

Abstract
We have designed, synthesized and tested conjugates of chemically modified luciferase siRNA (Luc-siRNA) with bi-, tri- and tetravalent cyclic(arginine-glycine-aspartic) peptides (cRGD) that selectively bind to the αvβ3 integrin. The cellular uptake, subcellular distribution and pharmacological effects of the cRGD conjugated Luc-siRNAs as compared to un-conjugated controls were examined using a luciferase reporter cassette stably transfected into αvβ3 positive M21+ human melanoma cells. The M21+ cells exhibited receptor-mediated uptake of cRGD-siRNA conjugates but not of unconjugated control siRNA. The fluorophore-tagged cRGD-siRNA conjugates were taken up by a caveolar endocytotic route and primarily accumulated in cytosolic vesicles. The bi-, tri- and tetravalent cRGD conjugates were taken up by M21+ cells to approximately the same degree. However, there were notable differences in their pharmacological effectiveness. The tri- and tetravalent versions produced progressive, dose-dependent reductions in luciferase expression, while the bivalent version had little effect. The basis for this divergence of uptake and effect is currently unclear. Nonetheless the high selectivity and substantial ‘knock down’ effects of the multivalent cRGD-siRNA conjugates suggest that this targeting and delivery strategy deserves further exploration.
INTRODUCTION
Antisense and siRNA oligonucleotides have displayed considerable promise for treatment of a variety of diseases. Advancement of new chemistries and design features have led to oligonucleotides that provide improved stability against nucleases, enhanced potency, reduced immune responses and reduced toxicity profiles compared to early generations of oligonucleotides.1–3 Excellent results for both antisense and siRNA have been attained in cell culture and animal models, and this has driven the initiation of multiple clinical trials.1,4 However, despite important progress, a key impediment to the wider success of this approach is poor delivery of oligonucleotides to their sites of action within cells, due to the need to cross several biological barriers subsequent to administration.5,6
The endogenous regulatory mechanisms involved in RNA interference (RNAi) can be activated by the delivery of short double stranded RNAs (siRNA) to the cell interior.7 The mechanism of targeted mRNA degradation by siRNA is highly complex and not completely understood. It includes the formation of an RNA-induced silencing complex (RISC) that contains the Argonaute 2 protein, while the Dicer RNase and other accessory proteins are also important in loading of siRNA on to the RISC complex.8 The potency and specificity of siRNAs can be improved and their impact on the innate immune system can be reduced via chemical modification guided by design algorithms.3 Nonetheless, effective delivery of siRNA to its intracellular sites of action remains a challenge.
One important modality for delivery of both antisense and siRNA molecules has been the use of polymeric or lipid nanocarriers.9 Lipid based carriers have proven to be particularly efficacious in the delivery of siRNA to the liver.10 A variety of polymeric11 or other types of nanoparticles12 have also been employed for siRNA delivery. Effective deployment of siRNA to tumors has been challenging, but recently targeted lipid based nanoparticles have displayed substantial activity.13
An alternative approach for delivery of antisense and siRNA moieties involves covalent conjugation of high affinity and receptor specific ligands that bind to cell surface receptors.14–17 This strategy can be used to provide cell-type selective targeting and delivery, and thereby to enhance tissue specific intracellular accumulation of oligonucleotides. For example, we have utilized peptide ligands with high affinity to integrins,18,19 to a G Protein-Coupled Receptor,20 or to the sigma receptor,21 to enhance antisense oligonucleotide delivery into tumour cells that over-express these receptors.
In the present report we extend the strategy for tissue specific delivery of siRNAs by conjugating high affinity cRGD ligands that are known to bind to αvβ3 integrin to luciferase siRNA. The high affinity bi-, tri-, and tetravalent cRGD were conjugated to the 3′-end of the sense strand of the siRNA. We demonstrate enhanced receptor-specific cellular uptake of cRGD-siRNA conjugates and dose dependent inhibition of a luciferase reporter gene in the absence of transfection agents. We also demonstrate uptake inhibition of cRGD-siRNA conjugates by preincubation of the αvβ3 integrin positive cells22 with excess RGD peptide, and associations of biological effect with the valency or cRGD content of the siRNA conjugate studied.
MATERIALS AND METHODS
Reagents
Bivalent, trivalent and tetravalent of cyclic RGD (cRGD) peptides were obtained from Peptides International (Louisville, Kentucky). The amino acid residues of the bi-, tri- and tetravalent cRGD peptides respectively are: H-Cys-Aca-Glu-[cyclo(Arg-Gly-Asp-D-Phe-Lys)]2, H-Cys-Aca-Glu-[cyclo(Arg-Gly-Asp-D-Phe-Lys)]-Glu [cyclo(Arg-Gly-Asp-D-Phe-Lys)]2 and H- Cys-Aca-Glu-{Glu-[cyclo(Arg-Gly-Asp-D-Phe-Lys)]2}2. A thiol group was introduced to the cRGDs by attaching a cysteine residue via the spacer – 6-aminocaproic acid, to the α-amino group of glutamic acid to enable conjugation of the peptide to a maleimide containing oligonucleotide under Michael addition conditions (Figure 1).
FIGURE 1. Chemical structure of cysteine-bearing cyclic RGD (cRGD) peptide.

The cysteine is connected to a 6-aminohexanoic acid linker that joins two, three and four cyclic RGD moieties respectively for the bi-, tri and tetravalent versions. Only the trivalent peptide is shown here. Arrow indicates the site of conjugation of the peptide (thiol) to the maleimide functionality on the luciferase siRNA sense strand.
Solid-Phase Synthesis of Modified siRNA Targeting the Firefly Luciferase Gene
All oligonucleotides used in this study were synthesized on an ABI 394 DNA synthesizer using standard phosphoramidite chemistry with commercially available phosphoramidite monomers. The oligonucleotides were de-protected with methylamine solution at 65 °C for 10 min (40 wt % in water) followed by DMSO wash and cooled on dry ice for 10 min, then treated with TEA.3HF at 65 °C for 12 min. All oligonucleotides were purified by anion-exchange HPLC and characterized by LC-MS analysis. For peptide conjugation an amino linker was introduced at the 3′-end of the sense strand by initiating the oligonucleotide synthesis on a trans-4-hydroxyproline based support 4 under standard oligonucleotide synthesis conditions (Scheme 1).
Scheme 1a.

Conjugation of cRGD peptides to siRNA
a (i) Solid phase oligonucleotide synthesis and deprotection; (ii) PBS buffer, pH 7.2, rt, 20 min;(iii) 0.4M KCl, 40% MeCN in H2O, rt, overnight; (iv) antisense strand, annealing at 95 °C in H2O;(v) Alexa 488 NHS ester, PBS buffer, pH 7.2, 20 min.
Postsynthetic Conjugation of cRGD Peptides to Oligonucleotide
The HPLC purified amino linked sense strand 5 (23.6mg, 3.35μmole) was reacted with 6-maleimidohexanoic acid N-hydroxysuccinimide ester (EMCS) (21mg, 68μmol) in 1x PBS buffer, at pH 7.2 for 20min at RT to obtain maleimido modified sense strand 6 (Scheme 1). The reaction mixture was then diluted with HPLC grade water (15mL) and directly loaded onto a Waters XTerra RP-18 column on an AKTA purifier (GE Healthcare, USA) and purified using a gradient of buffers A and B. Buffer A: 0.1M TEAA at pH 7.0 and buffer B: CH3CN; gradient: 0% B for 1 column volume, then 10% to 50% of B for 5 column volumes and 50% to 100% of B for 2 column volumes; flow rate:15mL/min, UV: 260nm. The HPLC fractions with the right mass were collected (22.6mg, 3.11μmol, 93%), aliquoted into the desired quantity and treated immediately with peptides 1–3 individually to obtain the desired cRGD conjugated sense strands 7–9 (Scheme 1). The products formed were analyzed by analytical HPLC and LC-MS to confirm integrity.
Bivalent cRGD peptide 1 (1.6mg, 1.02μmol) was dissolved in water (1mL) and added to the solution of maleimido oligonucleotide 6 (3mg, 0.41μmol) in 0.4M KCl with 40% CH3CN, and the mixture was incubated overnight at room temperature. The reaction mixture was dialyzed in water and loaded onto a Waters XTerra RP-18 column on the AKTA purifier and purified using buffers A and B under same conditions used for the purification of maleimido oligonucleotide 6. Fractions containing the bivalent conjugate 7 were pooled and lyophilized (3.4mg, 0.39μmol, 95%). The tri- and tetravalent cRGDs (2 and 3) were conjugated to maleimido oligonucleotide 6 under similar conditions to obtain the corresponding oligonucleotide conjugates 8 and 9 (Scheme 1). Post-synthetic conjugation of trivalent cRGD peptide 3 (2.3mg, 1.02μmol) to maleimido oligonucleotide 6 (3mg, 0.41μmol) yielded the conjugate 8 (3.3mg, 0.348μmol, 84%). Conjugation of the peptide 3 (2.0mg, 0.68μmol) to oligonucleotide 6 (2mg, 0.27μmol) gave the desired conjugate 9 (1.8mg, 0.18μmol, 67%). The trivalent cRGD conjugated sense strand 10 (Table 1) of a more potent luciferase siRNA was also synthesized according to similar conditions. All conjugated oligonucleotides were analyzed by HPLC and LC-MS to confirm the integrity of the products formed (Table 1).
Table 1.
cRGDs conjugated sense strands of luciferase siRNA
Similarly antisense strand 15 with the fluorophore Alexa 488 at the 3′-end was obtained from the corresponding amino linked antisense strand synthesized from the solid support 4 (Scheme 1). The amino linked oligonucleotide obtained from 4 was post-synthetically reacted with commercially available Alexa 488 NHS ester to obtain the fluorophore tagged antisense strand 15 under similar conditions described for the synthesis of maleimido oligonucleotide 6.
Preparation of Luciferase cRGD-siRNA Conjugates
cRGD-siRNA conjugates with and without the fluorophore Alexa 488 were prepared for the present study. Annealing of cRGD conjugated oligonucleotides 7–9 individually with equimolar amount of antisense strand at 95 °C for 3 min in sterile water followed by slow cooling afforded the siRNAs 11, 12 and 13 (Scheme 1, Table 2). While our studies were in progress we identified a more potent luciferase siRNA sequence and a trivalent cRGD-siRNA conjugate 14 of the potent siRNA was also prepared to study conjugate mediated RNAi activity in vitro. Similarly annealing of conjugates 7–9 with Alexa 488 tagged antisense strand 15 afforded the fluorophore labeled siRNAs 16–18 (Scheme 1, Table 2). Each set contains the control siRNAs 19 and 20 with no cRGD moiety. Duplex formation was confirmed by capillary gel electrophoresis and LC-MS analysis.
Table 2.
Luciferase siRNAs conjugated to multivalent cRGD
| siRNA | S/ASa | Sequence (5′-3′)b | cRGD content |
|---|---|---|---|
| 19 | S | cuuuAcGcuGAGuAcuucGAdTdT | 0 |
| AS | UCGAAGuACUcAGCGuAAGdTdT | ||
| 11 | S | cuuuAcGcuGAGuAcuucGAdTdTX1 | 2 |
| AS | UCGAAGuACUcAGCGuAAGdTdT | ||
| 12 | S | cuuuAcGcuGAGuAcuucGAdTdTX2 | 3 |
| AS | UCGAAGuACUcAGCGuAAGdTdT | ||
| 13 | S | cuuuAcGcuGAGuAcuucGAdTdT X3 | 4 |
| AS | UCGAAGuACUcAGCGuAAGdTdT | ||
| 14 | S | AccGAAAGGucuuAccGGAdTsdTX2 | 3 |
| AS | UCCGGuAAGACCUUUCGGUdTsdT | ||
| 20 | S | cuuuAcGcuGAGuAcuucGAdTdT | 0 |
| AS | UCGAAGuACUcAGCGuAAGdTdTY | ||
| 16 | S | cuuuAcGcuGAGuAcuucGAdTdT X1 | 2 |
| AS | UCGAAGuACUcAGCGuAAGdTdTY | ||
| 17 | S | cuuuAcGcuGAGuAcuucGAdTdT X2 | 3 |
| AS | UCGAAGuACUcAGCGuAAGdTdTY | ||
| 18 | S | cuuuAcGcuGAGuAcuucGAdTdTX3 | 4 |
| AS | UCGAAGuACUcAGCGuAAGdTdTY | ||
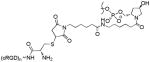 X1: n = 2; X2: n = 3; X3: n = 4 X1: n = 2; X2: n = 3; X3: n = 4 |
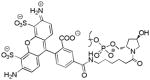 Y Y |
||
Sense/Antisense
Direction of sequence
Cell lines
M21+ melanoma cells with high expression of αvβ3 integrin,22 as well as M21- cells that lack this integrin, were obtained from Dr. D. Cheresh (University of California, San Diego) and were cultured in DMEM medium (Gibco/Invitrogen) supplemented with 10% FBS (Sigma). M21+ cells were transfected with a luciferase expression plasmid pGL3-promoter (Promega) and pcDNA3.1/hygro (Invitrogen) at a 10:1 ratio using an Amaxa Nucleoporation system as per manufacturer’s instructions. Selection was carried out in DMEM medium containing 500 μg/ml hygromycin B (Roche) and 10% FBS for two weeks. Individual clones were picked and screened for luciferase activity using a Luciferase assay kit (Promega). The single cell clone with the highest activity was referred to as M21+GL3 and used in further studies.
Cell uptake studies
Uptake studies of the fluorescently labeled siRNA conjugates were performed similarly to our previous studies on uptake of antisense oligonucleotides.20 Briefly, total cellular uptake of the Alexa 488-labeled siRNA was measured by flow cytometry using a LSR II cell analyzer (Becton-Dickenson, San Jose, CA, USA). After treatment with oligonucleotides for various times, the cells were trypsinized and analyzed by flow cytometry, with a 490 nm laser and 520 nm emission filters for Alexa 488 fluorescence. In all cases oligonucleotide concentrations used were normalized on Alexa 488 fluorescence.
Luciferase knockdown studies
M21+GL3 cells were treated with various concentrations of cRGD- conjugated or control siRNAs for 4 h. in serum free medium. At this point serum was added to 2% and the incubation continued to 96 h. Cells were lysed and luciferase activity and cell protein determined as previously described.20
Confocal fluorescence microscopy
Intracellular distribution of the Alexa 488 labeled siRNA conjugates in living cells was examined using an Olympus Confocal FV300 fluorescent microscope with 63×-oil immersion objectives. M21+GL3 cells were plated in 35 mm glass bottom microwell dishes (MatTek, Ashland, MA). Intracellular uptake of the siRNA conjugates (50 nM) was visualized by confocal microscopy after 4-hour or 24-hour treatment. Co-localization of the RGD-siRNA-Alexa 488 conjugates with Alexa 594 labeled transferrin (Tfn) or cholera toxin B (CTB) (Invitrogen), as markers for clathrin-coated vesicles and lipid rafts, respectively, was also done by confocal microscopy. Transferrin (20 μg/ml) and cholera toxin B (4μg/ml) were used for 15 minutes and 30 minutes, respectively, prior to live cell imaging.
RESULTS
Synthesis and characterization
The chemical structure of the trivalent cRGD peptide is illustrated in Figure 1; the bi- and tetravalent versions have analogous structures. Each peptide contains a cysteine residue attached to the 6-aminohexanoic acid that joins the cRGD moieties through the glutamic acid residue. The thiol (−SH) reactive group of the cysteine residue of each peptide undergoes chemical conjugation to the maleimide functionality of the sense strand of firefly luciferase siRNA via a Michael addition reaction. The amino linked sense strand 5 of luciferase RNA obtained from the solid support 4 as shown in Scheme 1 was reacted with 6-maleimidohexanoic acid N-hydroxysuccinimide ester to obtain maleimido functionalized sense strand 6. The reaction was performed under controlled conditions for only 20 min in 1x PBS buffer at pH 7.2 to avoid maleimide degradation. The product was immediately purified by RP-HPLC and the purified material was directly used for the conjugation reaction. The peptide conjugation reaction occurred between the maleimido functionalized sense strand 6 of luciferase siRNA and the reactive thiol group (−SH) of the multivalent cRGD peptides 1–3 (Scheme 1). Using 2.5 equivalents of peptide in each conjugation reaction, almost all starting maleimido oligonucleotide 6 was consumed and converted to the corresponding cRGD conjugated sense strand with an excellent yield (Table 1). The conjugates were purified by reverse-phase HPLC (RP-HPLC) using an AKTA purifier. The HPLC profiles for the purification of bivalent, trivalent and tetravalent cRGD conjugates are shown in Figure 2 (a), (b) and (c) respectively. The arrows in each RP-HPLC chart indicate the position of maleimido oligonucleotide 6 and the corresponding cRGD conjugate. Each HPLC profile showed that the conjugate peak was clearly resolved from its precursor 6. After purification, bivalent cRGD conjugate 7 was obtained at relatively higher yield (95%) than trivalent cRGD conjugate 8 (84%) which in turn was higher than the tetravalent conjugate 9 (67%). After lyophilization, the conjugates were re-dissolved in sterile water without any difficulty. We have not seen aggregation of these conjugates in water. For all cases, the structure of the final product was confirmed by LC-MS analysis.
FIGURE 2. The conjugation of multivalent cyclic RGD peptides to the maleimide containing sense strand of the luciferase siRNA.

The oligonucleotides (A-59841.1 and A-59842.1) were purified by HPLC after each step in the synthesis.
(I) 6-Maleimidohexanoic acid N-hydroxysuccinimide ester, 1x PBS buffer, pH 7.2, 20 min, room temperature (RT);
(II) Cysteine-bearing multivalent cRGD peptides, 0.4M potassium chloride, 40% aqueous acetonitrile, overnight, RT.
Annealing of siRNA-cRGD Conjugates
After conjugation of cRGD peptides to the 3′-end of the sense strand of luciferase siRNA, we initially attempted to anneal the cRGD conjugated sense strand 7–9 with the complementary antisense strand in 1x PBS buffer at pH 7.4 according to a standard annealing procedure.23 However we observed a degradation of the cRGD conjugated sense strand during annealing process with the extent of degradation depends on annealing temperature. The degraded products were identified as the corresponding cRGD peptide and the carboxylic acid 19 by LC-MS analysis. The plausible mechanism of degradation of the cRGD-siRNA conjugate at elevated temperature in PBS buffer at pH 7.4 is the intra-molecular amine assisted retro-Michael addition reaction shown in Scheme 2. Interestingly the degradation was not observed in metal free sterile water at elevated temperature. Therefore, the annealing was performed in sterile water to obtain the desired cRGD-siRNA conjugates 11–14 and 16–18 (Scheme 1). We prepared two sets of cRGD-siRNA conjugates in sterile water with fluorescently labeled antisense strand 15 or one without the fluorescent label (Table 2). The appendage of the reporter group to the antisense strand of cRGD-siRNA conjugates 16–18 enabled evaluation of cRGD assisted cellular uptake and intracellular trafficking of the conjugates. The conjugates 11–14 with no fluorescent tag were used for the determination of the efficacy of the conjugates by knockdown assay. After annealing, the integrity of double stranded cRGD-siRNA conjugates were analyzed and confirmed by capillary gel electrophoresis and LC-MS analysis.
Scheme 2a.

a Intra-molecular amine assisted retro Michael addition of cRGD-Oligonucleotide conjugate at elevated temperature (>90 °C) in PBS buffer at pH 7.4.
Cellular uptake
The conjugation of cRGD moieties dramatically enhanced the uptake of siRNA by M21+GL3 melanoma cells. As seen in Figure 3, the control non-conjugated siRNA 20 displayed little uptake at all concentrations tested. In contrast, the bi-, tri-, and tetravalent cRGD conjugates 16–18 each displayed enhanced uptake that increased progressively with concentrations ranging from 5 to 100 nM. No evidence of saturation of uptake was noted in the concentration range tested. Previous studies have indicated that the affinities of multivalent RGD conjugates for αvβ3 are in the range of 30–100nM.24 However, we did not seek to measure affinity in the current study. There were no major differences in the uptake of the siRNA with bi, tri or tetravalent conjugation. The uptake of the cRGD-siRNA conjugates clearly involved specific interactions of the RGD group with its receptor, since αvβ3 positive M21+ cells accumulated far more of the RGD-conjugated siRNA than the control non-conjugated siRNA while αvβ3 negative M21- cells did not (Figure 4A). Further, excess RGD peptide competitively blocked the cRGD-siRNA uptake process (Figure 4B). In contrast, uptake was only slightly affected by the presence of serum proteins even though some potential competitors such as vitronectin and fibronectin are present in low amounts in serum (Figure 4C). Uptake and RGD inhibition experiments were also conducted with compound 14, a trivalent conjugate with a more potent siRNA sequence. The results were similar to the other RGD conjugates and are shown in the Supplementary Material (Figure S1).
FIGURE 3. HPLC Analysis.

AKTA RP-HPLC purification profiles for the crude reaction mixtures of (a) bivalent (b) trivalent and (c) tetravalent RGD-sense oligonucleotide conjugates. Arrows indicate the relative change of retention time of the conjugates (A-69252.1, A-69253.1, and A-69254.1) from the maleimide-RNA sense strand (A-59842.1). HPLC analysis was performed as described in the materials and methods section.
FIGURE 4. Possible degradation product.

A possible mechanism for the degradation of cRGD-sense strand conjugate in 1X PBS buffer, pH 7.4 during annealing (95°C, 3min, then cooled to RT).
Inhibition of luciferase expression
The cRGD conjugates 11–13, as well as the control non-conjugated siRNA 19 were designed to inhibit luciferase expression. The control siRNA 19 had no activity unless it was complexed with a transfection agent. For example, when complexed with Lipofectamine 2000, the non-conjugated control at 100 nM caused approximately an 80% reduction in luciferase activity. The cRGD conjugated siRNAs were tested in the absence of any transfection agent. Interestingly the tri- and tetravalent cRGD conjugates 12 and 13 produced progressive dose-dependent reductions in luciferase activity while the bivalent version 11 had little effect (Figure 5). Using the original siRNA sequence, the trivalent cRGD conjugate 12 produced a 40% reduction in luciferase at 50nM. During the course of our studies we identified a more potent luciferase siRNA for cRGD conjugation and it was conjugated to the trivalent cRGD ligand to obtain the conjugate 14 (Table 2). The newly designed conjugate 14 produced a 70% reduction in luciferase at 25 nM (Figure 5). Interestingly the biological effect of luciferase knockdown was not well correlated with the total cell uptake. As shown in Figure 3, all three fluorophore labeled cRGD conjugates 16–18 displayed enhanced cell uptake, but as seen in Figure 5 only the tri- and tetravalent versions produced significant knockdown.
FIGURE 5. Uptake dose-response for control and cRGD- conjugated siRNAs.
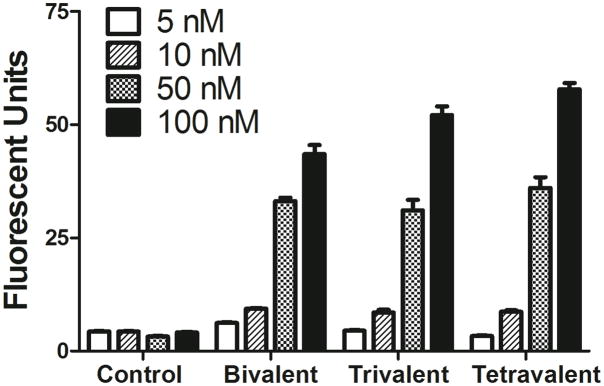
M21+GL3 cells were treated with various concentrations of control or RGD-conjugated Alexa 488 labeled siRNA for 4 h at 37°C in OPTI-MEM without serum. Thereafter the cells were harvested and analyzed for fluorescence levels by flow cytometry. Results are the means and standard errors of triplicate determinations.
Subcellular localization
We examined the subcellular localization of the Alexa 488 labeled siRNA at 4 and 24 h time points, comparing the unconjugated 20 and tetravalent cRGD conjugate 18. As seen in Figure 6A, the unconjugated labeled siRNA 20 was not detectable, while the cRGD conjugate 18 displayed substantial uptake into vesicular structures in the cytosol, these presumably being various types of endosomes. At 24 h the accumulated fluorescence was more intense and was now concentrated in perinuclear locations. Most of the material continued to be vesicular, although some slight cytosolic fluorescence was discernable. In order to more precisely identify the endomembrane compartments involved in the uptake and trafficking of the RGD-siRNA, we co-incubated the Alexa 488 labeled oligonucleotide (green) with Alexa 594 (red) labeled markers for well-known endocytotic pathways. Thus transferrin is known to be internalized via the clathrin coated pit pathway, while cholera toxin is substantially internalized via the caveolar pathway or other lipid raft dependent pathways.25 As seen in Figure 6B, at 4 h there was virtually no overlap of the labels for transferrin and siRNA; in contrast there was substantial overlap of the cholera toxin and the cRGD siRNA conjugate 18. This suggests that the initial uptake pathway for cRGD-siRNA involves calveolae; this observation is consistent with literature on the mechanism of internalization of the αvβ3 integrin.26 We further investigated the uptake pathways by using a series of well-known pharmacological inhibitors of various endocytotic processes. The trivalent cRGD-siRNA 14 was incubated with M21+ cells in the presence of non-toxic concentration of selected inhibitors and the results are summarized in Table 3. Methyl-β-cyclodextrin, an agent that affects lipid raft stability, had the greatest inhibitory effect. This observation supports the notion that lipid raft/caveolar pathways are important in the internalization of the RGD-conjugates.
FIGURE 6. (A) Uptake of cRGD-conjugated siRNA by M21+ or M21- cells.
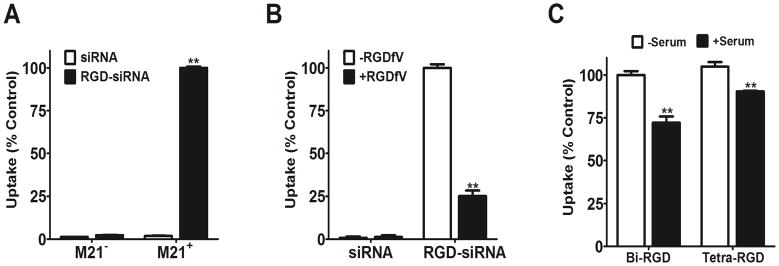
M21+GL3 cells (avb3 positive) or M21- cells (avb3 negative) were treated with 100 nM control or tetravalent RGD conjugated siRNA for 4 h. Uptake of Alexa 488 labeled siRNA was measured by flow cytometry. Data normalized on M21+ cells with RGD-siRNA as 100%. (B) Uptake of tetravalent RGD-siRNA conjugates plus or minus the presence of cycloRGD inhibitor. M21+GL3 cells were pretreated with 10 mg cycloRGD peptide for 15 min followed by treatment with 50 nM control siRNA or tetravalent cRGD-siRNA for 4 h at 37°C. Thereafter the cells were harvested and analyzed for Alexa 488 fluorescence levels by flow cytometry. Data normalized on RGD-siRNA (no inhibitor) as 100%. (C) Uptake of RGD-conjugated siRNA with or without serum. M21+GL3 cells were treated with 100 nM control or RGD conjugated siRNAs for 4 h at 37°C in OPTI-MEM with 5% FBS or without serum. Thereafter the cells were harvested and analyzed for fluorescence levels by flow cytometry. Data normalized on tetra-RGD-siRNA (no serum) as 100%. Results A–C are means and standard errors of triplicate or quadruplicate determinations.
Table 3.
Effects of Inhibitors on cRGD-siRNA Uptake
| siRNA 50 nM | Drug/Inhibitor | Conc. | Endocytosis Pathway Putatively Affected | cRGD-siRNA uptake |
|---|---|---|---|---|
| 14 | None | 100 ± 1.4 % | ||
| Chlorpromazine | 2.5 μM | Clathrin mediated endocytosis | 93.3 ± 1.9 % | |
| Methyl-β-cyclodextrin | 1 mM | Lipid-raft mediated endocytosis | 42.8 ± 1.0 % | |
| Amiloride | 100 μM | Macropinocytosis | 98.5 ± 2.3 % | |
| Cytochalasin D | 2 μM | Actin inhibitor | 37.9 ± 0.9 % |
M21+ cells were treated with the indicated non-toxic concentration of drugs and trivalent cRGD-siRNA 14. Uptake was measured after 3 h by flow cytometry. Results were normalized to uptake of the siRNA conjugate 14 by the drug/inhibitor free control cell lines.
Cytotoxicity
There was little toxicity associated with the use of the cRGD-siRNA conjugates at the concentrations examined. Thus in the ‘knockdown’ experiments of Figure 5 the cell protein recovered in each of the treated samples was essentially the same as for the untreated control. The only exceptions were the cases where Lipofectamine 2000 was used where some protein loss was observed (data not shown). Additionally the cells treated with the cRGD-siRNA conjugates maintained normal morphology as illustrated in the DIC images of Figure 6.
DISCUSSION
It would be of substantial value to be able to effectively target antisense or siRNA oligonucleotides to specific cell types by making use of differential expression of cell surface receptors. In this study we have focused on the αvβ3 integrin, a cell surface glycoprotein that is preferentially expressed in angiogenic endothelia and in certain tumor types including melanomas.27,28 The process of targeted oligonucleotide delivery using conjugates with high affinity ligands involves several steps. First there is selective association with the receptor followed by some form of endocytosis of the receptor-ligand complex. This is followed by trafficking through various endomembrane compartments such as early and late endosomes, the trans-Golgi apparatus, and lysosomes. Finally, there is release from membrane bound compartments into the cytosolic or nuclear sites of action, respectively, of siRNA and antisense oligonucleotides.5 It has become clear that there are important relationships between the intracellular trafficking of receptors and their functional activity.29 Likewise we have developed initial evidence that intracellular trafficking pathways can influence the pharmacological activity of oligonucleotides.19
The current report demonstrates that high affinity cRGD ligands can strongly and specifically enhance uptake of siRNA by cells that express the αvβ3 integrin. Uptake was blocked by excess unconjugated RGD peptide and receptor positive M21+ cells accumulated the cRGD-siRNA conjugates to a much greater degree than receptor negative M21- cells. Interestingly, increasing the valency from bi- to tetravalent had little effect on the magnitude of the overall uptake process. Confocal microscopy and inhibitor studies indicated that initial uptake of the cRGD-siRNA conjugates was primarily via caveolar endocytosis.
A surprising result was the difference in pharmacological effectiveness of conjugates with differing valencies. While the bivalent cRGD-siRNA conjugate displayed only a slight effect, the tri- and tetravalent versions produced substantial dose-dependent reduction in luciferase expression. This was especially true of the trivalent conjugate 14 made with the more potent luciferase siRNA sequence. Since all of the conjugates were taken up approximately equally well, this suggests that events downstream of the initial uptake are responsible for the different effects observed. One possibility is that the several conjugates follow different intracellular trafficking pathways or traverse those pathways at different rates. However, our current observations indicate that, at least at early time points, the tetravalent cRGD-siRNA conjugate follows the caveolar internalization pathway that we have previously described for bivalent cRGD-oligonucleotide conjugates.18 It is possible that the higher valency conjugates are retained more effectively within the cells at later time points, but this issue is difficult to study because of the inevitable degradation of the siRNA at longer times. Another possibility is that the presence of multiple cRGD peptides may cause a destabilization of the endosome membrane leading to more effective oligonucleotide release to the cytosol. However, at this point we do not have a clear-cut mechanistic explanation of the differing efficacy of the bi- and multivalent conjugates. Nonetheless the current results are encouraging in that they clearly demonstrate receptor specific targeting and significant pharmacological effects of multivalent ligand-siRNA conjugates. This could emerge as an interesting alternative to the delivery of siRNA using various forms of lipid or polymer nanoparticles. However, further evolution of the chemical conjugation approach will require both a better understanding of mechanism and also validation in an in vivo setting.
Supplementary Material
FIGURE 7. Knockdown dose-response for cRGD conjugated siRNAs.

M21+GL3 cells were treated with various concentrations of control siRNA or RGD-conjugated siRNA for 4 h at 37°C in OPTI-MEM without serum. The control siRNA complexed with Lipofectamine 2000 was used as a positive control. After 4 h serum was added to 2% to all wells with the exception of the Lipofectamine 2000 in which the medium was replaced with DMEM-H with 2% serum. After 96 h the cells were lysed and luminescence analyzed on a Fluostar Omega microplate reader and normalized to total cellular protein. Results are the means of pentuplicate determinations and are expressed as percent of untreated control cells (ordinate). The abcissa shows siRNA concentrations (nM). The unconjugated siRNA had no effect at any concentration tested (not shown) unless it was complexed with Lipofectamine 2000, in which case an 80% reduction was attained at 100 nM for the original sequence while a 99% reduction was obtained for the ‘B’ sequence (not shown).
FIGURE 8. (A) Cellular distribution of siRNAs.

M21+GL3 cells were incubated with control siRNA-Alexa 488 (50 nM) for 4 hours or Tetravalent RGD-siRNA-Alexa 488 (50 nM) for 4 hours and 24 hours. Live cells were observed by confocal fluorescence microscopy as described in the materials and methods section. B. Co-localization with endocytosis markers. Tetravalent RGD-siRNA-Alexa 488 (50 nM) was co-incubated with Alexa-594 labeled transferrin (Tfn) (20 μg/ml) and cholera toxin B (CTB) (4 μg/ml). Live cells were observed by confocal fluorescence microscopy as above. Selected vesicles showing co-localization are marked with white arrows.
Acknowledgments
We gratefully acknowledge Dr. D. Cheresh (University of California, San Diego) for providing M21+ and M21- melanoma cells and Drs. Meena and C Sherrill (Alnylam Pharmaceuticals) for providing amino functionalized oligonucleotides for conjugation. This work was supported by NIH grant P01GM059299 to RLJ and by Alnylam Pharmaceuticals.
Footnotes
Supporting Information: A supplementary figure is provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 2.Prakash TP, Bhat B. 2′-Modified oligonucleotides for antisense therapeutics. Curr Top Med Chem. 2007;7:641–9. doi: 10.2174/156802607780487713. [DOI] [PubMed] [Google Scholar]
- 3.Walton SP, Wu M, Gredell JA, Chan C. Designing highly active siRNAs for therapeutic applications. FEBS J. 2010;277:4806–13. doi: 10.1111/j.1742-4658.2010.07903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, de Fougerolles T, Maraganore J. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juliano R, Bauman J, Kang H, Ming X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol Pharm. 2009;6:686–95. doi: 10.1021/mp900093r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lares MR, Rossi JJ, Ouellet DL. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010;28:570–9. doi: 10.1016/j.tibtech.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623–32. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, Frank-Kamenetsky M, Yip KN, Alvarez R, Sah DW, de Fougerolles A, Fitzgerald K, Koteliansky V, Akinc A, Langer R, Anderson DG. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107:1864–9. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabanov AV, Vinogradov SV. Nanogels as pharmaceutical carriers: finite networks of infinite capabilities. Angew Chem Int Ed Engl. 2009;48:5418–29. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura A, Nagasaki Y. Smart siRNA delivery systems based on polymeric nanoassemblies and nanoparticles. Nanomedicine (Lond) 2010;5:1089–102. doi: 10.2217/nnm.10.76. [DOI] [PubMed] [Google Scholar]
- 13.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–31. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juliano R, Alam MR, Dixit V, Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158–71. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu L, Mahato RI. Targeted delivery of siRNA to hepatocytes and hepatic stellate cells by bioconjugation. Bioconjug Chem. 2010;21:2119–27. doi: 10.1021/bc100346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cesarone G, Edupuganti OP, Chen CP, Wickstrom E. Insulin receptor substrate 1 knockdown in human MCF7 ER+ breast cancer cells by nuclease-resistant IRS1 siRNA conjugated to a disulfide-bridged D-peptide analogue of insulin-like growth factor 1. Bioconjug Chem. 2007;18:1831–40. doi: 10.1021/bc070135v. [DOI] [PubMed] [Google Scholar]
- 17.Ming X. Cellular delivery of siRNA and antisense oligonucleotides via receptor-mediated endocytosis. Expert Opin Drug Deliv. 2011;8:435–49. doi: 10.1517/17425247.2011.561313. [DOI] [PubMed] [Google Scholar]
- 18.Alam MR, Dixit V, Kang H, Li ZB, Chen X, Trejo J, Fisher M, Juliano RL. Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis. Nucleic Acids Res. 2008;36:2764–76. doi: 10.1093/nar/gkn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alam MR, Ming X, Dixit V, Fisher M, Chen X, Juliano RL. The biological effect of an antisense oligonucleotide depends on its route of endocytosis and trafficking. Oligonucleotides. 2010;20:103–9. doi: 10.1089/oli.2009.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ming X, Alam MR, Fisher M, Yan Y, Chen X, Juliano RL. Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor. Nucleic Acids Res. 2010;38:6567–76. doi: 10.1093/nar/gkq534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa O, Ming X, Huang L, Juliano RL. Targeted intracellular delivery of antisense oligonucleotides via conjugation with small-molecule ligands. J Am Chem Soc. 2010;132:8848–9. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Felding-Habermann B, Mueller BM, Romerdahl CA, Cheresh DA. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J Clin Invest. 1992;89:2018–22. doi: 10.1172/JCI115811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25:1149–57. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 24.Li ZB, Cai W, Cao Q, Chen K, Wu Z, He L, Chen X. (64)Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor alpha(v)beta(3) integrin expression. J Nucl Med. 2007;48:1162–71. doi: 10.2967/jnumed.107.039859. [DOI] [PubMed] [Google Scholar]
- 25.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 26.Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic. 2006;7:14–21. doi: 10.1111/j.1600-0854.2005.00362.x. [DOI] [PubMed] [Google Scholar]
- 27.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eliceiri BP, Cheresh DA. Role of alpha v integrins during angiogenesis. Cancer J. 2000;6(Suppl 3):S245–9. [PubMed] [Google Scholar]
- 29.Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: location makes a difference. Exp Cell Res. 2009;315:1601–9. doi: 10.1016/j.yexcr.2008.09.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.