

Abstract
Purpose
The objective of this investigation was to assess whether common pharmaceutical excipients regulate the expression of drug-metabolizing enzymes in human colon and liver cells.
Methods
Nineteen commonly used excipients were evaluated using a panel of experiments including cell-based human PXR activation assays, real-time RT-PCR assays for CYP3A4 mRNA expression, and immunoblot analysis of CYP3A4 protein expression in immortalized human liver cells (HepG2 and Fa2N4), human primary hepatocytes, and the intestinal LS174T cell models.
Results
No excipient activated human PXR or practically induced CYP3A4. However, three excipients (polysorbate 80, pregelatinized starch, and hydroxypropyl methylcellulose) tended to decrease mRNA and protein expression across experimental models.
Conclusion
This study represents the first investigation of the potential role of excipients in the expression of drug-metabolizing enzymes. Findings imply that some excipients may hold potential for excipient-drug interactions by repression of CYP3A4 expression.
Keywords: Excipients, CYP3A4, PXR, Induction, Repression
INTRODUCTION
The Biopharmaceutics Classification System (BCS) is employed by various regulatory agencies to characterize drugs in terms of aqueous solubility and intestinal permeability in order to assess risk for bioinequivalence (1, 2). The US Food and Drug Administration has provided waivers of in vivo bioequivalence studies (i.e. “biowaivers”) for rapidly dissolving immediate-release sold oral dosage forms of Class 1 drugs, which exhibit high solubility and high permeability (3). It has been suggested that Class 3 drugs, which exhibit high aqueous solubility and low intestinal permeability, be considered for BCS-based biowaivers as well, with the further requirement that dissolution be very rapid, as rapidly dissolving Class 3 drug products essentially function as oral solutions (4–7).
BCS-based biowaivers of Class 3 drugs would assume that excipients do not modulate in vivo drug permeability, solubility, intestinal transit, or stability/metabolism, since test and reference products generally contain different excipients. Excipients are often described as inert constituents of drug formulations (e.g. filler in tablets or capsules). However, excipients have been shown to impact drug bioavailability of Class 3 drugs in vivo. For example, scintigraphic studies showed that sodium acid pyrophosphate reduced small intestinal transit time by 43%, thereby reducing ranitidine bioavailability (8). Similarly, cimetidine exhibited reduced bioavailability from a mannitol chewable tablet compared to its sucrose counterpart, due to reduced small intestinal transit time (9). However, such studies examining modulation of intestinal transit have not implicated common excipients at typical “excipient dose” levels in immediate release oral solid dosage forms as problematic.
One theoretical mechanism for excipients to modulate Class 3 drug absorption and metabolism is altering the expression of drug-metabolizing genes. Class 3 drugs possess biopharmaceutical properties that afford them to be susceptible to intestinal transporter effects (10), as well as transporters and possibly metabolic enzymes throughout the body (e.g. kidney, liver). Many drugs and environmental toxins are known to modulate gene expression of major drug-metabolizing enzymes and transporters (11, 12). Drug-mediated perturbation of these enzymes and transporters may affect the efficacy and toxicity of comedicated drugs, and results in pharmacokinetic-based drug-drug interactions (13, 14). Recently, Wasan and colleagues have reported that several lipid excipients and their components significantly decreased P-glycoprotein expression and activity in Caco-2 cells (15, 16).
The objective of this investigation was to assess whether several common pharmaceutical excipients were able to regulate the expression of drug-metabolizing genes. Cytochrome P450 3A4 (CYP3A4) was chosen as a candidate gene because it accounts for approximately 40% of total hepatic P450 protein in human and is involved in the metabolism of more than 50% of clinically used drugs (17, 18). Moreover, as a highly inducible P450 isozyme, drug-mediated induction of CYP3A4 is predominantly regulated at the transcription level by the xenobiotic receptor, pregnane X receptor (PXR), of which cell-based promoter reporter assays are well accepted for in vitro prediction of CYP3A4 induction (19, 20). Class 3 drugs possess biopharmaceutical properties that make them susceptible to intestinal transporter effects. Therefore, the multidrug-resistance (MDR1, ABCB1) gene was also included as a candidate gene in the intestinal LS174 cells.
MATERIALS AND METHODS
Cell Culture and Reagents
HepG2 cells from American Type Culture Collection (ATCC) were grown in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. LS174T cells (a kind gift from Dr. Douglas Ross, University of Maryland Greenebaum Cancer Center, Baltimore, MD) were maintained in the same media conditions. Fa2N4 cells (XenoTech; Lenexa, KS) were grown in multi-function enhancing (MFE) Support Media F supplemented with MFE Supplement A (XenoTech; Lenexa, KS). All cell cultures were maintained in a humidified incubator at 37°C and 5% CO2. Human primary hepatocyte cultures were derived from liver tissues acquired from the University of Maryland Medical Center following donor consent and prior approval from the Institutional Review Board at the University of Maryland Baltimore. Hepatocytes were isolated by a modified two-step collagenase digestion as previously described (21). Primary hepatocytes were seeded at 1.5 × 106 cells/well in six-well BioCoat plates in DMEM supplemented with 5% GBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 4 μg/ml insulin, and 1 μM dexamethasone. After 4 to 6 hours of attachment at 37°C in a humidified atmosphere of 5% CO2, cells were overlaid with Matrigel (0.25 mg/ml) in serum-free Williams’ E medium supplemented with ITS+ (6.25 μg/ml insulin, 6.25 μg/ml transferring, and 6.25 ng/ml selenium) and 0.1 μM dexamethasone. Hepatocytes were maintained with a daily medium change, and chemical treatments start 36–48 hours after seeding as described previously (22, 23).
Excipients
Table 1 lists studied excipients, including abbreviation, dose, functionality and source of each excipient. Nineteen excipients were studied. Dose of each excipient used here is defined as the typical amount of excipient in a tablet or capsule in a fasting gastric volume of 25 ml. However, several excipients used lower concentrations than this general approach as indicated in Table 1. Given their acidity, citric acid (CA) and malic acid (MA) used a 10-fold greater dilution (i.e. 0.4 mg/ml). Sodium lauryl sulfate (SLS) used a 100-fold greater dilution, as SLS is a potent surfactant. Polysorbate 80 (PS-80) was tested over a range of concentrations as indicated in figures. All excipients were dissolved/dispersed in 1X PBS. As Table 1 notes, not all excipients were soluble, as expected.
Table 1.
Information of excipients used in the current studies
| Abbr. | Excipient name | Dosea | Functionalityb | Source |
|---|---|---|---|---|
| SDc | Amorphous fumed silicon dioxide | 0.1 mg/ml | Glidant | Degussa Corp.; Ridgefield, NJ |
| CA | Citric acid | 0.4 mg/ml | Acidifier | Spectrum; Gardena, CA |
| CCSc | Croscarmellose sodium | 2.0 mg/ml | Disintegrant | FMC; Belgium |
| X-PVPc | Crospovidone | 2.0 mg/ml | Disintegrant | ISP Technologies; Wayne, NJ |
| DCPc | Dicalcium phosphate dehydrate | 20 mg/ml | Filler/binder | JRS Pharma; Germany |
| FAc | Fumaric acid | 4.0 mg/ml | Acidifier | Sigma; St. Louis, MO |
| HPMC | Hydroxypropyl methylcellulose | 20 mg/ml | Filler/binder | Dow Chemical; Midland, MI |
| LAC | Lactose | 20 mg/ml | Filler/binder | DMV International; The Netherlands |
| MgStrc | Magnesium stearate | 0.1 mg/ml | Lubricant | Mallincrodt; St. Louis, MO |
| MA | Malic acid | 0.4 mg/ml | Acidifier | Spectrum; Gardena, CA |
| MCCc | Microcrystalline cellulose | 20 mg/ml | Filler/binder | FMC; Philadelphia, PA |
| PEG-3350 | Polyethylene glycol 3350 | 10 mg/ml | Plasticizer | Dow Chemical; Midland, MI |
| PS-80 | Polysorbate-80 | 1.0 – 0.0001 mg/ml | Surfactant | Spectrum; Gardena, CA |
| PVP | Povidone | 2.0 mg/ml | Filler/binder | BASF; Germany |
| PgSc | Pregelatinized starch | 3.0 mg/ml | Filler/binder | National Starch Food; Bridgewater, NJ |
| PG | Propylene glycol | 0.4 mg/ml | Plasticizer | Dow Chemical; Midland, MI |
| SLS | Sodium lauryl sulfate | 2.0 mg/ml | Surfactant | Sigma; St. Louis, MO |
| SSGc | Sodium starch glycolate | 1.0 mg/ml | Disintegrant | Mendell; Patterson, NY |
| SUC | Sucrose | 10 mg/ml | Filler/binder | JRS Pharma; Patterson, NY |
Dose is defined as the amount of each excipient estimated to be in a tablet or capsule containing a total of 500mg of excipient dissolved in 25ml luminal volume.
Individual excipient compounds are assigned to one functionality group, but may have more than one function.
Excipient concentration was not completely soluble in PBS.
Data in all graphs are presented as fold change of vehicle control and excipients are organized by functionality groups: fillers/binders (DCP, PVP, MCC, HPMC, PgS, SUC and LAC); lubricants/glidant (MgStr and AmSD); disintegrants (SSG, CCS, and X-PVP); plasticizers (PEG-3350 and PG); acidifiers (CA, MA and FA); and surfactants (SLS and PS-80).
Luciferase Reporter Assay
HepG2 cells were seeded in 24-well plates at a density of 1.5 × 105 cells per well. After overnight culture, cells were transiently transfected with hPXR expression vector pSG5-hPXR obtained from Dr. Steve Kliewer (University of Texas Southwestern Medical Center, Dallas, TX), and CYP2B6-PBREM/XREM reporter construct (24) by Fugene 6 (Roche) following the manufacturer’s instruction. Twenty-four hours after transfection, cells were treated with DMSO (0.1%), rifampicin (10uM in 0.1% DMSO) (Sigma; St. Louis, MO), or excipients (in 1X PBS) at concentrations indicated in Table 1 for 24hr. Cell lysates were harvested in 1X Reporter Lysis Buffer and assayed for Firefly luciferase activity using Promega luciferase assay (Promega; Madison, WI). Transfection efficiency was normalized to total protein concentrations, which were measured by BCA assay (Thermo Scientific; Williston, VT).
Real-time PCR Analysis
LS174T cells were plated in 12-well tissue culture plates at a density of 1.5 × 105 cells per well and grown to approximately 80% confluence before treatment. Fa2N4 cells were plated in collagen-coated 6-well plates (BD; San Jose, CA) at the density of 2.5 × 105 cells per well and grown to approximately 80% confluence before treatment. HPH cultures were plated in collagen-coated 6-well (HL#014) and 12-well (HL#017) plates at a confluent density and covered with Matrigel overlay. All cells were treated for 24 hrs with excipients at indicated concentrations (Table 1), vehicle control (PBS) [denoted none], 10 μM rifampicin (RIF), or 20 μM ketaconazole (KTC). RIF and KTC served as induction and repression controls, respectively, and employed 0.1% DMSO co-solvent. Treated cells were harvested using TRIzol Reagent (Invitrogen; Calsbad, CA). Total RNA was isolated following manufacturer’s instructions and reverse transcribed using the High Capacity cDNA Archive kit (Applied Biosystems Inc; Foster, CA). Primers for CYP3A4 and MDR1 genes were designed using Primer Express Version 2.0 and entered into the NCBI Blast to ensure specificity as described previously (25) (CYP3A4-forward: 5′-GTGGGGCTTTTATGATGGTCA-3′; CYP3A4-reverse: 5′-GCCTCAGATTTCTCACCAACACA-3′; and MDR1-forward: 5′-GTGGTGGGAACTTTGGCTG-3′; MDR1-reverse: 5′-TACCTGGTCATGTCTTCCTCC-3′). The mRNA expression of CYP3A4 and MDR1 was normalized against that of GAPDH (GAPDH-forward: 5′-CCCATCACCATCTTCCAGGAG-3′, and GAPDH-reverse: 5′-GTTGTCATGGATGACCTTGGC-3′). SYBR green PCR assays were performed in 96-well optical plates on an ABI 7000 Sequence Detection System (Applied Biosystems Inc; Foster City, CA). Data is represented as fold induction calculated by the equation 2−ΔΔCt, where ΔΔCt is the relative change in threshold cycle between control and treated samples.
Western Blot Analysis
Fa2N4 cells were plated in collagen-coated 6-well plates at a density of 2.5×105 cells per well. HPH cultures (HL#017) were plated in collagen-coated 6-well tissue culture plates at a confluent density and covered with Matrigel overlay. All cultures were treated for 72 hrs with excipient, vehicle control, RIF (10 μM) or KTC (20 μM) as described above, and harvested in lysis buffer [20 mM Tris, 100 mM NaCl, 0.5% NP-40, 0.5 mM PMSF and 1X cocktail of protease inhibitors (Roche; Basel, Switzerland)]. Samples (20 μg) were separated on 10% polyacrylamide gel and electropheretically transferred to IPVDF membranes. Subsequently, membranes were incubated with CYP3A4 antibody (Millipore-Chemicon, CA) at 1:5000. B-actin (Sigma, St. Louis) was used for normalization of protein loading. After incubating with horseradish peroxidase goat anti-rabbit IgG antibody (1:4000), membranes were developed using SuperSignal ECL reagent (Pierce) and exposed to film.
Statistical Analysis
All data represent three independent experiments and are expressed as the mean ± S.D. Statistical comparisons were made using Students ‘t-test. Statistical significance was set at p values <0.05 (*, ^), or <0.01 (**, ^^).
RESULTS
Excipient activation of PXR in promoter reporter assays
The ability of excipients to activate human PXR was evaluated by promoter reporter assay in HepG2 cells. The CYP2B6-2.2kb construct, containing both the proximal phenobarbital responsive enhance module (PBREM) and the distal xenobiotic response enhancer module (XREM), was used to detect PXR activation as described previously (24). Excipient-mediated increases in CYP2B6 reporter activities were compared with vehicle control as percentage of the increase achieved by the positive control RIF (10 μM), which was normalized against DMSO (0.1%, v/v). The criterion from Faucette et al (23), which utilized 30% of RIF response as the threshold, was applied to evaluate the extent of excipient-mediated activation of PXR. Excipients exhibiting less than 30% RIF activation of hPXR were classified as weak or nonactivators of hPXR. As demonstrated in Figure 1, in contrast to the robust RIF activation of PXR, none of the 19 excipients showed marked ability to activate the CYP2B6 promoter.
Figure 1.

Activation of human PXR in cell-based promoter reporter assays. HepG2 cells were cotransfected with PXR expression vector and CYP2B6-PBREM/XREM reporter construct as described in “Materials and Methods”. Twenty-four hours after transfection, cells were treated with excipients at concentrations indicated in Table 1, RIF (10 μM), DMSO (0.1%), or PBS (none) for 24 hrs. After harvest, firefly luciferase activities were measured using luciferase reagent normalized to protein concentration. Data represent mean ± SD (n = 3) expressed as fold over vehicle control. The dot line indicates 30% of RIF activation.
Effect of excipients on the expression of CYP3A4 in immortalized hepatocytes
Immortalized hepatocyte Fa2N4 cells provide a more relevant hepatocyte system than immortalized hepatoma cell-line such as HepG2 cells and are more readily available than human primary hepatocyte cultures. Figure 2A illustrates the effect of 17 excipients on CYP3A4 mRNA induction in Fa2N4 cells after 24 hr treatment, where RIF (10 μM) and KTC (20 μM) served as induction and repression control, respectively. Fumaric acid (FA) and sodium lauryl sulfate (SLS) were not evaluated since they were cytotoxic in an MTT assay using HepG2 or Fa2N4 cells, where cytotoxicity was concluded if 60% or more mortality was observed at 24 hrs. The other 17 excipients were non-toxic in the MTT assays (data not shown).
Figure 2.


Excipients affect CYP3A4 expression in immortalized hepatocytes Fa2N4. Cultured Fa2N4 were treated with excipients at concentrations indicated in Table 1, positive control (RIF 10 μM) or PBS (none) and DMSO (0.1%) as respective solvent controls. (A) After treatment for 24 hrs, total RNA was isolated and realtime RT-PCR was used to detect CYP3A4 mRNA expression as outlined in “Materials and Methods”. (B) After treatment for 72 hrs, cell homogenates (20 μg) from each group were subjected to immunoblot analysis of CYP3A4 protein expression.
As expected, RIF treatment led to a robust induction of CYP3A4 mRNA (29-fold), while KTC repressed CYP3A4 mRNA expression by 70% (Fig. 2A). Excipients showed limited capacity to upregulate CYP3A4 expression. The hydrophilic polymers hydroxypropyl methylcellulose (HPMC) and croscarmellose sodium (CCS) caused a moderate but statistically significant 1.8-fold and 2.4-fold increase, respectively. Meanwhile, the water-insoluble excipients magnesium stearate (MgStr) and silicon dioxide (SD), and the surfactant PS-80 (1 mg/ml) decreased CYP3A4 mRNA expression by more than 40%, 70%, and 50%, respectively. Repression by MgStr, SD, and PS-80 was not due to cell toxic, as these excipients were not cytotoxic in MTT assays using HepG2 or Fa2N4 cells for a period of 24 hrs.
Figure 2B shows CYP3A4 protein expression in Fa2N4 cells treated for 72 hr with excipients and controls. As expected, RIF increased and KTC decreased CYP3A4 protein, compared to DMSO vehicle control. No excipient induced the expression of CYP3A4 protein. However, excipients HPMC, crospovidone (X-PVP), polyethylene glycol 3350 (PEG-3350), propylene glycol (PG), citric acid (CA), malic acid (MA), and PS-80 (0.1 and 0.01 mg/ml) decreased CYP3A4 protein expression compared to PBS vehicle control. Excipient SD was highly toxic to the Fa2N4 cells at 72 hr and was not concluded to repress CYP3A4. PS-80 at 1 mg/ml was also toxic in Fa2N4 cells at 72 hr in this assay (data not shown).
Effect of excipients on the expression of CYP3A4 in human primary hepatocyte cultures
Human primary hepatocytes (HPH) were prepared from two liver donors and treated with 13 of the 19 excipients. In Figure 3A, treatment with RIF (10 μM) resulted in 20- and 47-fold induction of CYP3A4 mRNA from donor HL#14 and HL#17, respectively, reflecting the inter-individual variability of CYP3A4 induction. Excipients povidone (PVP), CCS and PEG-3350 yielded about 2-fold increase of CYP3A4 mRNA expression over vehicle control. Although PEG-3350 represents the highest induction of CYP3A4 (2.3-fold) among the tested excipients, this induction only accounts 4% of the RIF induction in the same liver (47-fold), indicating the test excipients showed practically no induction of CYP3A4. Meanwhile, dicalcium phosphate dehydrate (DCP), pregelatinized starch (PgS), and PS-80 decreased CYP3A4 mRNA expression by 30%, 90%, and 70%, respectively, compared to vehicle control. Additionally, among the excipients tested, only PS-80 markedly repressed CYP3A4 protein expression by 70% in HPH (Figure 3B) that was consistent with the observation in Fa2N4 cells.
Figure 3.

Excipients affect CYP3A4 expression in human primary hepatocytes. Human primary hepatocytes were prepared from two different donors (HL#14 and HL#17) and treated with RIF (10 μM), DMSO (0.1%), PBS (none) or selected excipients at concentrations indicated in Table 1 for 24 hrs mRNA (A) and 72 hrs protein (B) analysis, respectively. (A) Total RNA was isolated, reverse transcribed, then subjected to realtime RT-PCR analysis of CYP3A4 mRNA expression. (B) Whole cell homogenates (20 μg) from each group were subjected to immunoblot analysis of CYP3A4 protein expression as described in “Materials and Methods”.
Effect of excipients on the expression of CYP3A4 and MDR1 LS174T cells
Potential regulation of MDR1 mRNA expression by excipients was also investigated, since Class 3 drugs are susceptible to intestinal transporter effects (10). Colon adenocarcinoma-derived LS174T cells were employed as an in vitro model for intestine. Figure 4 illustrates the effect of 16 excipients on the expression of CYP3A4 and MDR1 in LS174T cells. RIF served as a positive control for induction of both CYP3A4 and MDR1. No excipient significantly upregulated CYP3A4 or MDR1 mRNA expression (Fig. 4A and 4B). HPMC, PgS, and X-PVP significantly decreased CYP3A4 expression, where PgS decreased expression by more than 65% compared to vehicle control (i.e. none). PgS, along with PEG-3350, also decreased MDR1 mRNA expression by greater than 40%.
Figure 4.
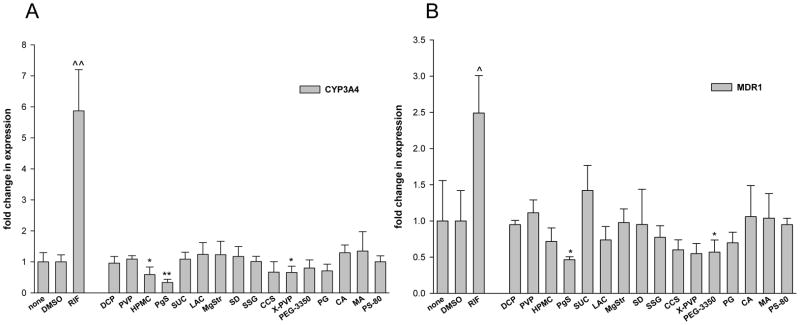
Excipients affect CYP3A4 and MDR1 mRNA expression in LS174T cells. Human colon carcinoma LS174T cells were treated for 24 hrs with RIF (10 μM), DMSO (0.1%), PBS (none) or excipients at concentrations indicated in Table 1. As indicated in “Material and Methods”, total RNA was isolated from each group, reverse transcribed, and subjected to realtime RT-PCR analysis of CYP3A4 (A) and MDR1 (B).
DISCUSSION
Excipients perform several critical functions in a formulated pharmaceutical product (26, 27). There are several categories of excipients (e.g. diluents, binders, disintegrants, surfactants, etc.), reflecting the varied roles of excipients to yield formulated drug products with necessary properties. Some excipients promote drug release within the gastrointestinal tract, while others allow for tablets or capsules to be manufactured on a large, commercial scale. Nevertheless, excipient behaviors can have unintended consequences. Gelatin, which is frequently used in formulation, can become cross-linked at elevated temperature and high humidity, but ultimately does not impact drug absorption in vivo (28). Fetzner et al. (29) observed that the excipient beta-cyclodextrin, used to aid drug solubilization, induced glycolytic enzymes in E. fergusonii bacteria, which can reside in the human gut. Although the question of whether specific excipients could be absorbed is largely unanswered, particularly for polymeric excipients such as PS-80 and HPMC, several excipients have been shown to affect drug transporters function both in vivo and in vitro. For instance, Yamagata et al., (30, 31) demonstrated that both Pluronic P85 and Tween 20 repressed the efflux function of breast cancer resistance protein (BCRP, ABCG2) in mice and BCRP-expressing cell line. Nevertheless, the literature appears practically devoid of reports of possible excipient effects on gene regulation. Common excipients, such as starch derivatives, polyethylene glycol, and sodium lauryl sulfate, have been utilized in tablets and capsules for several decades. It is the commonality and familiarity of these excipient compounds that has contributed to our current lack of evaluation.
Primary hepatocytes and hepatocyte-derived immortalized cell lines are optimal systems to investigate chemical-mediated regulation of drug metabolizing genes. These cell systems maintain in vivo pathways, such as activation of the PXR and downstream upregulation of phase I CYP enzymes, phase II enzymes and transporters which is a common mechanism for regulating drug metabolism (20, 32). Cotransfected with the PXR expression vector and a CYP2B6 luciferase reporter construct, the reporter assay in HepG2 cells demonstrates that none of the excipients activated PXR or increased the luciferase activity (Fig. 1). PXR activation and enzyme upregulation is a major concern in early drug discovery and failure to activate the PXR pathway minimizes the possibility of excipient-mediated drug interactions. However, regulation of human drug metabolism is not limited to PXR activation. Excipient treatment of immortalized hepatocyte cultures (Fa2N4) and HPHs illustrates the involvement of other regulatory mechanisms by altering the expression of CYP3A4 message and protein (Fig. 2 and 3).
Excipient effects on CYP3A4 mRNA and protein expression largely confirm the results of the reporter assays. Although, a few excipients were able to slightly but statistical significantly increase mRNA expression in Fa2N4 and HPH cultures. For instance, CCS was the strongest inducer of CYP3A4 in Fa2N4 at 2.4-fold; PEG-3350 was the strongest inducer in HPH at 2.3-fold. However, these inductions were marginal and only account 8% and 5% in comparison to that of the positive RIF-mediated CYP3A4 induction (defined as 100%). In light of the FDA guideline proposed in 2006 by which a drug that produces an increase that is equal to or greater than 40% of the positive control can be considered an enzyme inducer in vitro (33), none of the tested excipients could be identified as an inducer of CYP3A4.
On the contrary, the more striking observation of excipient effects was a decreased or repressed CYP3A4 expression at the mRNA and/or protein levels across multiple cell lines. Ten excipients decreased CYP3A4 mRNA or protein in Fa2N4 and three excipients decreased expression in HPH; the only common excipient between them was PS-80. The failure to activate PXR, combined with the observation of an overall decrease in CYP3A4 expression, suggests that these excipient compounds elicit their effects through alternate pathways. To identify excipient-mediated mechanisms, MTT cytotoxicity assays were done to eliminate cell death and/or toxicity as a mechanism of decreased gene and protein expression. With the exception of SD treatment in Figure 2B which displays obvious toxicity, we can conclude that excipient-mediated changes in gene and protein expression are not mediated by cytotoxic effects.
This repressive regulatory effect was also seen in the expression of CYP3A4 and MDR1 mRNA, following excipient treatment of LS174T cells (Fig. 4). In this in vitro model of small intestine, no excipient significantly increased CYP3A4 or MDR1 mRNA, but several decreased the expression of one or both genes. Specifically, PgS decreased CYP3A4 and MDR1 mRNA by 54% and 65%, respectively. In a more physiologically-relevant context, excipients in oral drug formulations are more likely to first encounter enterocytes of the gut and small intestine, as opposed to hepatocytes of the liver. Small intestine expresses its own complement of metabolizing enzymes, including CYP3A4, and possess the similar regulatory mechanisms found in hepatocytes (34, 35). Theoretically, the ability of excipients to decrease the expression of efflux transporters such MDR1 presents a concern for their inclusion in drug products where the active ingredient has limited intestinal absorption.
In total, as many as 19 excipeints were used to treat three different cell lines representing two related but distinct organ systems. The effects of an individual excipient were occasionally inconsistent from mRNA to protein or across cell lines. However, based on all the data presented, the three excipients HPMC, PgS, and PS-80 showed statistically significant effects over multiple cell lines.
HPMC and PgS are both hydrophilic polymers and categorized as a binder or filler in formulation. HPMC is not cross-linked and is water soluble, while PgS possess both a water soluble and a water insoluble structural component (36). The regulatory effects of HPMC illustrate some of the inconsistencies mentioned above. HPMC elicited the greatest promoter activation in the reporter assay (Fig. 1) and upregulated CYP3A4 mRNA in Fa2N4 cells (Fig. 2A). However, these observations were practically insignificant. HPMC also decreased CYP3A4 mRNA in LS174T cells and CYP3A4 protein in Fa2N4 cells; the decrease in MDR1 expression, however, was not statistical significance (Fig. 2B and 4). PgS had little effect on Fa2N4 cells, but strikingly decreased mRNA expression in HPH and LS174T cells. The decreased mRNA expression of CYP3A4 in HPH was greater than that seen with PS-80, which concomittantly decreased CYP3A4 protein expression as well. Based on existing knowledge, neither HPMC nor PgS is expected to be absorbed intact. Whereas, PgS is commonly derived from corn starch and is digestable.
PS-80 is a nonionic surfactant and a type of polyoxyethylene sorbitan fatty acid ester. Using Caco-2 cell monolayers, PS-80 can increase apical-to-basolateral transport, possibly through efflux inhibition, and increased membrane fluidity without compromising cell monolayer integrity (37, 38). In Table 2, PS-80 repressed CYP3A4 protein expression in Fa2N4 and HPH cells. However, PS-80 did not module CYP3A4 or MDR1 in LS174T cells. Given its water solubility and relatively high molecular weight, PS-80 may not be expected to be well absorbed from oral drug products, but its intestinal absorption is not known.
Table 2.
Effects of excipients on CYP3A4 and MDR1 expression
| Fa2N4 | HPH | LS174T | ||||
|---|---|---|---|---|---|---|
| Excipient | mRNA | Protein | mRNA | Protein | CYP3A4 | MDR1 |
| HPMC | ↑ | ↓ | = | X | ↓ | ↓a |
| PgS | = | = | ↓ | X | ↓ | ↓ |
| CCS | ↑ | = | ↑ | X | ↓a | ↓a |
| X-PVP | ↑a | ↓ | = | X | ↓ | ↓a |
| PS-80 | ↓ | ↓ | ↓ | ↓ | = | = |
↑: increased expression; ↓: decreased expression; = : no change; x: not measured
Change was not statistically significant.
It is widely held that excipients (particularly bulky or polymerized compounds, i.e. X-PVP) are not absorbed from the gut or intestinal lumen, but this assumption is ultimately somewhat belied by the observed changes in gene and protein expression presented here. Not attributable to cytotoxic-mediated events, these excipients may alter signaling or transcriptional events by other mechanisms that don’t require cellular uptake of the excipient. Mechanisms traditionally associated with decreased cytochrome P450 expression are regulated by mediators of inflammation and disease. Inflammatory cytokines have been identified as regulators of CYP3A4 expression in rodent and human hepatocyte cultures and circulating cytokine levels have been found to be inversely proportional to P450-dependent plasma drug clearance in patients with disease (39). NF-kB, nitric oxide synthase (NOS), PKA and PKC have been identified as protein mediators of decreased CYP3A4 expression in a PXR-dependent mechanism (40–42). Given similar regulatory networks between small intestine and liver, these protein mediators represent potential targets for excipient action to downregulate hepatic or intestinal CYP3A4 expression.
In summary, excipients failed to activate PXR or upregulate CYP3A4 expression. Instead, a number of excipients displayed a trend towards decreasing the CYP3A4 and MDR1 expression. Overall, nineteen excipients with varying degrees of solubility were used to treat three cell types representing two organ systems and subsequently, some inconsistencies in excipient effects were observed. HPMC, PgS and PS-80 demonstrate possible detrimental effects (Table 2). This study represents the first investigation of potential excipient effects on the regulation of drug metabolizing genes. Based on the data presented, it appears that most excipients could be inert. However, for a number of excipients, excipient-mediated repressive effects could be significant in susceptible populations, and merit further investigation.
Acknowledgments
The authors thank Dr. Steve Kliewer (University of Texas, Southwestern Medical Center, Dallas, TX) for providing the pSG5-hPXR expression vector. Human liver tissues were procured with the assistance of Jennifer Fuhrman from the University of Maryland Medical Center (Baltimore, MD). This research was partly supported by National Institute of Health Grant (R01, DK061652).
Abbreviations
- CA
Citric acid
- CCS
croscarmellose sodium
- CYP3A4
cytochrome P450 3A4
- DCP
dicalcium phosphate dehydrate
- DMSO
dimethyl sulfoxide
- FA
fumaric acid
- HPMC
hydroxypropyl methylcellulose
- KTC
ketaconazole
- LAC
lactose
- MA
malic acid
- MCC
microcrystalline cellulose
- MgStr
magnesium stearate
- MDR1
multidrug resistance
- PEG-3350
polyethylene glycol 3350
- PS-80
polysorbate-80
- PVP
povidone
- PXR
pregnane X receptor
- PgS
pregelatinized starch
- PG
propylene glycol
- RIF
rifampicin
- SD
amorphous fumed silicon dioxide
- SLS
sodium lauryl sulfate
- SSG
sodium starch glycolate
- SUC
sucrose
- X-PVP
Crospovidone
References
- 1.FDA. CDER, Guidance for Industry. 2000. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. [Google Scholar]
- 2.EMEA. Note for guidance on the investigation of bioavailability and bioequivalence. Committee for Proprietary Medicinal Products; 2001. [Google Scholar]
- 3.Polli JE, Yu LX, Cook JA, Amidon GL, Borchardt RT, Burnside BA, Burton PS, Chen ML, Conner DP, Faustino PJ, Hawi AA, Hussain AS, Joshi HN, Kwei G, Lee VH, Lesko LJ, Lipper RA, Loper AE, Nerurkar SG, Polli JW, Sanvordeker DR, Taneja R, Uppoor RS, Vattikonda CS, Wilding I, Zhang G. Summary workshop report: biopharmaceutics classification system--implementation challenges and extension opportunities. Journal of pharmaceutical sciences. 2004;93:1375–1381. doi: 10.1002/jps.20064. [DOI] [PubMed] [Google Scholar]
- 4.Polli JE, Abrahamsson BS, Yu LX, Amidon GL, Baldoni JM, Cook JA, Fackler P, Hartauer K, Johnston G, Krill SL, Lipper RA, Malick WA, Shah VP, Sun D, Winkle HN, Wu Y, Zhang H. Summary workshop report: bioequivalence, biopharmaceutics classification system, and beyond. The AAPS journal. 2008;10:373–379. doi: 10.1208/s12248-008-9040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, Shah VP, Lesko LJ, Chen ML, Lee VH, Hussain AS. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharmaceutical research. 2002;19:921–925. doi: 10.1023/a:1016473601633. [DOI] [PubMed] [Google Scholar]
- 6.Benet LZ, Amidon GL, Barends DM, Lennernas H, Polli JE, Shah VP, Stavchansky SA, Yu LX. The use of BDDCS in classifying the permeability of marketed drugs. Pharmaceutical research. 2008;25:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.EMEA. Guideline on the investigation of bioequivalence. Committee for Medicinal Products for Human Use; 2008. [Google Scholar]
- 8.Koch KM, Parr AF, Tomlinson JJ, Sandefer EP, Digenis GA, Donn KH, Powell JR. Effect of sodium acid pyrophosphate on ranitidine bioavailability and gastrointestinal transit time. Pharmaceutical research. 1993;10:1027–1030. doi: 10.1023/a:1018918907670. [DOI] [PubMed] [Google Scholar]
- 9.Adkin DA, Davis SS, Sparrow RA, Huckle PD, Wilding IR. The effect of mannitol on the oral bioavailability of cimetidine. Journal of pharmaceutical sciences. 1995;84:1405–1409. doi: 10.1002/jps.2600841205. [DOI] [PubMed] [Google Scholar]
- 10.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharmaceutical research. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 11.Klaassen CD, Slitt AL. Regulation of hepatic transporters by xenobiotic receptors. Current drug metabolism. 2005;6:309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- 12.Meyer UA. Overview of enzymes of drug metabolism. Journal of pharmacokinetics and biopharmaceutics. 1996;24:449–459. doi: 10.1007/BF02353473. [DOI] [PubMed] [Google Scholar]
- 13.Lin JH. Sense and nonsense in the prediction of drug-drug interactions. Current drug metabolism. 2000;1:305–331. doi: 10.2174/1389200003338947. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clinical pharmacokinetics. 2003;42:1331–1357. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 15.Sachs-Barrable K, Thamboo A, Lee SD, Wasan KM. Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells. J Pharm Pharm Sci. 2007;10:319–331. [PubMed] [Google Scholar]
- 16.Barta CA, Sachs-Barrable K, Feng F, Wasan KM. Effects of monoglycerides on P-glycoprotein: modulation of the activity and expression in Caco-2 cell monolayers. Molecular pharmaceutics. 2008;5:863–875. doi: 10.1021/mp800050q. [DOI] [PubMed] [Google Scholar]
- 17.Kumar GN, Surapaneni S. Role of drug metabolism in drug discovery and development. Medicinal research reviews. 2001;21:397–411. doi: 10.1002/med.1016. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Tompkins LM. CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Current drug metabolism. 2008;9:598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. The Journal of clinical investigation. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeCluyse EL, Alexandre E, Hamilton GA, Viollon-Abadie C, Coon DJ, Jolley S, Richert L. Isolation and culture of primary human hepatocytes. Methods in molecular biology (Clifton, NJ. 2005;290:207–229. doi: 10.1385/1-59259-838-2:207. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Chen T, Stanton JD, Sueyoshi T, Negishi M, Wang H. The peripheral benzodiazepine receptor ligand 1-(2-chlorophenyl-methylpropyl)-3-isoquinoline-carboxamide is a novel antagonist of human constitutive androstane receptor. Molecular pharmacology. 2008;74:443–453. doi: 10.1124/mol.108.046656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. The Journal of pharmacology and experimental therapeutics. 2006;317:1200–1209. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. The Journal of biological chemistry. 2003;278:14146–14152. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Stanton JD, Tolson AH, Luo Y, Wang H. Bioactive terpenoids and flavonoids from Ginkgo biloba extract induce the expression of hepatic drug-metabolizing enzymes through pregnane X receptor, constitutive androstane receptor, and aryl hydrocarbon receptor-mediated pathways. Pharmaceutical research. 2009;26:872–882. doi: 10.1007/s11095-008-9788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habib YS, Augsburger LL, Shangraw RF. Production of inert cushioning beads: effect of excipients on the physicomechanical properties of freeze-dried beads containing microcrystalline cellulose produced by extrusion-spheronization. International journal of pharmaceutics. 2002;233:67–83. doi: 10.1016/s0378-5173(01)00924-3. [DOI] [PubMed] [Google Scholar]
- 27.Shangraw RF. Emerging trends in the use of pharmaceutical excipients. Pharmaceutical Technology. 1997;21:36–42. [Google Scholar]
- 28.Digenis GA, Gold TB, Shah VP. Cross-linking of gelatin capsules and its relevance to their in vitro-in vivo performance. Journal of pharmaceutical sciences. 1994;83:915–921. doi: 10.1002/jps.2600830702. [DOI] [PubMed] [Google Scholar]
- 29.Fetzner A, Bohm S, Schreder S, Schubert R. Degradation of raw or film-incorporated beta-cyclodextrin by enzymes and colonic bacteria. Eur J Pharm Biopharm. 2004;58:91–97. doi: 10.1016/j.ejpb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Improvement of the oral drug absorption of topotecan through the inhibition of intestinal xenobiotic efflux transporter, breast cancer resistance protein, by excipients. Drug metabolism and disposition: the biological fate of chemicals. 2007;35:1142–1148. doi: 10.1124/dmd.106.014217. [DOI] [PubMed] [Google Scholar]
- 31.Yamagata T, Morishita M, Kusuhara H, Takayama K, Benameur H, Sugiyama Y. Characterization of the inhibition of breast cancer resistance protein-mediated efflux of mitoxantrone by pharmaceutical excipients. International journal of pharmaceutics. 2009;370:216–219. doi: 10.1016/j.ijpharm.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, Hamilton G, Rizzo C, Jolley S, Gilbert D, Downey A, Mudra D, Graham R, Carroll K, Xie J, Madan A, Parkinson A, Christ D, Selling B, LeCluyse E, Gan LS. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug metabolism and disposition: the biological fate of chemicals. 2002;30:795–804. doi: 10.1124/dmd.30.7.795. [DOI] [PubMed] [Google Scholar]
- 33.FDA. FDA Guidelines. 2006. In vitro evaluation of CYP induction. [Google Scholar]
- 34.Burk O, Koch I, Raucy J, Hustert E, Eichelbaum M, Brockmoller J, Zanger UM, Wojnowski L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR) The Journal of biological chemistry. 2004;279:38379–38385. doi: 10.1074/jbc.M404949200. [DOI] [PubMed] [Google Scholar]
- 35.Glaeser H, Drescher S, Eichelbaum M, Fromm MF. Influence of rifampicin on the expression and function of human intestinal cytochrome P450 enzymes. British journal of clinical pharmacology. 2005;59:199–206. doi: 10.1111/j.1365-2125.2004.02265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rane Y, Mashru R, Sankalia M, Sankalia J. Effect of hydrophilic swellable polymers on dissolution enhancement of carbamazepine solid dispersions studied using response surface methodology. AAPS PharmSciTech. 2007;8:Article 27. doi: 10.1208/pt0802027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–246. doi: 10.1016/s0928-0987(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 38.Rege BD, Yu LX, Hussain AS, Polli JE. Effect of common excipients on Caco-2 transport of low-permeability drugs. Journal of pharmaceutical sciences. 2001;90:1776–1786. doi: 10.1002/jps.1127. [DOI] [PubMed] [Google Scholar]
- 39.Aitken AE, Morgan ET. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug metabolism and disposition: the biological fate of chemicals. 2007;35:1687–1693. doi: 10.1124/dmd.107.015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu X, Ke S, Liu D, Sheng T, Thomas PE, Rabson AB, Gallo MA, Xie W, Tian Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. The Journal of biological chemistry. 2006;281:17882–17889. doi: 10.1074/jbc.M601302200. [DOI] [PubMed] [Google Scholar]
- 41.Aitken AE, Lee CM, Morgan ET. Roles of nitric oxide in inflammatory downregulation of human cytochromes P450. Free radical biology & medicine. 2008;44:1161–1168. doi: 10.1016/j.freeradbiomed.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pondugula SR, Dong H, Chen T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert opinion on drug metabolism & toxicology. 2009;5:861–873. doi: 10.1517/17425250903012360. [DOI] [PMC free article] [PubMed] [Google Scholar]