

Abstract
Approximately one third of patients with non-small cell lung cancer have unresectable stage IIIA or stage IIIB disease; combined cytotoxic chemotherapy and radiation therapy delivered concurrently has been established as the standard treatment for such patients. Despite many clinical trials testing many different radiochemotherapy combinations, it seems that a plateau of efficiencies at the acceptable risk of complications has been reached. Clinical studies indicate that the improved efficacy of radiochemotherapy is associated with the radiosensitizing effects of chemotherapy. Improvement of outcomes of this combined modality by developing novel radiosensitisers is a viable therapeutic strategy. In addition to causing cell death, ionizing radiation also induces a many-faceted signaling response, which activates many pro-survival pathways leading to enhanced proliferation in the surviving fraction of cells and increased vascularization in tumors. Radiation at doses used in the clinic activates cytoplasmic phospholipase A2 (cPLA2), leading to increased production of arachidonic acid (AA) and lysophosphatidylcholine (LPC). The former is the initial step in the generation of eicosanoids, while the later is the initial step in the formation of lysophosphaditic acid, leading to the activation of inflammatory pathways. The echinoderm microtubule associated protein-like 4 Anaplastic lymphoma kinase (EML4-ALK) is member of the insulin superfamily of receptor tyrosine kinases. The EML4-ALK fusion gene appears unique to lung cancer and signals through Erk and PI3K. Heat shock protein 90 (Hsp90) is often overexpressed and present in an activated multichaperone complex in cancer cells and it is now regarded as essential for malignant transformation and progression. In this review we focus on radiosensitizing strategies involving the targeting of membrane phospholipids, EML4-ALK and Hsp90 with specific inhibitors.
Introduction
It is estimated that in 2013 lung cancer will cause about 30% of all cancer deaths among men and 27 % of all cancer deaths among women in the United States. A total of 163,890 deaths which is more than the combined number of deaths from the next 3 most common causes of cancer deaths namely colon, breast, and prostate cancers1. Worldwide, in 2008, lung cancer was the leading cause of cancer deaths in males and the second leading cause of cancer deaths in females, about 1,400, 000, or 18% of all cancer deaths. Five-year survival for lung cancer patients ranges from 6% to 18%. The 5-year survival has changed only very little over the last 2 decades, with progress lagging significantly behind other common cancers. Conventionally, lung cancer is divided into small and non-small-cell subtypes, the latter accounting for around 85% of cases. (Thus this review will focus on NSCLC). The combination of standard chemotherapy (CT) with radiotherapy (RT), chemoradiotherapy (CRT), is used routinely for stage III NSCLC patients2–4. Concurrent CRT has been shown to be more effective than consequent CRT5. Unfortunately, the survival benefit of concurrent regimens above sequential regimes of CRT carries the risk of additional toxicity5. Two recent comprehensive meta-analyses of CRT clinical trials have shown that local disease control by RT is important and contributes to improved survival3,5.
An important conclusion from these results is that the success of CRT is a reflection of the radiosensitizing effect of CT due to its action on tumor biological factors that can affect the response to RT. Thus, improvement of the efficacy of RT would also lead to improvement of CRT outcomes. One strategy towards achieving this goal is the development of novel radiosensitizers, which is the focus of this review.
Three main aspects of tumor biology that are well known to affect RT outcome are: the extent of hypoxia, the ability of the surviving tumor cells to repopulate within treatment time and the intrinsic radioresistance of tumor cells6. Convincing evidence for a role for each of these radiobiological factors in RT has accumulated from many clinical studies at all treatment sites7. Evidence has also been accumulating that vasculature and stromal cells also play a crucial in response to cancer therapy, including RT8,9.
Radiotherapy
Currently, definitive radiotherapy is mainly used for Stage I inoperable NSCLC, where the advancement of stereotactic techniques has led to impressive outcomes10–12. The combination of CT with RT and CRT is used routinely for stage III NSCLC patients2–4,13. Concurrent CRT has been shown to be more effective than consequent CRT. Unfortunately, the survival benefit of concurrent regimens above sequential regimes of CRT carries the risk of additional toxicity5. Two recent comprehensive meta-analyses have shown that local disease control by RT is important and contributes to improved survival. This important outcome gives a strong impetus to the continued improvement of radiation treatments. The outcome of RT is dependent on accurate delineation of the tumor area and all involved lymph nodes7. Inadequate treatment planning strategies will increase the chance of local recurrences and/or result in a higher chance of normal tissue damage. Innovations such as the introduction of three-dimensional conformal planning, the use of multi- leaf collimators, four-dimensional planning CT scans, intensity modulated radiation therapy (IMRT) and image-guided radiation therapy (IGRT) techniques have dramatically improved targeting of the tumor volume14,15. These new radiation delivery techniques have allowed significant dose escalation without concurrent increases in normal tissue toxicity14,15. The use of IMRT can achieve a dose escalation of 20–35% compared to three-dimensional conformal radiotherapy by more conformal dose distributions and steeper dose fall off. Clinical trials determining the effect of higher doses archived with the advanced radiation techniques are being conducted currently. While the concurrent CRT schedule has been favored with an absolute survival benefit, but there is no difference in progression-free survival (PFS). The concurrent regimen primarily improved the local regional control but did not affect the rate of distant progression. An important conclusion from these results is that the success of CRT is a reflection of the radiosensitizing effect of CT. Such radiosensitization may be a reflection to a large extent, of the action of CT agents on tumor biological factors that can affect tumor response to RT.
Radiation induced signaling
Over the last two decades, it has been shown that ionizing radiation (IR) at doses used in the clinic, in addition to inducing DNA damage in the nucleus, also triggers the activation of a large network of intracellular signaling events16. These include the transient activation of central regulators of prosurvival response pathways, and the induction of inflammation.17. Many of the inflammatory cascades elicited by radiation are injurious to normal tissues, but they confer a survival advantage to tumor cells. Thus, specific targeting of downstream components of the radiation inducible inflammatory signaling response offer the possibility of simultaneously abrogating radioresistance within tumor cells and blocking deleterious inflammatory responses of normal tissues to RT18.
In general, intrinsic tumor radiosensitivity has been thought to mainly reflect the balance between radiation-induced DNA damage and its repair. Recent results indicate that the cascade of radiation-induced cytoplasmic signaling events described above play a salient role in tumor radiosensitivity19. The cellular signaling triggered by clinical doses of IR occurs at two discrete sites: the nucleus and the cytoplasm. In the nucleus, a DNA damage response (DDR), is elicited which coordinates DNA repair, cell cycle check points and cell death pathways20. The structure of this response involves molecules that sense the DNA damage which elicits the participation of several mediators, such as ATM, which in turn recruit an army of proteins that play key roles in several pivotal cellular processes. Thus, the DDR regulates physiological processes that involve multiple layers of decisions, including the determination to undergo apoptosis or enter terminal differentiation through senescence, the activation of heightened immune surveillance, as well as DNA repair itself.
In the cytoplasm, the radiation generated ionizing events in water are amplified in the mitochondria in a Ca++ dependent manner, leading to the generation of large amounts of reactive oxygen species (ROS) and reactive nitrogen species (RNS)21. The resulting redox imbalance leads to the inhibition of protein tyrosine phosphatase (PTPase) activities22. The relative activity of a PTPase is approximately one order of magnitude higher than that of the substrate (kinase) it dephosphorylates23. PTPase activity is sensitive to oxidation and/or nitrosylation of a key cysteine residue in its active site, and thus, any agent that generates ROS or RNS has the potential to lead to decreased PTPase activity and, hence, increased tyrosine phosphorylation of multiple proteins24. As a net result of the decrease PTPase activity, there is increased tyrosine phosphorylation resulting in the activation of receptor and non-receptor tyrosine kinases and the activation of down-stream signal transduction pathways.
In addition, radiation activates acidic sphingomyelinase and increases the production of ceramide. Radiation-induced ceramide has been shown to promote membrane-associated receptor activation by facilitating the clustering of receptors within lipid rafts25,26. Radiation also induces the eicosonoid inflammatory pathway by inducing increased activity of cytoplasmic phospholipase A2 (cPLA2), resulting in increased levels of arachidonic acid that is metabolized through cyclooxygenase-2 (COX-2), which is also induced by radiation, into various forms of prostaglandins27.
Eicosonoids
Inflammation is now recognized to be a critical component for tumor progression and one of the recently added hallmarks of cancer28,29. Epidemiological and genetic studies support the link between chronic inflammation and tumor progression30. Eicosanoid generating enzymes, such as COX-2 and 5-lipoxygenase (5-LOX), are over-expressed in several cancers including breast, lung, and pancreas31. Eicosanoids, including prostaglandins and leukotrienes, are generated by local cell type specific arachidonic acid metabolism and can be potent mediators of inflammation. The enzyme families COX and LOX are responsible for the metabolism of arachidonic acid, leading to the production of prostaglandins and leukotrienes, respectively. Both of these pathways have been implicated in cancer progression31,32. There are two types of COX activities in cells, COX-1 and COX-2. COX-1 is constitutive expressed, while COX-2 is inducible. Several different cancers are found to overexpress COX-2, including lung cancer33. Interestingly, overexpression of COX-2 alone causes tumorigenesis in transgenic mouse models34.
Ionizing radiation activates cPLA2, triggering the release of arachidonic acid from membrane phospholipids and subsequent production of eicosanoids through the cyclooxygenase pathways. COX-2 is also induced by radiation in various cell culture models, including NSCLC35,36. In many tissues, radiation exposure leads to increased eicosanoid production. Within hours after irradiation, increased levels of prostaglandins PGE1, PGE2, PGF2 and PGI2 are detectable in most tissues, and the increased levels of eicosanoid levels may persist for several days or weeks27. Tumor models of lung, brain, breast and sarcomas treated with pan-COX inhibitor indomethacin prior to irradiation, improved the therapeutic index, since the effects of radiation on tumors was enhanced, while those on normal tissue effects were not37. In mouse fibrosarcoma, daily treatment with indomethacin prior to radiotherapy improved the responses in both single and fractionated-dose regimens. Indomethacin treatment was associated with prolonged delays in tumor growth, an increased cure rate, and increased time to recurrence with no significant change in normal tissue response. Indometacin inhibits both COX-1 and COX-227. Several studies involving animal tumor models also demonstrated radiation-potentiating effects of selective COX-2 inhibition. No substantial increase in radiation induced normal tissue damage was seen, suggesting that selective COX-2 inhibition was associated with a true therapeutic gain for radiotherapy37. In several studies the antiangiogenic actions of COX-2 inhibitors have been implicated in the increased radioresponse37.
The expression of COX-2 has been documented in up to one-third of lung atypical adenomatous hyperplasia and carcinoma in situ, and is also overexpressed in 70% to 90% of NSCLCs, especially in adenocarcinomas38,39. Increased COX-2 mRNA levels indicate a worse overall survival rate and a more aggressive disease in NSCLC40,41. A recent meta analysis of 16 studies found that COX-2 overexpression seems to have no significant impact on survival of NSCLC patients42. The availability of specific COX-2 inhibitors such as Celecoxib, has led to clinical trials involving NSCLC patients. Several such clinical trials were reviewed recently, including the effect of celecoxib on first line treatment, second line treatment and in combination with EGFR inhibitors43. All 13 studies were phase I/II; increased PFS and OS was reported in one study, while in a second line study, using a doublet chemotherapy protocol, the patients receiving the specific COX-2 inhibitor celecoxib, fared worse. Two large randomized phase III trials studied the effect of celecoxib on salvage chemotherapy in stage IIIb/IV NSCLC; both studies failed to demonstrate a survival benefit of the addition of celecoxib to palliative chemotherapy44,45. Two clinical trials combining chemotherapy, radiation and celecoxib have also been published recently46,47. Although one study was too small, while one closed because it did not meet its response rate goals, both studies observed that in unselected patients, the addition of celecoxib to concurrent chemoradiotherapy with inoperable stage IIIA/B NSCLC does not improve survival. Several trials were halted because of the cardiovascular side effects of celecoxib.
Cytosolic phospholipase A2
The enzymatic action of cytosolic phospholipase A2 (cPLA2) on membrane releases both arachidonic acid and lysophospholipids (LPC). While the arachidonic acid is metabolized by COX-1 and COX-2 to generate eicosanoids, LPC is metabolized to lysophosphatidic acid (LPA). We have been studying the cPLA2 -Autotaxin (ATX) axis to improve the efficacy of radiotherapy in lung cancer and glioblastoma48–51. Phospholipase A2 (PLA2) are biologically active enzymes that catalyze the hydrolysis of membrane phospholipids at the sn-2 position to release lipid second messengers that play a vital role in cancer52. Three major classes of PLA2, namely secretory (sPLA2), Ca2+ independent (iPLA2) and cytosolic (cPLA2) PLA2 have been identified based on their biological roles in the cell. Ionizing radiation triggers the activation of cytosolic phospholipase A2 (cPLA2) which cleaves phosphatidylcholine (PC) to yield LPC, metabolism of which activates Akt and ERK1/251 (Fig 1). Inhibition of cPLA2 prevented radiation induced activation of ERK1/2 and decreased clonogenic survival of irradiated vascular endothelial cells49. cPLA2 has been shown to promote the growth and survival of endothelial cells after irradiation which lead to growth and survival of vascular endothelium, improved migration and tumor formation49,50,53. Activation of cPLA2 by irradiation leads to increased vasculature and enhanced tumorogenesis and invasion leading to radioresistance of the tumor and diminishing the efficacy of the radiotherapy of the tumor models54,55. Radiation induced cPLA2 dependent signaling has been identified to regulate cell viability. cPLA2 has been implicated in eliciting inflammatory response among the other cellular responses. Increased phospholipase signaling has been implicated to various cancers and disease pathogenesis55–57. In normal tissue vasculature pericytes are quiescent and provide important mechanical and physiological support. During tumor angiogenesis pericytes are activated and rapidly proliferate and differentiate and these detached pericytes aid endothelial cells to form new blood vessels58. cPLA2 knockout (cPLA2-KO) mice injected with lewis lung carcinoma (LLC) cells formed smaller tumors than wild type (WT) mice. The LLC tumors formed in the cPLA2-KO mice later regressed. Interestingly tumors in cPLA2-KO mice had dramatically decreased pericytes. Since pericytes regulate vascular integrity and maintenance58,59 regression of LLC tumors could be due to the lack of pericytes in cPLA2-KO mice50.
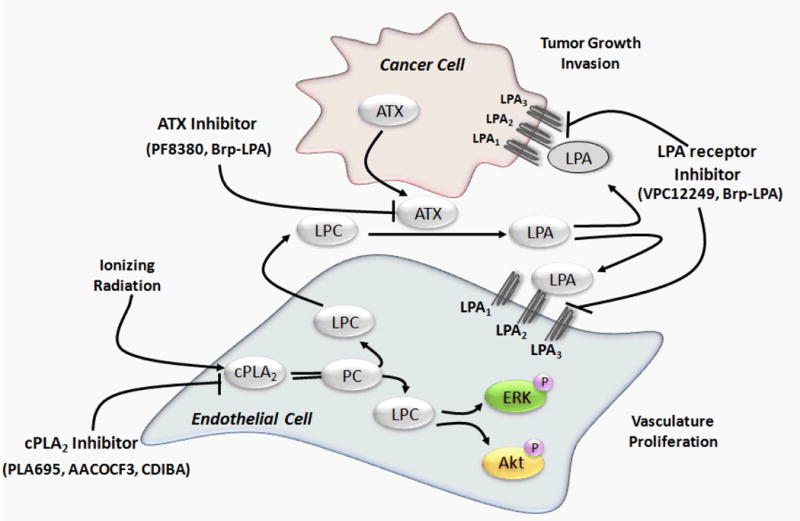
Figure 1. Phospholipase A signaling in response to ionizing radiation.
Ionizing radiation activates cytosolic phospholipase A2 (cPLA2) which cleaves phosphatidylcholine (PC) to yield lysophosphatidylcholine (LPC). LPC can phosphorylate Akt and ERK1/2. This activation leads to increased vasculature and enhanced tumorogenesis and invasion leading to radioresistance of the tumor. LPC is a secondary messenger to many signaling pathways that stimulate endothelial survival and proliferation by regulating the cytokine synthesis, endothelial growth factor expression and chemotaxis. Autotaxin that posses the lysophospholipase D (LysoPLD) activity catalyzes the reaction by cleaving the headgroup of LPC to form lysophosphatidic acid (LPA). LPA can then bind to lysophosphatidic acid receptors (LPA1–3). LPA1–3 belongs to the Endothelial differentiation gene family (EGD). LPA1 is highly expressed in the nervous system and is required for development of the brain. LPA2 is highly expressed in immune system organs such as the thymus and spleen. LPA3 is highly expressed in reproductive organs such as the testis and uterus and is linked with ovarian cancers.
Orthotopic mouse lung tumors (CMT167 and LLC) form a primary tumor followed by metastasis, which was reduced in cPLA2-KO mice. Wild type mice transplanted with cPLA2-KO bone marrow showed a better survival advantage compared to mice receiving the bone marrow of WT mice60.
Inhibition of cPLA2 using a chemical inhibitor, CDIBS delayed heterotopic LLC tumor growth in mice50. In WT mice the studies looking at the tumor micro environment (TME) revealed that cPLA2 recruited macrophages and stimulate the production of inflammatory cytokine interleukin-6 leading to tumor formation. Histological analysis of the LLC tumors grown in cPLA2-KO mice had fewer blood vessels in their tumors54. Combination of irradiation with inhibition of cPLA2 in preclinical lung cancer tumor models has been shown to suppressed growth and reduced blood flow61.
Autotaxin
Lysophosphatidylcholine (LPC) which is a second messenger in many lipid signaling pathways that stimulate endothelial cell survival and proliferation by regulating cytokine synthesis, endothelial growth factor expression and chemotaxis62. Autotaxin (ATX) converts extracellular LPC to LPA through its lysophospholipase D activity. ATX belongs to the ectonucleotide pyrophosphate/phosphodiesterase (ENPP) family and is encoded by the ENPP2 gene63. The cellular effects of LPA are mediated through the six distinct G-protein coupled receptors (GPCRs)64. Recent studies show that ATX is not only a lysoPLD enzyme it also is a lipid carrier protein that efficiently transports LPA to respective cognate GPCRs65. Receptor expression is cell type specific and this allows unique cellular responses to LPA depending upon the type of GPCR it is binding. GPCR mediate cellular effects such as migration and proliferation in cancer66. Autotaxin was originally identified as a tumor motility protein and is over expressed in various cancers including NSCLC63 and known to contribute to the tumor invasiveness67. There is direct evidence in transgenic mice indicating that ATX and LPA are involved in invasiveness and metastasis of breast cancer. Increased expression of ATX and its receptors LPA1, LPA2 and LPA3 in mammary epithelium of transgenic mice induced estrogen positive mammary cancer68. ATX has been shown to stimulate angiogenesis either by enhancing the expression of vascular endothelial growth factor (VEGF)69 or stimulating motility70 in endothelial cells. It has been shown that in Hodgkin’s lymphoma cell motility is dependent on the ATX expression and expression of LPA receptors71.
BrP-LPA, a pan-antagonist of LPA1–4 receptors and inhibitor of the lyosphospholipase D activity of autotaxin, was shown to inhibit cell migration and cell invasion of lung cancer cells72 and glioblastoma cells48. In a 3-D lung cancer xenograft model Brp-LPA inhibited tumor growth and reduced tumor vascularization72. Inhibition of ATX and LPA receptors by Brp-LPA diminished the radiation induced activation of pro survival kinase Akt. Brp-LPA treatment enhanced radiation-induced endothelial cell death, disrupted endothelial cell biological functions, reduced glioma cell viability and migration48. The specific ATX inhibitor PF8380 reduces the LPA levels in the tumor microenvironment and blocks LPA signaling51.
Anaplastic lymphoma kinase
The echinoderm microtubule associated protein-like 4 Anaplastic lymphoma kinase (EML4-ALK) fusion oncogene represents one of the newest molecular targets in NSCLC. EML4-ALK was first identified in 2007 by Soda and colleagues, by screening a cDNA library derived from the tumor of Japanese male patient with adenocarcinoma of the lung73. Chromosomal aberrations involving ALK have been identified in several other cancers, including anaplastic large cell lymphomas (ALCL), inflammatory myofibroblastic tumors (IMT), and neuroblastomas74. In cases of ALK translocations, including EML4-ALK, the fusion partner has been shown to mediate ligand-independent dimerization of ALK, resulting in constitutive kinase activity. In cell culture systems, EML4-ALK possesses potent oncogenic activity73. In transgenic mouse models, lung-specific expression of EML4-ALK leads to the development of numerous lung adenocarcinomas75. The growth of cell lines and tumors harboring the EML4-ALK translocation is inhibited by small molecule inhibitors targeting ALK76,75. These results support the concept that ALK-driven lung cancers are addicted to the kinase activity of the fusion EML4-ALK oncogene77.
ALK is thought to play a role during the development and function of the nervous system, and it is not expressed in most, if any, adult tissues, including lung. ALK knockout mice are completely viable without any obvious alterations74. The ligands involved in the activation of ALK are yet to be identified. The key downstream effectors of ALK are better understood than the putative upstream activators (Fig 2). A number of EML4-ALK variants have been identified in NSCLCs, all of which appear to confer gain-of-function properties78. Similar to epidermal growth factor (EGFR) mutations, EML4-ALK fusions result in constitutive tyrosine kinase activity, dependence of the cancer cell on the activated downstream mitogenic pathways, and exquisite sensitivity to ALK inhibition, and thus are another example of oncogene addiction79.
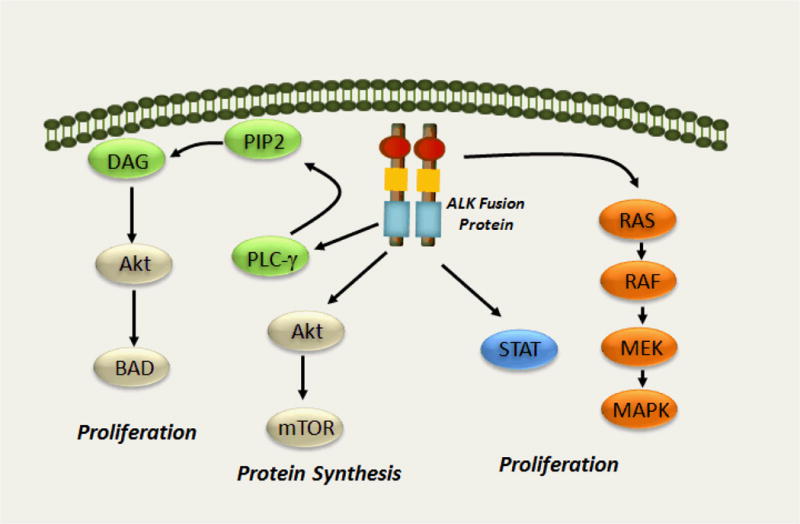
Figure 2. The Anaplastic Lymphoma Kinase (ALK) signaling pathway.
The downstream signaling pathways include the Rat sarcoma (Ras), mitogen activated protein (MAP), extracellular signal regulated kinase (ERK), phosphoinositide 3-kinase (PI3K), Akt, Janus activated kinase (JAK3) and Signal Transducer and Activator of Transcription (STAT3) signaling pathways. In general, the RAS/MEK/ERK pathway drives cell proliferation, and the PI3K/Akt and JAK3-STAT3 pathways play pro-survival roles. Although different ALK fusions may differentially activate downstream signaling pathways, EML4-ALK signals through ERK and PI3K. Activation of the RAS/MEK/ERK and PI3K/Akt pathways play key roles in EML4-ALK–mediated cellular oncogenesis.
Lung cancers harboring ALK rearrangements represent a unique subpopulation of lung cancer patients. The frequency of ALK rearrangements ranges from 3% to 7% in unselected NSCLC patients80,81. Similar to EGFR mutations, the frequency of this genetic alteration is higher in NSCLC patients with adenocarcinomas and in patients who have never smoked or are light cigarette smokers. ALK rearrangements tend to be mutually exclusive with EGFR and KRAS mutations and to have a lower frequency of p53 mutations76,81. In these tumors, ALK is the sole determinant of critical growth pathways, resulting in the activation of downstream canonical PI3K/AKT as well as MAPK/ERK pathways. Given the significant fraction of ALK related cancers, this population could represent more than 10,000 new cases in the US and more than 70,000 worldwide. The initial studies reporting the discovery of EML4-ALK raised the possibility that inhibiting the kinase activity of ALK may be an effective clinical therapy73,75. Treatment of EML4-ALK NSCLC cell lines with an ALK-kinase inhibitors, which were developed for non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase, led to the down-regulation of critical survival signaling pathways and apoptosis76,82,83, similar to the effect of EGFR inhibitors in EGFR mutant NSCLC84. In the xenograft models generated from EML4-ALK NSCLC cell lines ALK inhibitors regressed tumors effectively75,76. Presently, only one agent targeting ALK, PF-02341066 (crizonitib) which was initially designed as an inhibitor of MET, is in clinical use although others have been examined in pre-clinical model systems75,76,85,86.
The first phase I study of crizonitib demonstrated a remarkable 53% response rate and a disease control rate of 79%87,88. The clinical benefit of crizotinib therapy is limited by the development of acquired resistance, as is the case with EGFR TKI. At least eight different point mutations conferring resistance to ALK inhibitors have already been described in detail, and most of them shown to result in cross-resistance to other ALK inhibitors89,90. Multiple distinct mutations in the ALK kinase domain can abrogate the inhibitory capacity of crizotinib, in sharp contrast to EGFR-activating mutations, where the gate keeping T790M mutation essentially represents the sole resistance mutation91. Bypass signaling including the KIT and EGFR pathways, has been identified as potential resistance mechanisms92–94. From a therapeutic standpoint, the wide array of resistance alterations will make it challenging to develop strategies to overcome ALK inhibitor resistance95. The potential radiosensitizing effect of ALK inhibitors has been reported in two recent studies. Lu and coworkers ths crizonitib radiosensitized in NSCLC cells and xenografts expressing EML4-ALK96. Another study reported the lack of radiosensitization under similar experimental conditions97.
Heat shock protein 90
Heat shock protein 90 (Hsp90) is an ATP-dependent molecular chaperone that regulates the late-stage maturation, activation, and stability of a diverse range of client proteins (defined as proteins with demonstrated binding to Hsp90) There are more than 200 identified clients, many of such clients are involved in signal transduction and other key pathways that are especially important in malignancy98. Although it is highly expressed in normal cells, where it helps to maintain protein homeostasis, Hsp90 is exploited by cancer cells for at least 2 purposes: (i) to support the activated or metastable forms of oncoproteins, including many kinases and transcription factors and (ii) to buffer cellular stresses induced by the malignant lifestyle99,100. The classical driver-mutation view of oncogenesis and cancer progression has inspired the design of novel therapeutics targeting driver oncogenes as susceptible nodes in the complex signaling networks regulating the hallmark traits of malignancy29,101. However, the addiction of individual cancers to specific oncogenes is only a single component of molecularly multifaceted aspects of cancer. The maintenance of the conditions required to sustain tumor growth, requires regulation of the microenvironment and also significantly alter intracellular homeostasis in order to deal with the metabolic burden of rapid clonal expansion in low nutrient conditions resulting from deregulated cellular proliferation29,102. Meeting these demands may occur by the upregulation of a repertoire of stress responses found in normal and cancer cells. Cancer cells are thought to be particularly dependent on the upregulation of such non-oncogene pathways for survival99,100. Recently, it has also been established that the genomic instability associated with cancer cells results in aneuploidy, gene copy number variation, chromosomal translocations and missense mutations, results in increased cellular concentrations of proteins with suboptimal stability, leading to proteotoxic stress. Thus an increased requirement for proteostasis is now an additional hallmark feature of cancer102. Hsp90 is at the hub of oncogenic proteostasis, which entails the functional and structural stabilization of a host of known oncoproteins103,104.
Hsp90 is often overexpressed105 and present in an activated multi-chaperone complex in cancer cells106 and it is now regarded as essential for malignant transformation and progression100. It has been demonstrated that a number of mutated oncoproteins, including, for example, BRAF and EGFR, are much more dependent on HSP90 than the corresponding wild-type proteins100. The combination of the observations outline above generate a strong case for targeting the HSP90 molecular chaperone for cancer treatment, since cancer cells are expected to be more sensitive to Hsp90 inhibitors than normal cells.
Since the first study demonstrating that the chaperoning of an oncoprotein Hsp90 was required for transformation by using an targeted inhibitor of hsp90107, there has been an explosion in the development of novel inhibitors of hsp90 and efforts to bring them to the clinic for various cancers108,109. Two excellent perspectives on future directions have been published recently110,111.
The EMK4-ALKT fusion oncoprotein was shown to be associated with hsp90 and the new hsp90 inhibitor IP-504 was shown to lower EMK4-ATK levels in cells in culture and xenografts, leading to growth inhibitions112. Significantly, cells what were selected for AKL-kinase inhibitor resistance, retained their sensitivity to IPI-504. Patients with advanced NSCLC, prior treatment with EGFR TKIs, and tumor tissue available for molecular genotyping, were enrolled in a prospective, nonrandomized, multicenter, phase II study of IPI-504 monotherapy. Best outcomes for ORR were the patients carrying ALK gene rearrangements113. In one study, Ganetespib monotherapy demonstrated a manageable side effect profile as well as clinical activity in heavily pre-treated patients with advanced NSCLC, particularly in patients with tumors harboring ALK gene rearrangement114. A panel of lung cancer cell lines harboring a diverse spectrum of KRAS mutations was treated with ganetespib, leading to potent cytotoxicity with concomitant destabilization of KRAS signaling effectors. Combinations of low-dose ganetespib with MEK or PI3K/mTOR inhibitors resulted in superior cytotoxic activity than single agents alone in a subset of mutant KRAS cells115. Ganetespib induced loss of EML4-ALK expression and depletion of multiple oncogenic signaling proteins in ALK-driven NSCLC cells and xenografts116. Studies with NVP-AUY922, a novel potent resorcinylic isoxazole amide inhibitor of hsp90 have been reported recently. In one study, the effect on NSCLC cells lines were reported, while in the later study also included xenograft models117,118. Both studies reported excellent results, and the establishment of clinical trials is in progress.
Pretreatment of cancer cell with hsp90 inhibitors leads to the radiosensitization of cancer cells of various types including NSCLC. Studies were performed with Geldanamycin (GM) and two derivatives of GM, 17AAG and 17DMAG. These specific inhibitors of hsp90 are quite potent radiosensitisers, achieving radiation enhancement ratios ranging from 2.3 to 2.7. Interestingly normal fibroblasts were not radiosensitized under similar experimental conditions119. Recenly, inhibitors of hsp90 with improved bioavailability and lower toxicity have become available, including a series of pyrazole resorcinol compounds that have proven to be stronger inhibitors of hsp90 (NVP-AUY922)120. Treatment of cancer cells, including NSCLC, with NVP-AUY922 leads to radiosensitization under normoxic and hypoxic condition121,122. The radiosensitization was accompanied by effects on DNA repair, cell cycle progression123 and abrogation of homologous recombination leading to mitotic entry with unresolved DNA damage124.
Combination of CRT with agents targeting angiogenesis
Several therapies targeting angiogenesis are currently in development for NSCLC. Bevacizumab, an anti-VEGF antibody, is currently approved for the treatment of advanced NSCLC in combination with carboplatin and paclitaxel. Completed phase III trials evaluating bevacizumab plus chemotherapy have shown prolonged progression-free survival; however, not all trials showed significant improvement in overall survival125. Vascular targeting agents are considered to have a low toxicity level, as they should only affect the angiogenic vessels. However, toxicities have been observed in the clinical setting when angiostatic drugs were combined with cytotoxic therapies125. Phase III trials of the tyrosine kinase inhibitors (TKIs) targeting VEGFR, vandetanib and sorafenib and the vascular disrupting agent ASA404 also failed to improve survival compared with chemotherapy alone. Phase III clinical trials are ongoing involving several new angiostatic agents that have shown promising phase I/II results, including new antibodies and multiple kinase inhibitors126. Two updated on new single and multitargeted agents have been published recently126,127.
In considering the potential of combining CRT with angiostatic agents, some further background on the complex interaction between RT and the vascular system is helpful. A classic study demonstrated that radiation effects on endothelial cells and thus on angiogenesis are dose dependent128. While increased tumor vascularization and blood flow was observed after a single treatment of 2–3 Gy, treatment with 6 Gy caused a decrease in tumor vasculature and blood flow, suggesting that low dose irradiation might directly potentiate tumor angiogenesis128. Recent studies in several different preclinical systems have supported the differential effects of low and high doses of radiation on the vascular system129. While angiostatic drugs might inhibit induction of angiogenesis by RT, the disruption of the tumor vasculature by such agents would hinder proper perfusion of the tumor. Since hypoxic regions are resistant to RT, the combination of angiostatic therapy with RT seems paradoxical. However, several preclinical studies found a positive interaction between angiogenesis inhibition and RT130. Interestingly angiostatic therapy actually increased tumor oxygenation in some cases130. The concept of ‘vascular normalization’ attempts to reconcile this paradox131. Vascular normalization involves to the remodeling of a dysfunctional (tumor) vasculature to a more normal phenotype. It occurs when the angiostatic agents restore the balance between anti-angiogenesis and pro-angiogenesis factors in the tumor microenvironment. Few clinical studies have addressed the use of angiostatic agents in combination with CRT132. Despite the activity of bevacizumab combined with chemotherapy in advanced NSCLC125 strategies to combine this angiostatic agent with RT have proved disappointing due to toxicity. Phase II trials in NSCLC and in limited stage small cell lung cancer (SCLC) were abandoned due to higher than anticipated tracheo-esophageal fistulation and pulmonary hemorrhage. A phase I study to determine the safety of two dose levels of bevacizumab with RT alone has also been halted due to toxicity concerns. The approach of combining VEGF and EGFR inhibitors was tested using concurrent bevacizumab, erlotinib, carboplatin, paxlitaxel and RT but consolidation therapy with bevacizumab/erlotinib was determined not to be feasible, and the squamous histology cohort was closed due to pulmonary hemorrhage. Median overall survival was 19 months (non-squamous 19 months vs. squamous 17 months), which was similar to the results of concurrent chemoradiotherapy alone. The multi-targeted antiangiogenic TKI sunitinib efficiency in preclinic is currently being tested in phase I and phase II studies.
Summary
We have summarized two radiation inducible inflammation pathways, the inhibition of which increases the radioresponse of cells in culture and animal tumor models. Progress with the eicosanoid pathway has been hampered by the side effects of COX-2 inhibitors. Recently, there has been a focus on inhibiting, microsomal prostaglandin E synthase-1 mPGES-1, the terminal synthase responsible for the synthesis of the pro-tumorigenic prostaglandin E(2). (PGE-2). mPGES-1 is overexpressed in a wide variety of cancers and the effects of a variety of new specific inhibitors of this enzyme are subject to ongoing studies133. It has been well established that increased expression of cPLA2 and ATX in cancer cells and tumor microenvironment play a role in tumor progression, metastasis, migration and proliferation. Activation of cPLA2 by ionizing radiation leads to increased survival and viability of vascular endothelial cells16 and could contribute to poor outcomes in RT and CRT. Recent data using inhibitors of cPLA249,50,55 and inhibitors of ATX and LPA receptors48 show a great promise for the validation of cytosolic phospholipase A2 autotaxin and lysophosphatidic acid receptors as molecular targets for the development of novel radiosensitisers, for the improvement of lung cancer61 and malignant glioma48 treatment. The EML4-ALK fusion gene appears unique to NSCLC. In a remarkably short period of time, from its initial discovery to clinical validation, ALK targeted therapies are in advanced clinical development for EML4-ALK NSCLC134. EML4-ALK NSCLC represents a unique subset of NSCLC patients for whom ALK inhibitors may represent a very effective therapeutic strategy. Some recent efforts to circumvent ALK inhibitor resistance have involved novel inhibitors designed to inhibit hsp90116. There has been a considerable effort to determine the effect of hsp90 inhibitors in lung cancer, mainly focusing on the naturally occurring antibiotic Geldanamycin and its derivatives135. The prospect of developing combinatorial approaches using TKI and hsp90 inhibitors, and overcoming resistance to TKI with hsp90 inhibitors are exciting new advances in NSCLC treatment.
Acknowledgments
We would like to thank all of our colleagues whose work inspired us but we could not cite due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Anderson CS, Curran WJ. Combined modality therapy for stage III non-small-cell lung cancer. Semin Radiat Oncol. 2010;20:186–91. doi: 10.1016/j.semradonc.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Baas P, Belderbos JS, van den Heuvel M. Chemoradiation therapy in nonsmall cell lung cancer. Curr Opin Oncol. 2011;23:140–9. doi: 10.1097/CCO.0b013e328341eed6. [DOI] [PubMed] [Google Scholar]
- 4.Stinchcombe TE, Bogart JA. Novel approaches of chemoradiotherapy in unresectable stage IIIA and stage IIIB non-small cell lung cancer. Oncologist. 2012;17:682–93. doi: 10.1634/theoncologist.2012-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curran WJ, Jr, Paulus R, Langer CJ, et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst. 2011;103:1452–60. doi: 10.1093/jnci/djr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall EJ, Giaccia AJ. Radiobiology for the radiologist. 7. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2012. [Google Scholar]
- 7.Halperin EC, Perez CA, Brady LW. Perez and Brady’s principles and practice of radiation oncology. 5. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 8.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Chargari C, Clemenson C, Martins I, Perfettini JL, Deutsch E. Understanding the functions of tumor stroma in resistance to ionizing radiation: Emerging targets for pharmacological modulation. Drug Resist Updat. 2013 doi: 10.1016/j.drup.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Robinson CG, Bradley JD. The treatment of early-stage disease. Semin Radiat Oncol. 2010;20:178–85. doi: 10.1016/j.semradonc.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Palma D, Senan S. Stereotactic radiation therapy: changing treatment paradigms for stage I nonsmall cell lung cancer. Curr Opin Oncol. 2011;23:133–9. doi: 10.1097/CCO.0b013e328341ee11. [DOI] [PubMed] [Google Scholar]
- 12.Stephans K. Stereotactic body radiotherapy for stage I non-small cell lung cancer. Cleve Clin J Med. 2012;79(Electronic Suppl 1):eS26–31. doi: 10.3949/ccjm.79.s2.06. [DOI] [PubMed] [Google Scholar]
- 13.Salama JK, Vokes EE. New Radiotherapy and Chemoradiotherapy Approaches for Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 doi: 10.1200/JCO.2012.44.5064. [DOI] [PubMed] [Google Scholar]
- 14.Haasbeek CJ, Slotman BJ, Senan S. Radiotherapy for lung cancer: clinical impact of recent technical advances. Lung Cancer. 2009;64:1–8. doi: 10.1016/j.lungcan.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Thariat J, Hannoun-Levi JM, Sun Myint A, Vuong T, Gerard JP. Past, present, and future of radiotherapy for the benefit of patients. Nat Rev Clin Oncol. 2013;10:52–60. doi: 10.1038/nrclinonc.2012.203. [DOI] [PubMed] [Google Scholar]
- 16.Valerie K, Yacoub A, Hagan MP, et al. Radiation-induced cell signaling: inside-out and outside-in. Mol Cancer Ther. 2007;6:789–801. doi: 10.1158/1535-7163.MCT-06-0596. [DOI] [PubMed] [Google Scholar]
- 17.Dent P, Yacoub A, Contessa J, et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res. 2003;159:283–300. doi: 10.1667/0033-7587(2003)159[0283:sariao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 18.Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol. 2010;80:1904–14. doi: 10.1016/j.bcp.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szumiel I. Intrinsic radiation sensitivity: cellular signaling is the key. Radiat Res. 2008;169:249–58. doi: 10.1667/RR1239.1. [DOI] [PubMed] [Google Scholar]
- 20.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–54. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 22.Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer research. 2001;61:3894–901. [PubMed] [Google Scholar]
- 23.Dent P, Reardon DB, Wood SL, et al. Inactivation of raf-1 by a protein-tyrosine phosphatase stimulated by GTP and reconstituted by Galphai/o subunits. The Journal of biological chemistry. 1996;271:3119–23. doi: 10.1074/jbc.271.6.3119. [DOI] [PubMed] [Google Scholar]
- 24.Tonks NK. Protein tyrosine phosphatases and the control of cellular signaling responses. Adv Pharmacol. 1996;36:91–119. doi: 10.1016/s1054-3589(08)60578-5. [DOI] [PubMed] [Google Scholar]
- 25.Gulbins E, Kolesnick R. Raft ceramide in molecular medicine. Oncogene. 2003;22:7070–7. doi: 10.1038/sj.onc.1207146. [DOI] [PubMed] [Google Scholar]
- 26.Corre I, Niaudet C, Paris F. Plasma membrane signaling induced by ionizing radiation. Mutat Res. 2010;704:61–7. doi: 10.1016/j.mrrev.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Choy H, Milas L. Enhancing radiotherapy with cyclooxygenase-2 enzyme inhibitors: a rational advance? J Natl Cancer Inst. 2003;95:1440–52. doi: 10.1093/jnci/djg058. [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677–706. doi: 10.1146/annurev-immunol-020711-075008. [DOI] [PubMed] [Google Scholar]
- 31.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–93. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pidgeon GP, Lysaght J, Krishnamoorthy S, et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–24. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- 33.Agarwal S, Reddy GV, Reddanna P. Eicosanoids in inflammation and cancer: the role of COX-2. Expert Rev Clin Immunol. 2009;5:145–65. doi: 10.1586/1744666X.5.2.145. [DOI] [PubMed] [Google Scholar]
- 34.Liu CH, Chang SH, Narko K, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. The Journal of biological chemistry. 2001;276:18563–9. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- 35.Steinauer KK, Gibbs I, Ning S, French JN, Armstrong J, Knox SJ. Radiation induces upregulation of cyclooxygenase-2 (COX-2) protein in PC-3 cells. Int J Radiat Oncol Biol Phys. 2000;48:325–8. doi: 10.1016/s0360-3016(00)00671-4. [DOI] [PubMed] [Google Scholar]
- 36.Yang HJ, Kim N, Seong KM, Youn H, Youn B. Investigation of Radiation-induced Transcriptome Profile of Radioresistant Non-small Cell Lung Cancer A549 Cells Using RNA-seq. PLoS One. 2013;8:e59319. doi: 10.1371/journal.pone.0059319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao Z, Mason KA, Milas L. Cyclo-oxygenase-2 and its inhibition in cancer: is there a role? Drugs. 2007;67:821–45. doi: 10.2165/00003495-200767060-00001. [DOI] [PubMed] [Google Scholar]
- 38.Petkova DK, Clelland C, Ronan J, et al. Overexpression of cyclooxygenase-2 in non-small cell lung cancer. Respir Med. 2004;98:164–72. doi: 10.1016/j.rmed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer research. 1998;58:3761–4. [PubMed] [Google Scholar]
- 40.Laga AC, Zander DS, Cagle PT. Prognostic significance of cyclooxygenase 2 expression in 259 cases of non-small cell lung cancer. Arch Pathol Lab Med. 2005;129:1113–7. doi: 10.5858/2005-129-1113-PSOCEI. [DOI] [PubMed] [Google Scholar]
- 41.Brabender J, Park J, Metzger R, et al. Prognostic significance of cyclooxygenase 2 mRNA expression in non-small cell lung cancer. Ann Surg. 2002;235:440–3. doi: 10.1097/00000658-200203000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang H, Wang J, Zhao W. Cox-2 in non-small cell lung cancer: A meta-analysis. Clin Chim Acta. 2013;419:26–32. doi: 10.1016/j.cca.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Horn L, Backlund M, Johnson DH. Targeting the eicosanoid pathway in non-small-cell lung cancer. Expert Opin Ther Targets. 2009;13:675–88. doi: 10.1517/14728220902915567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch A, Bergman B, Holmberg E, et al. Effect of celecoxib on survival in patients with advanced non-small cell lung cancer: a double blind randomised clinical phase III trial (CYCLUS study) by the Swedish Lung Cancer Study Group. Eur J Cancer. 2011;47:1546–55. doi: 10.1016/j.ejca.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Groen HJ, Sietsma H, Vincent A, et al. Randomized, placebo-controlled phase III study of docetaxel plus carboplatin with celecoxib and cyclooxygenase-2 expression as a biomarker for patients with advanced non-small-cell lung cancer: the NVALT-4 study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4320–6. doi: 10.1200/JCO.2011.35.5214. [DOI] [PubMed] [Google Scholar]
- 46.Mutter R, Lu B, Carbone DP, et al. A phase II study of celecoxib in combination with paclitaxel, carboplatin, and radiotherapy for patients with inoperable stage IIIA/B non-small cell lung cancer. Clin Cancer Res. 2009;15:2158–65. doi: 10.1158/1078-0432.CCR-08-0629. [DOI] [PubMed] [Google Scholar]
- 47.Gore E, Bae K, Langer C, et al. Phase I/II trial of a COX-2 inhibitor with limited field radiation for intermediate prognosis patients who have locally advanced non-small-cell lung cancer: radiation therapy oncology group 0213. Clin Lung Cancer. 2011;12:125–30. doi: 10.1016/j.cllc.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Schleicher SM, Thotala DK, Linkous AG, et al. Autotaxin and LPA receptors represent potential molecular targets for the radiosensitization of murine glioma through effects on tumor vasculature. PLoS One. 2011;6:e22182. doi: 10.1371/journal.pone.0022182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linkous A, Geng L, Lyshchik A, Hallahan DE, Yazlovitskaya EM. Cytosolic phospholipase A2: targeting cancer through the tumor vasculature. Clin Cancer Res. 2009;15:1635–44. doi: 10.1158/1078-0432.CCR-08-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linkous AG, Yazlovitskaya EM, Hallahan DE. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. J Natl Cancer Inst. 2010;102:1398–412. doi: 10.1093/jnci/djq290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yazlovitskaya EM, Linkous AG, Thotala DK, Cuneo KC, Hallahan DE. Cytosolic phospholipase A2 regulates viability of irradiated vascular endothelium. Cell Death Differ. 2008;15:1641–53. doi: 10.1038/cdd.2008.93. [DOI] [PubMed] [Google Scholar]
- 52.Chakraborti S. Phospholipase A(2) isoforms: a perspective. Cell Signal. 2003;15:637–65. doi: 10.1016/s0898-6568(02)00144-4. [DOI] [PubMed] [Google Scholar]
- 53.Herbert SP, Odell AF, Ponnambalam S, Walker JH. Activation of cytosolic phospholipase A2-{alpha} as a novel mechanism regulating endothelial cell cycle progression and angiogenesis. J Biol Chem. 2009;284:5784–96. doi: 10.1074/jbc.M807282200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Linkous AG, Yazlovitskaya EM, Hallahan DE. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. J Natl Cancer Inst. 2010;102:1398–412. doi: 10.1093/jnci/djq290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linkous A, Yazlovitskaya E. Cytosolic phospholipase A2 as a mediator of disease pathogenesis. Cell Microbiol. 2010;12:1369–77. doi: 10.1111/j.1462-5822.2010.01505.x. [DOI] [PubMed] [Google Scholar]
- 56.Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–5. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 57.Han C, Demetris AJ, Michalopoulos G, Shelhamer JH, Wu T. 85-kDa cPLA(2) plays a critical role in PPAR-mediated gene transcription in human hepatoma cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G586–97. doi: 10.1152/ajpgi.00305.2001. [DOI] [PubMed] [Google Scholar]
- 58.Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol. 2010;85:593–8. doi: 10.1002/ajh.21745. [DOI] [PubMed] [Google Scholar]
- 59.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–64. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiser-Evans MC, Wang XQ, Amin J, et al. Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res. 2009;69:1733–8. doi: 10.1158/0008-5472.CAN-08-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Linkous A, Geng L, Lyshchik A, Hallahan DE, Yazlovitskaya EM. Cytosolic phospholipase A2: targeting cancer through the tumor vasculature. Clin Cancer Res. 2009;15:1635–44. doi: 10.1158/1078-0432.CCR-08-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prokazova NV, Zvezdina ND, Korotaeva AA. Effect of lysophosphatidylcholine on transmembrane signal transduction. Biochemistry (Mosc) 1998;63:31–7. [PubMed] [Google Scholar]
- 63.Lee HY, Murata J, Clair T, et al. Cloning, chromosomal localization, and tissue expression of autotaxin from human teratocarcinoma cells. Biochem Biophys Res Commun. 1996;218:714–9. doi: 10.1006/bbrc.1996.0127. [DOI] [PubMed] [Google Scholar]
- 64.Choi JW, Herr DR, Noguchi K, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–86. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 65.Nishimasu H, Okudaira S, Hama K, et al. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat Struct Mol Biol. 2011;18:205–12. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- 66.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Progress in lipid research. 2007;46:145–60. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 67.Kishi Y, Okudaira S, Tanaka M, et al. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J Biol Chem. 2006;281:17492–500. doi: 10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 68.Liu S, Umezu-Goto M, Murph M, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15:539–50. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.So J, Wang FQ, Navari J, Schreher J, Fishman DA. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2) Gynecol Oncol. 2005;97:870–8. doi: 10.1016/j.ygyno.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Nam SW, Clair T, Kim YS, et al. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer research. 2001;61:6938–44. [PubMed] [Google Scholar]
- 71.Baumforth KR, Flavell JR, Reynolds GM, et al. Induction of autotaxin by the Epstein-Barr virus promotes the growth and survival of Hodgkin lymphoma cells. Blood. 2005;106:2138–46. doi: 10.1182/blood-2005-02-0471. [DOI] [PubMed] [Google Scholar]
- 72.Xu X, Prestwich GD. Inhibition of tumor growth and angiogenesis by a lysophosphatidic acid antagonist in an engineered three-dimensional lung cancer xenograft model. Cancer. 2010;116:1739–50. doi: 10.1002/cncr.24907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 74.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 75.Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105:19893–7. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17:2081–6. doi: 10.1158/1078-0432.CCR-10-1591. [DOI] [PubMed] [Google Scholar]
- 78.Choi YL, Takeuchi K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–6. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 79.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 80. [DOI] [PubMed] [Google Scholar]
- 80.Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. European journal of cancer. 2010;46:1773–80. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 83.Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 84.Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64:7241–4. doi: 10.1158/0008-5472.CAN-04-1905. [DOI] [PubMed] [Google Scholar]
- 85.Xie H, Yang F, Deng L, et al. The performance of a bone-derived scaffold material in the repair of critical bone defects in a rhesus monkey model. Biomaterials. 2007;28:3314–24. doi: 10.1016/j.biomaterials.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 86.Bang YJ. The potential for crizotinib in non-small cell lung cancer: a perspective review. Ther Adv Med Oncol. 2011;3:279–91. doi: 10.1177/1758834011419002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 90.Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heuckmann JM, Holzel M, Sos ML, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17:7394–401. doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun Y, Nowak KA, Zaorsky NG, et al. ALK Inhibitor PF02341066 (Crizotinib) Increases Sensitivity to Radiation in Non-Small Cell Lung Cancer Expressing EML4-ALK. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-12-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tumati V, Kumar S, Yu L, Chen B, Choy H, Saha D. Effect of PF-02341066 and radiation on non-small cell lung cancer cells. Oncology reports. 2013;29:1094–100. doi: 10.3892/or.2012.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–53. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 99.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–16. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 100.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weinstein IB. Cancer Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 102.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 104.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 105.Pick E, Kluger Y, Giltnane JM, et al. High HSP90 expression is associated with decreased survival in breast cancer. Cancer research. 2007;67:2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 106.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 107.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Whitesell L, Lin NU. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim Biophys Acta. 2012;1823:756–66. doi: 10.1016/j.bbamcr.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 109.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–55. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Travers J, Sharp S, Workman P. HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today. 2012;17:242–52. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 112.Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30:2581–6. doi: 10.1038/onc.2010.625. [DOI] [PubMed] [Google Scholar]
- 113.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:4953–60. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Socinski MA, Goldman J, El-Hariry I, et al. A multicenter Phase II study of ganetespib monotherapy in patients with genotypically-defined advanced non-small cell lung cancer. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Acquaviva J, Smith DL, Sang J, et al. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol Cancer Ther. 2012;11:2633–43. doi: 10.1158/1535-7163.MCT-12-0615. [DOI] [PubMed] [Google Scholar]
- 116.Sang J, Acquaviva J, Friedland JC, et al. Targeted Inhibition of the Molecular Chaperone Hsp90 Overcomes ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Cancer Discov. 2013;3:430–43. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ueno T, Tsukuda K, Toyooka S, et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer. 2012;76:26–31. doi: 10.1016/j.lungcan.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 118.Garon EB, Finn RS, Hamidi H, et al. The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther. 2013 doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Camphausen K, Tofilon PJ. Inhibition of Hsp90: a multitarget approach to radiosensitization. Clin Cancer Res. 2007;13:4326–30. doi: 10.1158/1078-0432.CCR-07-0632. [DOI] [PubMed] [Google Scholar]
- 120.Eccles SA, Massey A, Raynaud FI, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer research. 2008;68:2850–60. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 121.Hartmann S, Gunther N, Biehl M, et al. Hsp90 inhibition by NVP-AUY922 and NVP-BEP800 decreases migration and invasion of irradiated normoxic and hypoxic tumor cell lines. Cancer Lett. 2013;331:200–10. doi: 10.1016/j.canlet.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 122.Djuzenova CS, Blassl C, Roloff K, et al. Hsp90 inhibitor NVP-AUY922 enhances radiation sensitivity of tumor cell lines under hypoxia. Cancer Biol Ther. 2012;13:425–34. doi: 10.4161/cbt.19294. [DOI] [PubMed] [Google Scholar]
- 123.Stingl L, Stuhmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS. Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br J Cancer. 2010;102:1578–91. doi: 10.1038/sj.bjc.6605683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zaidi S, McLaughlin M, Bhide SA, et al. The HSP90 inhibitor NVP-AUY922 radiosensitizes by abrogation of homologous recombination resulting in mitotic entry with unresolved DNA damage. PLoS One. 2012;7:e35436. doi: 10.1371/journal.pone.0035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aggarwal C, Somaiah N, Simon G. Antiangiogenic agents in the management of non-small cell lung cancer: where do we stand now and where are we headed? Cancer Biol Ther. 2012;13:247–63. doi: 10.4161/cbt.13.5.19594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ellis PM, Al-Saleh K. Multitargeted anti-angiogenic agents and NSCLC: clinical update and future directions. Crit Rev Oncol Hematol. 2012;84:47–58. doi: 10.1016/j.critrevonc.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 127.Blumenschein GR., Jr Developmental antiangiogenic agents for the treatment of non-small cell lung cancer (NSCLC) Invest New Drugs. 2012;30:1802–11. doi: 10.1007/s10637-011-9750-1. [DOI] [PubMed] [Google Scholar]
- 128.Geng L, Donnelly E, McMahon G, et al. Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer research. 2001;61:2413–9. [PubMed] [Google Scholar]
- 129.Kleibeuker EA, Griffioen AW, Verheul HM, Slotman BJ, Thijssen VL. Combining angiogenesis inhibition and radiotherapy: a double-edged sword. Drug Resist Updat. 2012;15:173–82. doi: 10.1016/j.drup.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 130.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 131.Yap TA, Workman P. Exploiting the cancer genome: strategies for the discovery and clinical development of targeted molecular therapeutics. Annu Rev Pharmacol Toxicol. 2012;52:549–73. doi: 10.1146/annurev-pharmtox-010611-134532. [DOI] [PubMed] [Google Scholar]
- 132.Koh PK, Faivre-Finn C, Blackhall FH, De Ruysscher D. Targeted agents in non-small cell lung cancer (NSCLC): clinical developments and rationale for the combination with thoracic radiotherapy. Cancer Treat Rev. 2012;38:626–40. doi: 10.1016/j.ctrv.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 133.Chang HH, Meuillet EJ. Identification and development of mPGES-1 inhibitors: where we are at? Future Med Chem. 2011;3:1909–34. doi: 10.4155/fmc.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roskoski R., Jr Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013;68:68–94. doi: 10.1016/j.phrs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 135.Shimamura T, Shapiro GI. Heat shock protein 90 inhibition in lung cancer. J Thorac Oncol. 2008;3:S152–9. doi: 10.1097/JTO.0b013e318174ea3a. [DOI] [PMC free article] [PubMed] [Google Scholar]