, for example, transforms human cells efficiently only when mutations are introduced into its replication origin to prevent viral replication77. The discovery of EBV in Burkitt’s lymphoma was fortuitous as most of the tumour cells harbour EBV DNA in a non-transmissible episomal form. Rare herpesvirus-like structures were also seen by electron microscopy in Kaposi’s sarcoma tumours as early as 1984 (REF. 78) but the vast majority of KSHV-related tumours silently harbour KSHV as latent genomes79-81
The most likely explanation for the connection between virus latency and tumorigenesis is that productively replicating viruses initiate cell death, which has long been known to virologists as the cytopathic effect (CPE). Counter-intuitively, from the point of view of tumour virology, virus-induced CPE can be harnessed to kill cancer cells in viral oncolytic therapies, illustrating the anticancer activity of active lytic viral replication82,83. Although CPE is frequently thought of as a virus-induced event, it is actually a stereotypical and nonspecific innate immune response of cells to infection by many types of viruses. When latent viruses switch to producing virions, virus replication generates pathogen-associated molecular patterns from partially synthesized viral chromosomes, double-stranded RNAs and empty capsids that trigger cellular DNA damage responses and innate immune signalling84-86. For some viruses, lytic replication generates a linear viral chromosome that can be recognized as a DNA fragment87 unless either the DNA ends are structurally hidden from DNA damage response sensors by encapsidation or these sensors are inactivated. Activation of toll-like receptor and interferon signalling by virus infection initiates and amplifies this innate immune response88. Together, these cellular responses generally kill infected cells that are undergoing productive virus replication — hence the term lytic replication. Once triggered, lytic viral replication is largely irreversible and initiates a race between the virus to successfully reproduce itself and the death of the host cell.
Among viruses, latency is best understood for the herpesvirus family (particularly for EBV and KSHV that have latent viral tissue culture systems), in which it is tightly regulated by transcriptional repression89. Latent herpesviral protein expression is limited to a few crucial, non-structural viral products that include oncogenic proteins and microRNAs (miRNAs). During herpesviral latency the viral genome is not packaged into virions but instead the viral genome replicates in tandem with the host cell using the replication machinery of the cell and is tethered to chromosomes as a naked circular genome90. Lytic replication to produce infectious virions is initiated through a highly stereotypical series of viral transactivator cascades, which are cued by cellular environmental signalling pathways, that leads to host cell death and the release of infectious virions91. Although not fully described, these cellular environmental triggers might tell the persistent virus when to initiate lytic virus replication to optimally achieve transmission to a new host and survive.
Viral control of lytic and latent replication is less well-understood for the RNA and small DNA tumour viruses, but these agents also show a similar absence of productive viral replication during malignancy. HTLV-I is maintained as an integrated DNA provirus that is largely transcriptionally silent within ATLL cells22,65. In these tumours, the viral oncoprotein TAX is thought to promote early precancerous cell expansion and survival, but it may not be required in the fully malignant T cell. However, another candidate HTLV-I oncoprotein, HBZ, continues to be expressed in mature ATLL cells and might have a role in maintaining cell transformation92. Among the small DNA tumour viruses, fragmentation and integration of viral DNA into the nascent tumour cell eliminates their ability to replicate as virions, a state that we have termed ‘pseudo-latency’. Integration provides primary evidence for cancer causation if the integrated virus is clonal within individual tumours, as occurs in some of the HPV-, MCV-, HBV- and HTLV-I-related malignancies30,34,61,93. Although herpesviruses do not generally integrate into the host genome, recombination patterns for their terminal repeat sequences can also be used as markers for tumour cell clonality94,95. In such a scenario, clonality is a piece of molecular evidence placing the suspect virus at the ‘scene of the crime’.
As viruses in tumours are generally latent, antiviral drugs targeting the viral replication machinery are ineffective in treating mature tumours. However, antiviral therapy can in some instances prevent the development of new tumours. Randomized clinical trial data show, for example, that targeting the KSHV thymidine kinase and phosphotransferase proteins96 can prevent >90% of new Kaposi’s sarcomas from forming97 but this targeting has no effect on established tumours98. A possible exception to the rule for viruses being non-replicative and silent in human malignancy is HBV. This virus can infect nearly all hepatocytes in the liver during acute HBV hepatitis but most cells survive infection, eventually clearing the viral genomes without cell death99. Intriguingly, non-cancerous liver tissues from patients with HBV-associated HCC show patterns of microclonality63. It is not known whether this results from re-expanding clones of hepatocytes or whether it represents a premalignant change.
Origins of viral oncogenes
Cancers caused by viruses — such as non-infectious cancers — are biological accidents. Tumours do not increase transmissibility of viruses or enhance their replication fitness. A common misperception is that cancer viruses cause cancer to increase viral burden and transmission. Instead, tumours are ‘dead-end’ events for viruses. Only a small proportion of people infected with any of the human tumour viruses develop tumours and, of those people who do, they rarely (if ever) serve as sources for ongoing transmission. Instead, most human tumour virus transmissions are asymptomatic or mildly symptomatic but do not lead to neoplasia.
If we discard the idea that viruses are evolutionarily programmed to cause cancer, then why do tumour viruses encode oncogenes? There is strong selection to maintain viral genes that can initiate tumorigenesis, as diverse viruses (including, non-tumour viruses) show remarkable convergence to target the same tumour suppressor pathways (FIG. 2). For example, most of the human tumour viruses encode oncoproteins that target RB1 and p53, although they do so through different and unique mechanisms100. Other common targets that have roles in tumorigenesis for tumour viruses include telomerase reverse transcriptase (TERT101-105), cytoplasmic PI3K–AKT–mTOR106, nuclear factor-κB (NF-κB)59,64,107-109, β-catenin (also known as CTNNB1)110 and interferon signalling pathways111.
Figure 2. Common cellular targets for unrelated tumour virus oncoproteins.
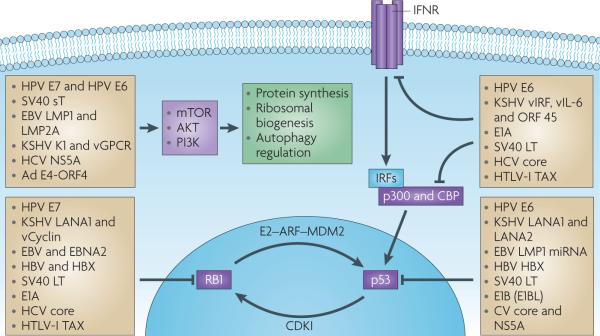
An incomplete but diverse list of animal and human tumour virus proteins that target RB1, p53, interferon and PI3K–mTOR signalling pathways. Most of these viral proteins are evolutionarily distinct from each other and have unique mechanisms for regulating or ablating these signalling pathways. Convergent evolution of tumour viruses to target these (and other cellular signalling pathways (not shown), including interleukin-6 (IL-6)–signal transducer and activator of transcription 3 signalling, telomerase and nuclear factor-κB (NF-κB) signalling pathways) reveals commonalities among the cancer viruses in tumour supressor and oncoprotein targeting. CBP, cAMP-response element binding protein; CDKI, cyclin-dependent kinase inhibitor; EBV, Epstein–Barr virus; HCV, hepatitis C virus; HPV, human papillomavirus; HTLV, human T-lymphotropic virus; IFNR, interferon receptor; IRF, interferon regulatory factor; KSHV, Kaposi’s sarcoma herpesvirus; LMP, latent membrane protein; miRNA, microRNA.
Two widely held views exist for the teleology of viral oncogenes (FIG. 3). The first hypothesis originated from the biology of small DNA tumour viruses (such as HPV and SV40) and was based on the presumed need for these viruses to re-initiate the cell cycle entry of differentiated cells to set conditions for viral replication100,112. Because the host replication machinery and nucleotide pools are limited in the G0 cell cycle phase of differentiated cells, these viruses might force unscheduled S phase entry to generate the cellular resources that are needed for viral genome replication. Disruption of cell cycle regulation, however, also activates cell death signalling pathways, such as p53, and so apoptotic signalling should also be inhibited to allow the efficient manufacture and export of viruses before cell death.
Figure 3. Two views for the origins of viral oncoproteins.

a | The tumour virus proteins target RB1 and p53 to drive a quiescent G0 cell into S phase of the cell cycle, allowing viral access to the nucleotide pools and replication machinery that are needed for replication and transmission100. Viral tumourigenesis is a by-product of the molecular parasitism by viruses to promote their own replication. Cells respond to virus infection by activating RB1 and p53 to inhibit virus replication as part of the innate immune response86. To survive, tumour viruses have evolved the means for inactivating these and other immune signalling pathways that place the cell at risk for cancerous transformation. This view holds that many tumour suppressor proteins have dual functions in preventing cancer formation and virus infection. b | An illustration of the overlap between intracellular innate immune and tumour suppressor signalling. Under typical circumstances, viruses do not cause cancers except in the settings of immunosuppression and/or complementing host cell mutations. Non-tumorigenic viruses, which constitute the overwhelming majority of viruses, target many of the same innate immune and tumour suppressor pathways as tumour viruses but do so in ways that do not place the host at risk for carcinogenesis. Apart from p53, RB1 and p300, additional proteins are likely to have both tumour suppressor and innate immune functions.
This is illustrated by the lifecycle of HPV, which infects basal epithelial cells that differentiate into arrested squamous epithelium. A commonly held view of HPV targeting of tumour suppressor pathways is that as the infected keratinocyte differentiates, the HPV E7 oncoprotein inactivates RB1 signalling to drive quiescent, infected cells back into a proliferative state, thus allowing viral genome replication113. Simultaneously, the HPV E6 protein induces ubiquitin-mediated degradation of p53, preventing the premature apoptosis that would otherwise limit the efficiency of virus production114. Under normal circumstances, these virus-deregulated cells will typically be sloughed off together with infectious virions. Rare mutations, however, that disrupt this lifecycle (such as an HPV integration event that results in the loss of early viral gene regulation) can set the stage for this molecular parasitism to turn into cancer cell transformation. By targeting the cell cycle checkpoints and anti-apoptotic machinery that are involved in genomic proofreading, viral oncogenes also induce cellular genomic instability and aneuploidy, which in turn contribute to carcinogenesis115,116. In summary, the most commonly held view for the function of viral oncogenes is that these genes target cellular tumour suppressor pathways to promote productive viral replication and only contribute to cancer when random mutations disrupt this equilibrium.
However, studies on the large DNA tumour viruses (such as EBV and KSHV) suggest a more complex interaction between the viral oncogenes and the host cell that may have more to do with evading immune responses during latency than ensuring viral genome replication86,111,117. During lytic viral replication, these viruses also hijack the cell cycle regulation machinery to promote their own genomic replication. The oncogenic herpesviruses encode proteins to inhibit p53, RB1 and other tumour suppressor checkpoints during active lytic viral replication118-121, and also possess virally encoded DNA synthesis enzymes95. These viral genes, like their counterparts among the small DNA tumour viruses, set the stage for the rapid replication and amplification of viral genomes by generating an S phase-like cellular state that can replicate viral DNA once lytic replication is initiated.
But the viral proteins and virus-encoded miRNAs that drive herpesviral tumours are expressed during latency, and these viral oncogenes can not directly contribute to productive viral replication108,122-126. Herpesvirus oncoproteins are expressed at the wrong time for them to be involved in generating the cellular resources needed for virus genome replication. The KSHV LANA1 oncoprotein suppresses lytic replication to maintain virus latency while it simultaneously targets RB1, p53 and interferon signalling responses127-129. The KSHV-encoded cyclin is another latent viral oncoprotein130,131 that is expressed in a cell cycle-dependent manner132. Similar to HPV E7, it targets RB1 and inactivates the G1/S checkpoint but it does not promote virus replication. For EBV, at least three classes of viral latency have been established, which are distinguished by different groups of oncogenic non-coding RNAs, Epstein—Barr virus nuclear antigens (EBNAs) and latent membrane proteins (LMPs)133. These EBV latent products target cell cycle and apoptotic signalling pathways (for example, p53-upregulated mediator of apoptosis (PUMA) is targeted by a latent EBV miRNA125), but are also not directly involved in generating EB virions or amplifying EBV genomes during productive virus replication. Studies from the herpesviruses raise the question: what are the viral oncoproteins doing if they are not preparing the cell for virus replication?
Viral oncogenes and immune evasion
Over the past decade, increasing evidence has indicated that the evasion of innate immunity also plays a fundamental part in viral tumorigenesis. Humans, as well as most complex metazoans, are chimeric for numerous viruses. In some cases, mammals may even exploit latent viral infections to beneficially regulate their own innate immune systems134. So, it is not surprising that major portions of the eukaryote cell are devoted to protecting the host genome from foreign viral sequences. Innate immune signalling shares many similarities to tumour suppressor signalling, as both processes initiate cell cycle arrest and prime apoptotic pathways. Key effector proteins such as the p21 cyclin-dependent kinase inhibitor135 and p53 (REF. 136) are shared by both tumour suppressor and innate immune surveillance signalling networks. This suggests that targeting of tumour suppressor pathways by viruses may actually represent an immune evasion response that disables antiviral pathways but inadvertently places the infected cell at risk for cancerous transformation (known as the anti-antivirus hypothesis)86,111.
The dual nature of innate immune signalling in antivirus and anticancer functions is illustrated by interferon regulatory factors (IRFs), a family of induced and immediate-early transcription factors that regulate interferon transcriptional responses137-139. KSHV encodes four IRF homologues95,140, including vIRF1 (REF. 141), which behaves similarly to IRF2 (REF. 139) by inhibiting interferon signalling and initiating cell transformation. Most of the other established KSHV oncoproteins, including interleukin-6 (vIL-6; also known as K2)142, FLICE inhibitory protein (vFLIP; also known as ORF71)109, ORF K1 protein143, latent nuclear antigen 1 (LANA1)129 and LANA2 (REFS 144,145), also have well defined innate immunomodulatory roles86,117. For EBV, RNA-dependent protein kinase (PKR) immune signalling is targeted by latent small non-coding EBV RNAs (EBERs)146 that might have a role in EBV-induced tumorigenesis. This implies that infected cells can sense latent virus infection and must deactivate cell cycle arrest and pro-apoptotic pathways to survive in the hostile environment of the cell.
Although a role for oncoproteins in innate immune evasion is best characterized for herpesviruses, other viral oncoproteins, such as the human adenovirus E1A oncoprotein that causes cancer in rodents, also dually inhibit interferon signalling and tumour suppressor pathways by targeting the histone acetyltransferases p300 and CBP147,148, which participate in interferon-induced transcription. The relationship between tumour suppression and cellular antiviral activity was described by Takaoka and colleagues who showed that knock out of Trp53 (encoding p53) causes immune deficiency to virus infection, and that virus-induced inflammatory cytokines prime cellular pro-apoptotic signalling pathways136. p53 seems to not only be the ‘guardian of the genome’ (REF. 149) but also a guardian against viral infection.
Other cellular pathways (FIG. 3a) with roles traditionally ascribed to preventing tumour cell formation might also play a part in immunity to viral infection150. Cellular sensors for DNA and RNA ends are generally studied as triggers for the repair of somatic mutations but they also have a role in sensing viral nucleic acids151,152. DNA damage responses are activated during viral uncoating and replication65,153-155 that can lead to cell cycle arrest and inflammatory signalling activation156. Disarming the antiviral targeting of tumour suppressor signalling may allow the prolonged persistence of viral infection but also carries obvious risks for generating a cancer. Despite evidence for a relationship between innate immune and tumour suppressor signalling (FIG. 3b), the mechanisms that cells might use to sense episomal viral genomes, as occurs during latent herpesvirus infection, remain unknown.
The newest member of the club: MCV
The examples we describe above mainly come from the first six human tumour viruses. How does the most recently discovered virus, MCV, compare? Similar to SV40 and murine polyomaviruses, MCV encodes a multiply spliced tumour (T) antigen protein complex that targets several tumour suppressor proteins157. As with EBV and Burkitt’s lymphoma, MCV is present in most MCCs and it is a near-ubiquitous infection of adults158-162. Within infected tumours, the T antigen is only expressed in tumour cells, as predicted for a direct viral carcinogen163. Given these features, the strongest evidence to support MCV causing MCC comes from its random clonal integration into Merkel cell tumours34,164 and knockdown studies showing that T antigen expression is required for the survival of virus-positive Merkel cell lines58. Although skin carriage of the virus is common165, no other tumours except MCC have yet been convincingly linked to MCV infection. As SV40 and related polyomaviruses have been workhorses for cancer research from the early 1960s, studies on these viruses can be directly applied to MCV, allowing rapid progress in understanding its biology in humans.
MCV is intriguing because the precise molecular events leading to cancer have been described and they help to explain why this common childhood infection can lead to a rare cancer that is associated with sun exposure (FIG. 4). The initial event in MCV-driven MCC carcinogenesis is likely to be the loss of immune surveillance for the virus. MCC principally occurs in the elderly and immunocompromised, and those people developing MCV-related MCC have greatly increased antibodies to MCV structural proteins, suggesting that the loss of cellular immune control over the virus may allow viraemia before the development of the tumour159,161,162.
Figure 4. The molecular evolution of a human tumour virus.
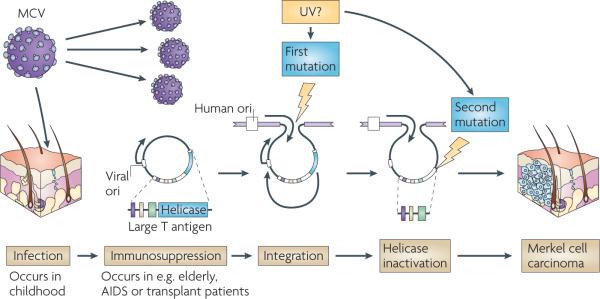
Merkel cell polyomavirus (MCV), which has tumour-specific truncation mutations, illustrates common features among the human tumour viruses involving immunity, virus replication and tumour suppressor targeting. Although MCV is a common infection, loss of immune surveillance through ageing, AIDS or transplantation and subsequent treatment with immunosuppressive drugs may lead to resurgent MCV replication in skin cells161. If a rare integration mutation into the host cell genome occurs34, the MCV T antigen can activate independent DNA replication from the integrated viral origin that will cause DNA strand breaks in the proto-tumour cell157. A second mutation that truncates the T antigen, eliminating its viral replication functions but sparing its RB1 tumour suppressor targeting domains, is required for the survival of the nascent Merkel tumour cell. Exposure to sunlight (possibly ultraviolet (UV) irradiation) and other environmental mutagens may enhance the sequential mutation events that turn this asymptomatic viral infection into a cancer virus.
The virus then undergoes at least two mutations, the first being non-homologous recombination with the host chromosome. As with other tumour viruses, clonality of integration in primary tumours and their metastases shows that this occurs before the tumour cell begins to proliferate34. As MCV has no mechanism to excise its genome, the virus cannot replicate and is no longer transmissible, and this is analogous to HPV in most cervical carcinomas.
MCV integration, however, generates a problem for the nascent tumour cell. The viral large T antigen not only targets tumour suppressor molecules, such as RB1, but it is also required for productive virus replication. During a typical infection by MCV, the large T antigen protein binds to the viral genome and its helicase domain unwinds the viral origin to allow DNA replication166. If the full-length large T antigen protein is expressed in tumour cells with an integrated virus, it initiates unlicensed DNA synthesis at the integration site causing replication fork collisions and DNA break responses that could lead to cytopathic cell death157. All MCV genomes that have been obtained from tumours so far, however, have inactivating secondary mutations in the T antigen gene that eliminate its DNA replication capacity. Whether additional cellular mutations are required for the successful outgrowth of MCV-infected MCC tumours is currently unknown.
Molecular evolution of MCV in MCC tumours illustrates many of the common features that have been described for the other human tumour viruses. Most notably, tumour formation is a rare, accidental occurrence in the lifecycle of this otherwise innocuous virus. The recent discovery of additional new human polyomaviruses165,167,168 provides the opportunity to determine whether other members of this group share a similar potential for contributing to human viral cancers.
Future directions
The reduced cost and increased accuracy of sequencing technologies has created the opportunity for most research groups to search for cancer viruses. Only a snippet of unique nucleic acid sequence is needed to discover a new human tumour virus and to begin characterizing it, so the pool of cancer-causing candidates is almost certain to grow in the coming decade. Equally importantly, the reliability of human sequence databases has matured to a level at which certain classes of cancer agents might be excluded when none is found. Identifying a new virus, however, is only the beginning in determining whether it causes human cancer. Cancer causation theories work well for uncommon viruses that are uniformly present in a particular type of cancer64,108. Head and neck squamous cell carcinomas169 and MCCs34 are examples of cancers that were previously assumed to be homogenous cancers but are now recognized as likely to be caused by both infectious and non-infectious (or at least not identified infectious) aetiologies. Epidemiologists will be increasingly pressed to determine whether a candidate viral agent might cause only a small but important portion of a type of tumour. New epidemiological methods that make better use of molecular biologic data will be key to resolving the causes for these cancers.
Viruses have had a chequered history in cancer biology over the past century. Depending on the time and the fashion, viruses have been either sought out as the primary cause for cancer, or ignored as inconsequential to this disease. We are now entering a more mature phase of research with the realization that a considerable proportion of cancers are indeed caused by viruses. For these cancers, infection is only one component in their ultimate cause. But failure to recognize the importance of viral cancers has led to overlooked opportunities in cancer control. Despite EBV being the first discovered human tumour virus, there is no EBV vaccine and little enthusiasm for its development. KSHV has emerged as a leading cause of adult cancer in sub-Saharan Africa170 but no movement has yet been made in developing clinical interventions against this virus or its cancers.
The development of anti-latent viral drugs and immunological therapies against cancer virus antigens are achievable goals that have not yet been pursued in modern cancer control. The real measure of success for the past century of tumour virus research will be the future exploitation of existing research to effectively diagnose, treat and prevent cancers that are caused by viruses.
Acknowledgments
The authors would like to thank G. Klein and O. Gjoerup for helpful comments and corrections to the manuscript; M. Melbye at the Danish Statens Serum Institut, and T. Söderqvist and his staff at the Medical Museion, University of Copenhagen, Denmark, for materials; and F. Zappa for help in preparing the manuscript. The authors are supported by NIH CA136363, CA120726, the Al Copeland Foundation, and American Cancer Society Research Professorships.
Glossary
- Antibody panning
cDNA from a tumour is used to express proteins in bacteria and transferred to replicate filters. Antibody screening of the filters can then be used to identify colonies expressing the specific cDNA encoding an antigen.
- Bayesian reasoning
A scientific approach developed from Bayes theorem, combining features of the Logical Positivist and Kuhnian schools of science philosophy, and describing how the probability of a hypothesis (in this case, virus A causes cancer B) changes with new evidence. In simple terms, it can be described as the repeated application of the scientific method to falsify a hypothesis such that the hypothesis has a high probability of being either true or false.
- Digital transcriptome subtraction
DTS. Method to discover new viruses by exhaustively sequencing cDNA libraries and aligning known human sequences by computer leaving a smaller candidate pool of potential viral sequences for analysis36.
- Endogenous retrovirus
ERV. Retrovirus that has inserted into the metazoan germline genome over evolutionary timescales and is now transmitted to offspring as a genetic element through Mendelian inheritance. Approximately 8% of the human genome is estimated to be derived from retroviral precursors.
- High-risk papillomaviruses
More than 160 different genotypes or strains of HPV have been described but only a few genotypes belonging to a high-risk carcinogenic clade of the α-HPV genus are responsible for invasive HPV-related anogenital cancers211.
- Longitudinal study
Virus infection is measured initially in a cohort of patients who are then followed over time to determine cancer occurrence.
- Prodromal phase
An early set of nonspecific symptoms that occur before the onset of specific disease symptoms.
- Representational difference analysis
A PCR-based subtractive hybridization technique that can subtract common human sequences from a tumour genomic library using a control human tissue genomic library35.
- Serology
The measurement of antibodies against viruses in blood or bodily fluids. This usually does not distinguish ongoing infections from past viral infections.
References
- 1.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 2.Bouvard V, et al. A review of human carcinogens-part B: biological agents. Lancet Oncol. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- 3.Chang MH, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997;336:1855–1859. doi: 10.1056/NEJM199706263362602. [DOI] [PubMed] [Google Scholar]
- 4.Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004;11:97–107. doi: 10.1046/j.1365-2893.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- 5.Goldie SJ, et al. Projected clinical benefits and cost-effectiveness of a human papillomavirus 16/18 vaccine. J. Natl Cancer Inst. 2004;96:604–615. doi: 10.1093/jnci/djh104. [DOI] [PubMed] [Google Scholar]
- 6.Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nature Rev. Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vennervald BJ, Polman K. Helminths and malignancy. Parasite Immunol. 2009;31:686–696. doi: 10.1111/j.1365-3024.2009.01163.x. [DOI] [PubMed] [Google Scholar]
- 8.Rous P. A transmissible avian neoplasm. (Sarcoma of the common fowl) J. Exp. Med. 1910;12:696–705. doi: 10.1084/jem.12.5.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911;13:397–411. doi: 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellerman V, Bang O. Experimentelle leukämie bei hühnern. Centralbl. f. Bakteriol. 1908;46:595–609. [Google Scholar]
- 11.Beard JW. Avian virus growths and their etiologic agents. Adv. Cancer Res. 1963;7:1–127. doi: 10.1016/s0065-230x(08)60982-3. [DOI] [PubMed] [Google Scholar]
- 12.Becsei-Kilborn E. Scientific discovery and scientific reputation: the reception of Peyton Rous’ discovery of the chicken sarcoma virus. J. Hist. Biol. 2010;43:111–157. doi: 10.1007/s10739-008-9171-y. [DOI] [PubMed] [Google Scholar]
- 13.Vogt PK. Peyton Rous: homage and appraisal. FASEB J. 1996;10:1559–1562. doi: 10.1096/fasebj.10.13.8940303. [DOI] [PubMed] [Google Scholar]
- 14.Gross L. Oncogenic Viruses. Pergamon; Oxford: 1970. [Google Scholar]
- 15.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;15:702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 16.Epstein-Barr virus and Kaposi’s sarcoma herpesvirus/human herpesvirus 8. IARC Monogr. Eval. Carcinog. Risks Hum; Proceedings of the IARC working group on the evaluation of carcinogenic risks to humans; Lyon, France. 17-24 June 1997.1997. pp. 1–492. [Google Scholar]
- 17.Kelly GL, Rickinson AB. Burkitt lymphoma: revisiting the pathogenesis of a virus-associated malignancy. Hematology Am. Soc. Hematol. Educ. Program. 2007;2007:277–284. doi: 10.1182/asheducation-2007.1.277. [DOI] [PubMed] [Google Scholar]
- 18.Bunge M. Causality: The Place of the Causal Principle in Modern Science. Meridian Books; Cleveland and New York: 1959. [Google Scholar]
- 19.Epstein MA, Henle G, Achong BG, Barr YM. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J. Exp. Med. 1965;121:761–770. doi: 10.1084/jem.121.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poiesz BJ, et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl Acad. Sci. USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyoshi I, et al. Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature. 1981;294:770–771. doi: 10.1038/294770a0. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida M. Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis. Oncogene. 2005;24:5931–5937. doi: 10.1038/sj.onc.1208981. [DOI] [PubMed] [Google Scholar]
- 23.Gallo RC. History of the discoveries of the first human retroviruses: HTLV-1 and HTLV-2. Oncogene. 2005;24:5926–5930. doi: 10.1038/sj.onc.1208980. [DOI] [PubMed] [Google Scholar]
- 24.Vahlne A. A historical reflection on the discovery of human retroviruses. Retrovirology. 2009;6:40. doi: 10.1186/1742-4690-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blumberg BS, Alter HJ, Visnich S. A “new” antigen in leukemia sera. JAMA. 1965;191:541–546. doi: 10.1001/jama.1965.03080070025007. [DOI] [PubMed] [Google Scholar]
- 26.Prince AM, Fuji H, Gershon RK. Immunohistochemical studies on the etiology of anicteric hepatitis in Korea. Am. J. Hyg. 1964;79:365–381. doi: 10.1093/oxfordjournals.aje.a120391. [DOI] [PubMed] [Google Scholar]
- 27.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet. 1981;2:1129–1133. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 28.zur Hausen H. Condylomata acuminata and human genital cancer. Cancer Res. 1976;36:794. [PubMed] [Google Scholar]
- 29.Durst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl Acad. Sci. USA. 1983;80:3812–3815. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boshart M, et al. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984;3:1151–1157. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choo QL, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 32.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang Y, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;265:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 34.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 36.Feng H, et al. Human transcriptome subtraction by using short sequence tags to search for tumor viruses in conjunctival carcinoma. J. Virol. 2007;81:11332–11340. doi: 10.1128/JVI.00875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, et al. Pathogen discovery from human tissue by sequence-based computational subtraction. Genomics. 2003;81:329–335. doi: 10.1016/s0888-7543(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 38.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 39.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 40.Hahn WC, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahn WC, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 42.Purtilo DT, Cassel CK, Yang JP, Harper R. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease) Lancet. 1975;1:935–940. doi: 10.1016/s0140-6736(75)92004-8. [DOI] [PubMed] [Google Scholar]
- 43.Kaposi M. Idiopathic multiple pigmented sarcoma of the skin. CA Cancer J. Clin. 1982. 1872;32:340–347. doi: 10.3322/canjclin.32.6.340. [DOI] [PubMed] [Google Scholar]
- 44.McGeoch DJ, Gatherer D, Dolan A. On phylogenetic relationships among major lineages of the Gammaherpesvirinae. J. Gen. Virol. 2005;86:307–316. doi: 10.1099/vir.0.80588-0. [DOI] [PubMed] [Google Scholar]
- 45.Miller G, et al. Antibodies to butyrate-inducible antigens of Kaposi’s sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med. 1996;334:1292–1297. doi: 10.1056/NEJM199605163342003. [DOI] [PubMed] [Google Scholar]
- 46.Gao SJ, et al. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. N. Engl. J. Med. 1996;335:233–241. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- 47.Simpson GR, et al. Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet. 1996;348:1133–1138. doi: 10.1016/S0140-6736(96)07560-5. [DOI] [PubMed] [Google Scholar]
- 48.Gao SJ, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nature Med. 1996;2:925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- 49.Kedes DH, et al. The seroepidemiology of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nature Med. 1996;2:918–924. doi: 10.1038/nm0896-918. [DOI] [PubMed] [Google Scholar]
- 50.Pellett PE, et al. Multicenter comparison of serologic assays and estimation of human herpesvirus 8 seroprevalence among US blood donors. Transfusion. 2003;43:1260–1268. doi: 10.1046/j.1537-2995.2003.00490.x. [DOI] [PubMed] [Google Scholar]
- 51.Engels EA, et al. Trends in cancer risk among people with AIDS in the United States 1980–2002. AIDS. 2006;20:1645–1654. doi: 10.1097/01.aids.0000238411.75324.59. [DOI] [PubMed] [Google Scholar]
- 52.zur Hausen H. Oncogenic DNA viruses. Oncogene. 2001;20:7820–7823. doi: 10.1038/sj.onc.1204958. [DOI] [PubMed] [Google Scholar]
- 53.Parsonnet J. In: Microbes and Malignancy. Parsonnet J, editor. Oxford Univ. Press; New York: 1999. pp. 3–18. [Google Scholar]
- 54.Steele C, Cowsert LM, Shillitoe EJ. Effects of human papillomavirus type 18-specific antisense oligonucleotides on the transformed phenotype of human carcinoma cell lines. Cancer Res. 1993;53:2330–2337. [PubMed] [Google Scholar]
- 55.Tan TM, Ting RC. In vitro and in vivo inhibition of human papillomavirus type 16 E6 and E7 genes. Cancer Res. 1995;55:4599–4605. [PubMed] [Google Scholar]
- 56.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl Acad. Sci. USA. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wies E, et al. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood. 2008;111:320–327. doi: 10.1182/blood-2007-05-092288. [DOI] [PubMed] [Google Scholar]
- 58.Houben R, et al. Merkel cell polyomavirus infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010;84:7064–7072. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood. 2004 doi: 10.1182/blood-2004-08-3052. [DOI] [PubMed] [Google Scholar]
- 60.Dirmeier U, et al. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene. 2005;24:1711–1717. doi: 10.1038/sj.onc.1208367. [DOI] [PubMed] [Google Scholar]
- 61.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai WL, Chung RT. Viral hepatocarcinogenesis. Oncogene. 2010;29:2309–2324. doi: 10.1038/onc.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mason WS, Liu C, Aldrich CE, Litwin S, Yeh MM. Clonal expansion of normal appearing human hepatocytes during chronic HBV infection. J. Virol. 2010;84:8308–8315. doi: 10.1128/JVI.00833-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yasunaga J, Matsuoka M. Leukaemogenic mechanism of human T-cell leukaemia virus type I. Rev. Med. Virol. 2007;17:301–311. doi: 10.1002/rmv.548. [DOI] [PubMed] [Google Scholar]
- 65.Jeang KT, Giam CZ, Majone F, Aboud M. Life, death, and tax: role of HTLV-I oncoprotein in genetic instability and cellular transformation. J. Biol. Chem. 2004;279:31991–31994. doi: 10.1074/jbc.R400009200. [DOI] [PubMed] [Google Scholar]
- 66.Hatakeyama M. Helicobacter pylori CagA - a bacterial intruder conspiring gastric carcinogenesis. Int. J. Cancer. 2006;119:1217–1223. doi: 10.1002/ijc.21831. [DOI] [PubMed] [Google Scholar]
- 67.Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67. doi: 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 68.Chen LP, Thomas EK, Hu SL, Hellstrom I, Hellstrom KE. Human papillomavirus type 16 nucleoprotein E7 is a tumor rejection antigen. Proc. Natl Acad. Sci. USA. 1991;88:110–114. doi: 10.1073/pnas.88.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guihot A, et al. Low T cell responses to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J. Infect. Dis. 2006;194:1078–1088. doi: 10.1086/507648. [DOI] [PubMed] [Google Scholar]
- 70.Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123–128. doi: 10.1016/0140-6736(90)90001-l. [DOI] [PubMed] [Google Scholar]
- 71.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. 2002;359:497–498. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 72.Vajdic CM, van Leeuwen MT. Cancer incidence and risk factors after solid organ transplantation. Int. J. Cancer. 2009;125:1747–1754. doi: 10.1002/ijc.24439. [DOI] [PubMed] [Google Scholar]
- 73.Schulz TF. Cancer and viral infections in immunocompromised individuals. Int. J. Cancer. 2009;125:1755–1763. doi: 10.1002/ijc.24741. [DOI] [PubMed] [Google Scholar]
- 74.Sugarbaker DJ, et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc. Natl Acad. Sci. USA. 2008;105:3521–3526. doi: 10.1073/pnas.0712399105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.zur Hausen H. SV40 in human cancers-an endless tale? Int. J. Cancer. 2003;107:687. doi: 10.1002/ijc.11517. [DOI] [PubMed] [Google Scholar]
- 76.Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392:1–10. doi: 10.1016/j.virol.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 77.Small MB, Gluzman Y, Ozer HL. Enhanced transformation of human fibroblasts by origin-defective simian virus 40. Nature. 1982;296:671–672. doi: 10.1038/296671a0. [DOI] [PubMed] [Google Scholar]
- 78.Walter PR, Philippe E, Nguemby-Mbina C, Chamlian A. Kaposi’s sarcoma: presence of herpes-type particles in a tumor specimen. Human Pathol. 1984;15:1145–1146. doi: 10.1016/s0046-8177(84)80309-3. [DOI] [PubMed] [Google Scholar]
- 79.Zhong W, Wang H, Herndier B, Ganem D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl Acad. Sci. USA. 1996;93:6641–6646. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parravicini C, et al. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 2000;156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1) J. Virol. 1998;72:1005–1012. doi: 10.1128/jvi.72.2.1005-1012.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bischoff JR, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 83.Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2010 Aug 26; doi: 10.1038/gt.2010.121. (doi: 10.1038/gt.2010.121) [DOI] [PubMed] [Google Scholar]
- 84.Jacquemont B, Roizman B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975;15:707–713. doi: 10.1128/jvi.15.4.707-713.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15:119–126. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 86.Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 2003;57:609–639. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tauer TJ, Schneiderman MH, Vishwanatha JK, Rhode SL. DNA double-strand break repair functions defend against parvovirus infection. J. Virol. 1996;70:6446–6449. doi: 10.1128/jvi.70.9.6446-6449.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bauer S, Muller T, Hamm S. Pattern recognition by Toll-like receptors. Adv. Exp. Med. Biol. 2009;653:15–34. doi: 10.1007/978-1-4419-0901-5_2. [DOI] [PubMed] [Google Scholar]
- 89.Lieberman PM, Hu J, Renne R. In: Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Arvin A, Campardelli-Fiome G, Mocarski AE, Moore PS, Roizman B, Whitley RJ, Yamanishi K, editors. Cambridge Univ. Press; Cambridge, UK: 2007. pp. 379–402. [PubMed] [Google Scholar]
- 90.Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency- associated nuclear antigen. Science. 1999;284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- 91.Roizman B. In: TheHumanHerpeviruses. Roizman B, Whitley RJ, Lopez C, editors. Raven Press, Ltd.; New York: 1993. pp. 1–9. [Google Scholar]
- 92.Matsuoka M, Green PL. The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology. 2009;6:71. doi: 10.1186/1742-4690-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc. Natl Acad. Sci. USA. 1984;81:2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raab-Traub N, Flynn K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell. 1986;47:883–889. doi: 10.1016/0092-8674(86)90803-2. [DOI] [PubMed] [Google Scholar]
- 95.Russo JJ, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc. Natl Acad. Sci. USA. 1996;93:14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambinder RF. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 1999;73:4786–4793. doi: 10.1128/jvi.73.6.4786-4793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin DF, et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 1999;340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 98.Little RF, Yarchoan R. Treatment of gammaherpesvirus-related neoplastic disorders in the immunosuppressed host. Semin. Hematol. 2003;40:163–171. doi: 10.1053/shem.2003.50016. [DOI] [PubMed] [Google Scholar]
- 99.Mason WS, et al. The amount of hepatocyte turnover that occurred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J. Virol. 2009;83:1778–1789. doi: 10.1128/JVI.01587-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses: the inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285–293. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 101.Oh ST, Kyo S, Laimins LA. Telomerase activation by human papillomavirus type 16 E6 protein: induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001;75:5559–5566. doi: 10.1128/JVI.75.12.5559-5566.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- 103.Verma SC, Borah S, Robertson ES. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 2004;78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kataoka H, et al. Immortalization of immunologically committed Epstein-Barr virus-transformed human B-lymphoblastoid cell lines accompanied by a strong telomerase activity. Differentiation. 1997;62:203–211. doi: 10.1046/j.1432-0436.1998.6240203.x. [DOI] [PubMed] [Google Scholar]
- 105.Terrin L, et al. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 2008;82:10175–10187. doi: 10.1128/JVI.00321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nature Rev. Microbiol. 2008;6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mosialos G, et al. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 108.Sarid R, Olsen SJ, Moore PS. Kaposi’s sarcoma-associated herpesvirus: epidemiology, virology and molecular biology. Adv. Virus Res. 1999;52:139–232. doi: 10.1016/s0065-3527(08)60299-7. [DOI] [PubMed] [Google Scholar]
- 109.Liu L, et al. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Iκ B kinase complex. J. Biol. Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 110.Fujimuro M, et al. A novel viral mechanism for dysregulation of β-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nature Med. 2003;9:300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- 111.Moore PS, Chang Y. Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends Genet. 1998;14:144–150. doi: 10.1016/s0168-9525(98)01408-5. [DOI] [PubMed] [Google Scholar]
- 112.McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc. Natl Acad. Sci. USA. 1988;85:7169–7173. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Munger K, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 115.Duensing S, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl Acad. Sci. USA. 2000;97:10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hein J, et al. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J. Virol. 2009;83:117–127. doi: 10.1128/JVI.01515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liang C, Lee JS, Jung JU. Immune evasion in Kaposi’s sarcoma-associated herpes virus associated oncogenesis. Semin. Cancer Biol. 2008;18:423–436. doi: 10.1016/j.semcancer.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakamura H, Li M, Zarycki J, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 2001;75:7572–7582. doi: 10.1128/JVI.75.16.7572-7582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gwack Y, et al. Kaposi’s sarcoma-associated herpesvirus open reading frame 50 represses p53-induced transcriptional activity and apoptosis. J. Virol. 2001;75:6245–6248. doi: 10.1128/JVI.75.13.6245-6248.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Park J, et al. The K-bZIP protein from Kaposi’s sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 2000;74:11977–11982. doi: 10.1128/jvi.74.24.11977-11982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Q, Gutsch D, Kenney S. Functional and physical interaction between p53 and BZLF1: implications for Epstein-Barr latency. Mol. Cell. Biol. 1994;14:1929–1938. doi: 10.1128/mcb.14.3.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sarid R, Klepfish A, Schattner A. Virology, pathogenetic mechanisms, and associated diseases of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) Mayo Clin. Proc. 2002;77:941–949. doi: 10.4065/77.9.941. [DOI] [PubMed] [Google Scholar]
- 123.Pfeffer S, et al. Identification of microRNAs of the herpesvirus family. Nature Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 124.Gottwein E, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Choy EY, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008;205:2551–2560. doi: 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nature Rev. Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 127.Friborg J, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 128.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene hras transforms primary rat cells. Nature Med. 2000;6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 129.Cloutier N, Flamand L. Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits interferon (IFN) β expression by competing with IFN regulatory factor-3 for binding to IFNB promoter. J. Biol. Chem. 2010;285:7208–7221. doi: 10.1074/jbc.M109.018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chang Y, et al. Cyclin encoded by KS herpesvirus. Nature. 1996;382:410. doi: 10.1038/382410a0. [DOI] [PubMed] [Google Scholar]
- 131.Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2:229–241. doi: 10.1016/s1535-6108(02)00123-x. [DOI] [PubMed] [Google Scholar]
- 132.Sarid R, Wiezorek JS, Moore PS, Chang Y. Characterization and cell cycle regulation of the major Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 1999;73:1438–1446. doi: 10.1128/jvi.73.2.1438-1446.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shah KM, Young LS. Epstein-Barr virus and carcinogenesis: beyond Burkitt’s lymphoma. Clin. Microbiol. Infect. 2009;15:982–988. doi: 10.1111/j.1469-0691.2009.03033.x. [DOI] [PubMed] [Google Scholar]
- 134.Barton ES, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 135.Chin YE, et al. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science. 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 136.Takaoka A, et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 137.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nature Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Senger K, et al. Gene repression by coactivator repulsion. Mol. Cell. 2000;6:931–937. doi: 10.1016/s1097-2765(05)00081-x. [DOI] [PubMed] [Google Scholar]
- 139.Harada H, et al. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science. 1993;259:971–974. doi: 10.1126/science.8438157. [DOI] [PubMed] [Google Scholar]
- 140.Lee HR, Kim MH, Lee JS, Liang C, Jung JU. Viral interferon regulatory factors. J. Interferon Cytokine Res. 2009;29:621–627. doi: 10.1089/jir.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gao SJ, et al. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–1985. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- 142.Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science. 2002;298:1432–1435. doi: 10.1126/science.1074883. [DOI] [PubMed] [Google Scholar]
- 143.Lee H, et al. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma- associated herpesvirus. Nature Med. 1998;4:435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- 144.Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001;75:429–438. doi: 10.1128/JVI.75.1.429-438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wies E, et al. The Kaposi’s Sarcoma-associated Herpesvirus-encoded vIRF-3 Inhibits Cellular IRF-5. J. Biol. Chem. 2009;284:8525–8538. doi: 10.1074/jbc.M809252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iwakiri D, Takada K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010;107:119–136. doi: 10.1016/S0065-230X(10)07004-1. [DOI] [PubMed] [Google Scholar]
- 147.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 148.Bhattacharya S, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-α. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 149.Lane DP. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 150.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hornung V, Latz E. Intracellular DNA recognition. Nature Rev. Immunol. 2010;10:123–130. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 152.Ranjan P, et al. Cytoplasmic nucleic acid sensors in antiviral immunity. Trends Mol. Med. 2009;15:359–368. doi: 10.1016/j.molmed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 153.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 154.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl Acad. Sci. USA. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shin YC, et al. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006;80:2257–2266. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu ZH, Miyamoto S. Many faces of NF-κB signaling induced by genotoxic stress. J. Mol. Med. 2007;85:1187–1202. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- 157.Shuda M, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl Acad. Sci. USA. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tolstov YL, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer. 2009;125:1250–1256. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carter JJ, et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J. Natl Cancer Inst. 2009;101:1510–1522. doi: 10.1093/jnci/djp332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pastrana DV, et al. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009;5:e1000578. doi: 10.1371/journal.ppat.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Touze A, et al. Generation of Merkel cell polyomavirus (MCV)-like particles and their application to detection of MCV antibodies. J. Clin. Microbiol. 2010;48:1767–1770. doi: 10.1128/JCM.01691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shuda M, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer. 2009;125:1243–1249. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sastre-Garau X, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009;218:48–56. doi: 10.1002/path.2532. [DOI] [PubMed] [Google Scholar]
- 165.Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kwun HJ, et al. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009;83:12118–12128. doi: 10.1128/JVI.01336-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Allander T, et al. Identification of a third human polyomavirus. J. Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gaynor AM, et al. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.D’Souza G, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007;356:1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 170.Parkin DM, et al. Part I: cancer in Indigenous Africans-burden, distribution, and trends. Lancet Oncol. 2008;9:683–692. doi: 10.1016/S1470-2045(08)70175-X. [DOI] [PubMed] [Google Scholar]
- 171.Rous P. The possible role of viruses in cancer. Opening remarks. Cancer Res. 1960;20:672–676. [PubMed] [Google Scholar]
- 172.Temin HM, Rubin H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology. 1958;6:669–688. doi: 10.1016/0042-6822(58)90114-4. [DOI] [PubMed] [Google Scholar]
- 173.Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226:1209–1211. doi: 10.1038/2261209a0. [DOI] [PubMed] [Google Scholar]
- 174.Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211–1213. doi: 10.1038/2261211a0. [DOI] [PubMed] [Google Scholar]
- 175.Chang HW, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 176.Bishop JM. Nobel Lecture. Retroviruses and oncogenes II. Biosci. Rep. 1990;10:473–491. doi: 10.1007/BF01116609. [DOI] [PubMed] [Google Scholar]
- 177.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 178.Coffin JM, Hughes SH, Varmus HE. Retroviruses. CSHL Press; Cold Spring Harbor: 1997. [PubMed] [Google Scholar]
- 179.Weiss RA. The discovery of endogenous retroviruses. Retrovirology. 2006;3:67. doi: 10.1186/1742-4690-3-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ebbesen P. In: Viruses Associated with Human Cancer. Phillips LA, editor. Marcel Dekker; New York: 1983. pp. 369–409. [Google Scholar]
- 181.Klein G. The Athiest and the Holy City: Encounters and Reflections. MIT Press; 1992. [Google Scholar]
- 182.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 183.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 184.Arnaud F, Varela M, Spencer TE, Palmarini M. Coevolution of endogenous betaretroviruses of sheep and their host. Cell. Mol. Life Sci. 2008;65:3422–3432. doi: 10.1007/s00018-008-8500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Contreras-Galindo R, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J. Virol. 2008;82:9329–9336. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Urisman A, et al. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoSPathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 187.Hohn O, et al. Lack of evidence for xenotropic murine leukemia virus-related virus(XMRV) in German prostate cancer patients. Retrovirology. 2009;6:92. doi: 10.1186/1742-4690-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Weiss RA. A cautionary tale of virus and disease. BMC Biol. 2010;8:124. doi: 10.1186/1741-7007-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Koch R. In: SourceBookofMedicalHistory. Clark DH, editor. Dover Publications, Inc; New York: 1942. pp. 392–406. [Google Scholar]
- 190.Fredericks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin. Microbiol. Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hill AB. Environment and disease: association or causation? Proc. R. Soc. Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science. 1967;157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- 193.Ernberg I, Klein G. In: Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Arvin A, et al., editors. Cambridge Univ. Press; Cambridge, UK: 2007. pp. 514–539. [PubMed] [Google Scholar]
- 194.Saemundsen AK, et al. Documentation of Epstein-Barr virus infection in immunodeficient patients with life-threatening lymphoproliferative diseases by Epstein-Barr virus complementary RNA/DNA and viral DNA/DNA hybridization. Cancer Res. 1981;41:4237–4242. [PubMed] [Google Scholar]
- 195.Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8. World Health Organization; 1997. [Google Scholar]
- 196.Shope RE, Hurst EW. Infectious papillomatosis of rabbits: with a note on the histopathology. J. Exp. Med. 1933;58:607–624. doi: 10.1084/jem.58.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bittner JJ. Some possible effects of nursing on the mammary gland tumor incidence in mice. Science. 1936;84:162. doi: 10.1126/science.84.2172.162. [DOI] [PubMed] [Google Scholar]
- 198.Gross L. “Spontaneous” leukemia developing in C3H mice following inoculation in infancy, with AK-leukemic extracts, or AK-embrvos. Proc. Soc. Exp. Biol. Med. 1951;76:27–32. [PubMed] [Google Scholar]
- 199.Gross L. A filterable agent, recovered from Ak leukemic extracts, causing salivary gland carcinomas in C3H mice. Proc. Soc. Exp. Biol. Med. 1953;83:414–421. doi: 10.3181/00379727-83-20376. [DOI] [PubMed] [Google Scholar]
- 200.Eddy BE, Borman GS, Grubbs GE, Young RD. Identification of the oncogenic substance in rhesus monkey kidney cell culture as simian virus 40. Virology. 1962;17:65–75. doi: 10.1016/0042-6822(62)90082-x. [DOI] [PubMed] [Google Scholar]
- 201.Trentin JJ, Yabe Y, Taylor G. The quest for human cancer viruses. Science. 1962;137:835–841. doi: 10.1126/science.137.3533.835. [DOI] [PubMed] [Google Scholar]
- 202.Kaposi’s sarcoma and Pneumocystis pneumonia among homosexual men-New York City and California. MMWR Morb. Mortal. Wkly Rep. 1981;30:305–308. [PubMed] [Google Scholar]
- 203.Barre-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 204.Baer R, et al. DNA sequence and expression of the B95–98 Epstein-Barr virus genome. Nature. 1984;310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 205.DeCaprio JA, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 206.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 207.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 208.Albrecht J-C, et al. Primary structure of the herpesvirus saimiri genome. J. Virol. 1992;66:5047–5058. doi: 10.1128/jvi.66.8.5047-5058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 210.Cesarman E, et al. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi’s sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86:2708–2714. [PubMed] [Google Scholar]
- 211.Schiffman M, Clifford G, Buonaguro FM. Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline. Infect. Agent. Cancer. 2009;4:8. doi: 10.1186/1750-9378-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]