

Abstract
Centromeres are chromosomal loci required for accurate segregation of sister chromatids during mitosis. The location of the centromere on the chromosome is not dependent on DNA sequence, but rather it is epigenetically specified by the histone H3 variant centromere protein A (CENP-A). The N-terminal tail of CENP-A is highly divergent from other H3 variants. Canonical histone N termini are hotspots of conserved posttranslational modification; however, no broadly conserved modifications of the vertebrate CENP-A tail have been previously observed. Here, we report three posttranslational modifications on human CENP-A N termini using high-resolution MS: trimethylation of Gly1 and phosphorylation of Ser16 and Ser18. Our results demonstrate that CENP-A is subjected to constitutive initiating methionine removal, similar to other H3 variants. The nascent N-terminal residue Gly1 becomes trimethylated on the α-amino group. We demonstrate that the N-terminal RCC1 methyltransferase is capable of modifying the CENP-A N terminus. Methylation occurs in the prenucleosomal form and marks the majority of CENP-A nucleosomes. Serine 16 and 18 become phosphorylated in prenucleosomal CENP-A and are phosphorylated on asynchronous and mitotic nucleosomal CENP-A and are important for chromosome segregation during mitosis. The double phosphorylation motif forms a salt-bridged secondary structure and causes CENP-A N-terminal tails to form intramolecular associations. Analytical ultracentrifugation of phospho-mimetic CENP-A nucleosome arrays demonstrates that phosphorylation results in greater intranucleosome associations and counteracts the hyperoligomerized state exhibited by unmodified CENP-A nucleosome arrays. Our studies have revealed that the major modifications on the N-terminal tail of CENP-A alter the physical properties of the chromatin fiber at the centromere.
Keywords: epigenetics, kinetochore, mass spectrometry
Centromeres are epigenetically inherited chromosomal loci that direct chromosome segregation. Kinetochores form atop centromeres to couple microtubules to sister chromatids. Nucleosomes harboring the centromere-specific histone-H3 variant, centromere protein A (CENP-A), are a conserved and essential component of the eukaryotic centromere. CENP-A is the best candidate for epigenetically marking the location of the centromere (1, 2). Centromeres in flies and humans are comprised of discontinuous patches of CENP-A nucleosomes, interspersed with H3 nucleosomes (3). CENP-A nucleosomes directly recruit CENP-C and CENP-N, which are part of a large constitutive centromere associated network (CCAN) of proteins recruited to mediate the processes of kinetochore formation and microtubule attachment necessary for mitosis (4–6).
CENP-A nucleosomes are assembled by the CENP-A–specific assembly factor Holliday Junction Recognition Protein (HJURP) in early G1 phase (7–10). The CENP-A–histone H4 heterodimer is recognized by HJURP via the CENP-A targeting domain, forming a prenucleosomal complex that exists during G2 and mitosis (8, 9, 11).
Human CENP-A and histone H3.1 are 56% identical within the histone fold; however, the unstructured N-terminal tail domains of these proteins are highly divergent, sharing only 24% identity. Several lysine residues in the canonical H3 N terminus are highly conserved targets of acetylation and methylation that mediate epigenetic regulation of local chromatin activity (12, 13). Posttranslational modification (PTM) of histones is usually combinatorial, and the net PTM status of these histones can negatively or positively influence transcription or direct global condensation of chromatin. The CENP-A N-terminal tail is enriched in arginines and lacks most of the well-characterized lysines that are targets for modification in histone H3, so the CENP-A tail simply cannot share many of the modifications to which H3 is subjected. In contrast, human CENP-A and histone H3 share similar phosphorylation sites (Ser10 in H3 and Ser7 in CENP-A) that are modified by the Aurora B kinase during mitosis (14–16). Phosphorylation by Aurora B does not appear to be a conserved feature of CENP-A–containing nucleosomes because Ser7 is very poorly conserved even within mammals.
We provide a comprehensive analysis of the posttranslational modifications of human CENP-A. The initiator methionine (Metinit) is cleaved from CENP-A, similar to canonical histones. The major form of the N-terminal tail of human CENP-A is posttranslationally modified by combinations of three modifications: trimethylation of the α-amino group of Gly1 and phosphorylation at Ser16 and Ser18 . In vitro data show that CENP-A N-terminal methylation can be catalyzed by N-terminal RCC1 methyltransferase (NRMT). CENP-A phosphorylated on Ser16/Ser18 forms a salt-bridged secondary structure within the N terminus that allows for intra- and intermolecular interactions and influences the conformation of reconstituted CENP-A nucleosome arrays. Expression of a version of CENP-A that cannot be phosphorylated results in mitotic errors.
Results
CENP-A Is Trimethylated on the α-N Position of Gly1.
Human CENP-A nucleosomes were affinity purified from micrococcal nuclease (MNase)-digested chromatin from an asynchronously growing stable cell line expressing tagged CENP-A (Fig. S1). Affinity purification yielded ∼50 pmol of CENP-A nucleosomes in a soluble form (Fig. S1 C and D).
Purified nucleosomes were digested into peptides using the endoproteinases trypsin, LysC, or GluC, and then analyzed using online reversed phase separation coupled to high-resolution MS (Fig. S1E). We readily detected CENP-C and histones H2A and H2B, which are known to tightly associate with CENP-A when in its nucleosomal form (Dataset S1A). Ninety-eight percent of the CENP-A primary sequence was covered by peptides that were detected at greater than 500:1 signal-to-noise. This level of sensitivity allows detection of modified forms of CENP-A peptides present at less than 0.2% of the most abundant peptide form (Dataset S1B).
The initiating methionine (Metinit) was removed in all forms observed in a GluC digest (Fig. 1 A and B). Removal of the initiating methionine occurs cotranslationally by methionine amino-peptidase (MetAP) activity. Because the Metinit of CENP-A is removed, similar to all other histone H3 variants, we have adopted conventional histone nomenclature to refer to positions of CENP-A amino acids where the first amino acid after the cleaved initiating methionine is designated as position 1 (17). Four different species of the CENP-A G1–E10 peptide were observed with masses differing by ∼14 Da, ranging from unmodified to an increased shift of 42 Da. High-resolution MS data show the Δ42 Da species is due to the addition of three methyl groups (Δ42.0470 Da, 3.12 ppm mass accuracy). The accurate mass measurement is not consistent with the addition of a single acetyl group (Δ42.0106 Da, 31.47 ppm mass accuracy) (Fig. S2 A and B). Fragmentation of the Δ42 Da species using election transfer dissociation (ETD) produced an MS2 spectrum that revealed Gly1 as the trimethylated amino acid. Glycine lacks a side-chain, so the N-terminal primary amine (α-N) is the only possible site of trimethylation (Fig. 1 A–C). CENP-A N-terminal trimethylation differs from known histone methylation that occurs on the side chains of arginine and lysine residues.
Fig. 1.

CENP-A is trimethylated on the α-amino position of the N-terminal glycine. (A) CENP-A GluC digestions produced an ETD MS2 spectrum of an N-terminal peptide trimethylated on the α-N position of glycine 1. Value above bracket indicates magnification. (B) Sequence coverage of α-N trimethylated CENP-A ETD MS2 spectrum. (C) GluC cleavages at E10 to produce an N-terminal peptide that had been subjected to proteolytic initiating methionine removal in vivo. A conventional histone nomenclature is adopted for CENP-A where the initiator methionine is “Met0.” (D) Integrated chromatographic peak areas of CENP-A G1–E10 methylated forms. Orange hexagons, α-N methylation. (E) A fusion protein containing amino acids 1–10 of CENP-A was engineered to reveal Gly1 by Factor X cleavage to produce a substrate for NRMT modification. (F) CENP-A fusion protein was methylated using recombinant NRMT and 3H-SAM. Wild-type CENP-A N termini (GPRR-) and mutants in which R3 and R4 were replaced by glutamine (GPQR- or GPQQ-) were tested. P < 0.01, Student’s t test, n = 3 independent experiments ± SD.
NRMT directly methylates the α-amino terminus of regulator of chromosome condensation 1 (RCC1) (18). Affinity purified CENP-A shows a pattern of complete N-terminal methylation (Fig. 1D). Almost 86% of the observed CENP-A G1–E10 peptides are trimethylated on Gly1 and incompletely methylated CENP-A (mono- and dimethylated) peptides constitute only 0.6% of the CENP-A G1–E10 peptides derived from nucleosomes. The remaining 13% of CENP-A nucleosomes are unmodified on Gly1. This predominant trimethylation is consistent with NRMT-mediated methylation, which transfers single methyl groups in a processive manner (19).
To determine whether the CENP-A N terminus was a direct substrate for NRMT in vitro, we engineered a recombinant CENP-A tail (G1–E10) in which Gly1 was exposed for modification by NRMT by Factor X cleavage to recapitulate the CENP-A tail following Metinit removal (Fig. 1E). The wild-type CENP-A amino tail (GPRR) was robustly methylated by NRMT in vitro, using 3H-S-adenosyl methionine (SAM) as a substrate, whereas CENP-A mutant sequences, which should be defective substrates for α-N methylation (GPQR and GPQQ), showed little or no methylation (Fig. 1F) (18, 19). MS analysis of the in vitro NRMT-methylated peptide shows that Gly1 is the only substrate and is completely trimethylated, consistent with the predominant trimethylation observed from in vivo-purified CENP-A (Fig. S2 C and D).
CENP-A N-Terminal Tail Is Doubly Phosphorylated at Ser16 and Ser18.
Purification of CENP-A from interphase cells and subsequent MS analysis identified two phosphorylations of the CENP-A N-terminal tail. Trypsin digestion produced a CENP-A fragment that encompassed residues R14–R27. The observed [M + 3H]+3 ion species (m/z 551.5814) was 159.9320 Da larger than the calculated mass of the unmodified peptide (m/z 498.2708), which is consistent with a doubly phosphorylated peptide (−0.54 ppm).
Two large-scale proteomic studies reported detection of a similar doubly phosphorylated CENP-A peptide; however, automated MS2 spectra interpretation was unable to distinguish which of the five possible sites within the CENP-A peptide were modified (20, 21). Our analysis of c and z ions in the R14–R27 ETD MS2 spectra was of insufficient coverage to confidently assign the phosphates to the correct amino acids due to the high concentration of prolines, which do not produce c and z fragments using ETD. To identify the sites of phosphorylation, we took advantage of a and y ions, which are produced at prolines using ETD, although these ions are less abundant than c and z ions (22). The total ion fragment data definitively demonstrated that Ser16 and Ser18 of CENP-A are the sites of phosphorylation (Fig. 2 A and B). Singly phosphorylated and unmodified species were observed at much lower frequency, 10% and 1%, respectively, relative to double phosphorylated peptides (Fig. 2C). In the 10% of cases where CENP-A contained only a single phosphate, ∼80% of the CENP-A peptides were phosphorylated only on Ser16, and 20% were phosphorylated on Ser18.
Fig. 2.

The CENP-A N-terminal tail is doubly phosphorylated at Ser16 and Ser18. (A) ETD MS2 spectrum of doubly phosphorylated CENP-A R14–R27. Value at bracket indicates magnification level for region of spectrum. (B) Sequence coverage of an MS2 spectrum of a trypsin-generated CENP-A peptide R14–R27. Signature a, c, y, and z ions indicate the presence of phosphate at Ser16 and Ser18. (C) CENP-A is unmodified and singly or doubly phosphorylated in a ratio of ∼1:10:100, respectively. Green ovals represent degree of phosphorylation on peptide.
CENP-A Is Combinatorially α-N Methylated and Phosphorylated.
Combinatorial patterns of PTMs form complex recognition signals that underlie a wide range of epigenetic phenomena. To determine what combinations of CENP-A α-N methylation and phosphorylation exist on the same protein, we used LysC to generate a peptide of the entire N-terminal tail of CENP-A. The most abundant of the CENP-A G1-K48 peptide was a combinatorially modified peptide that included α-N-trimethylation and phosphorylation of Ser16 and Ser18 (Fig. 3 A and B). We confirmed the identity of this peptide by performing a targeted ETD MS2 analysis (Fig. S3 A–C). CENP-A trimethylated, doubly phosphorylated peptides were threefold more abundant than the unmethylated, phosphorylated peptides in three independent CENP-A nucleosome purifications. Modifications reside in regions of the CENP-A tail that are well conserved in vertebrates (Fig. 3C) suggesting that these modifications may occur in several species.
Fig. 3.

CENP-A N-terminal tails are marked with combinations of stable phosphorylation and α-N methylation. (A) LysC digestion yields a CENP-A G1–K48 peptide. A full MS spectrum (17 averaged scans) shows combinatorial α-N trimethylation and phosphorylations of the same peptides in asynchronously cycling cells. Accurate mass and charge state is reported for the 13C x 3 isotopes. The most abundant form is trimethylated at Gly1 and doubly phosphorylated at Ser16 and Ser18. Green oval, phosphorylation; orange hexagon, methylation. (B) Integrated chromatographic peak areas of CENP-A G1–K48 PTM forms. n = 3 independent biological replicates. Error bars represent SEM. (C) Comparison of vertebrate CENP-A protein sequences reveals conservation at observed PTM sites: N termini (blue), proline–arginine (red), and serine (green). (D) Soluble and chromatin fractions of cells blocked in mitosis were used to purify populations of (circle 1) prenucleosomal, and nucleosomal CENP-A from (circle 2) asynchronously-dividing cells and (circle 3) mitotic cells. Integrated chromatographic peak area quantified CENP-A tail PTM forms: (E) prenucleosomal containing Metinit; (F) prenucleosomal and mitotic nucleosomal fractions lacking Metinit. Acetylation (blue square) was observed only when Metinit was present.
CENP-A N-Terminal Tail Is Combinatorially Modified Before Nucleosome Deposition.
The CENP-A prenucleosomal complex accumulates during mitosis and is recruited to centromeres in G1 phase (Fig. 3D). To identify the modifications of prenucleosomal CENP-A, we purified CENP-A from the chromatin-free extract of mitotically enriched cells (Fig. S4 A–D). We found high levels of HJURP peptides and did not observe peptides derived from histones H2A and H2B. These results are consistent with exclusive purification of the CENP-A prenucleosomal complex (8) (Dataset S1C).
We generated intact N-terminal tail peptides from prenucleosomal CENP-A using LysC (Dataset S1D). Ten percent of prenucleosomal CENP-A tails retained the initiating methionine, and a small fraction of this group was α-N acetylated on Met0 (Fig. 3E and Figs. S5A and S6 A and B). The presence of M0 is in contrast to the nucleosomal CENP-A where all of the peptides were subjected to Metinit removal and no α-N group acetylations were observed. Because Metinit-containing forms were not observed in analysis of nucleosomal CENP-A, we conclude that these forms become degraded or are not stably incorporated in chromatin.
Prenucleosomal CENP-A was α-N trimethylated; however, the level of α-N trimethylation was lower in prenucleosomal CENP-A compared with asynchronous CENP-A nucleosome populations. Unmethylated forms were present in a ratio of ∼2:1 compared with trimethylated forms. This ratio is nearly inverse to the degree of α-N methylation found in asynchronous nucleosomal CENP-A. Phosphorylation levels of prenucleosomal CENP-A were identical to CENP-A within the nucleosome, with a ratio of 1:10:100 for unmodified and singly and doubly phosphorylated forms (Fig. 3F, Fig. S5B). The same pattern of phosphorylation was also detected for all Metinit-containing forms.
Fraction of α-N Trimethylated CENP-A Increases with Cell Cycle Progression.
Centromere-bound nucleosomal CENP-A in mitosis has existed for a minimum of one cell cycle. For this reason, mitotic nucleosomal CENP-A represents an “old” population of CENP-A protein (Fig. 3D). To determine the modification state of CENP-A during mitosis, we isolated CENP-A nucleosomes from nocodazole-treated cells (Fig. S4 A–D). Analysis of our mitotically enriched nucleosomal CENP-A sample identified significant amounts of H2A, H2B, H3, H4, and CENP-C, but no HJURP peptides (Dataset S1E), consistent with purification of an exclusively nucleosome population. N-terminal α-N methylation and phosphorylations on mitotic CENP-A was observed on the same sites as in mitotic prenucleosomal and asynchronous nucleosomal CENP-A populations. Mitotic nucleosomal CENP-A was almost entirely trimethylated; less than 10% of CENP-A forms were unmethylated (Fig. 3F, Fig. S5C, Dataset S1F).
Tandem Phosphorylation Induces Secondary Structure in CENP-A N Termini.
We noted a correlation between Ser16/Ser18 phosphorylation and trypsin missed cleavages within CENP-A peptides (Fig. 4 A and B). Unphosphorylated CENP-A peptides were efficiently cleaved by trypsin at the P1–P1′ cleavage site to produce the S16–R27 peptide. In contrast, singly phosphorylated peptides were observed as a mixture of zero-, one-, and two-missed-cleavage peptides. Peptides phosphorylated at both Ser16 and Ser18 were almost exclusively detected in the two-missed-cleavage form. The P1–P1′ bond of R15–S16ph should be cleaved at a lower rate because it is followed by an acidic residue in the P1′ position (23, 24), and the P2–P1 bond between should be the preferred site. Instead, we observed that the P3–P2 bond is the primary site of cleavage. Structural constraints can influence the efficiency of trypsin cleavage (25), and we hypothesized that one or more arginine–phosphate salt bridges formed by Ser16 and Ser18 phosphorylation contribute to the protection of the P1–P1′ and P2–P1 bonds. To determine whether trypsin inhibition was an intrinsic property of the peptide and dependent on phosphorylation, we digested a synthetic CENP-A E10–G24 peptide using trypsin. At 19 h, trypsin digestion of the unmodified peptide resulted in equal parts zero- and one-missed-cleavage forms (Fig. 4C). During the same amount of time, trypsin digestion of a doubly phosphorylated peptide produced only the two-missed-cleavage form, R14–G24.
Fig. 4.

CENP-A Ser16/Ser18 phosphorylation forms a compact secondary structure. (A) Nomenclature for proteolytic sites. (B) The percentage of trypsin cleavage at the P1–P1′, P1–P2, and P2-P3 sites for peptides derived from affinity-purified CENP-A. (C) The percentage of trypsin cleavage at the P1–P1′, P1–P2, and P2–P3 sites for unmodified and phosphorylated synthetic CENP-A peptides E10–G24 digested with trypsin over time were followed by liquid chromatography (LC)-MS. (D) IM-MS analysis of the monomeric [M + 3H]+3 ion of unmodified and doubly phosphorylated E10–G24 peptide is used to calculate the ion-neutral collision cross section (CCS) for each species. CCS values for the major- and minor-contributing conformers were calculated as Gaussian peaks from the sum of each peptide. (E) Tryptic digested BSA peptides analyzed by IM-MS are plotted according to CCS and molecular weight. Comparison of phosphorylated (square) and unmodified (triangle) E10–G24 peptide with the random coil trend line reveals that phosphorylation causes greater compactness relative to the increase in molecular weight. (F) AUC profiles of in vitro assembled chromatin arrays showing differential MgCl2-dependent folding between H3, CENP-A, and phospho-mimetic (S16D/S18D) CENP-A arrays. All array types (minus MgCl2) have overlapping sedimentation profiles, indicating that they are saturated to the same extent. Therefore, the observed differences upon array folding are exclusively due to the contributions of different proteins.
Arginine–phosphoserine electrostatic salt bridging is highly stable as both arginine and phosphoserine are ionized at physiological pH (25). We reasoned that arginine–phosphate salt bridges could form intramolecular interactions within CENP-A tails and intermolecular interactions between individual tails. Therefore, we analyzed the conformation of doubly phosphorylated E10–G24 synthetic peptide using ion mobility-mass spectrometry (IM-MS). IM-MS drift time measurements are used to calculate the ion-neutral collision cross-section (CCS) of peptide ion conformations (26). Unmodified E10–G24 CENP-A peptides ([M + 3H]+3 ions) exhibit CCS profiles that deviate from a trend line of randomly coiled peptides by −7.6% (major peak), which indicates that E10–G24 exists in an already-compact conformation (Fig. 4 D and E). Phosphorylated E10–G24 exhibits a CCS profile that deviates −11.0% (major peak) from randomly coiled peptides (Fig. 4 D and E). Therefore, the conformation of phosphorylated E10–G24 peptide is further compacted relative to the unmodified E10–G24, consistent with intramolecular salt bridging of phosphorylated CENP-A N termini producing a more compact structure.
To assess intermolecular interactions mediated by CENP-A tail phosphorylation, we analyzed synthetic E10–G24 peptide for potential dimers by MS. Noncovalent dimeric and monomeric species have identical m/z values but can be distinguished by their isotopic peak pattern. Separation of isotopic peaks of a species is dictated by charge state, and calculating the molecular weight at a given m/z value reveals stoichiometry. Doubly phosphorylated E10–G24 peptide was present as highly abundant monomeric species ([M + 2H]+2 and [M + 3H]+3) and lower abundance dimeric species ([2M + 4H]+4 and [2M + 5H]+5) (Fig. S7A). In contrast, no dimeric species were observed for the unmodified peptide (Fig. S7 A and B). Similarly, phosphorylated R14–R27 dimers were observed in analysis of trypsin-digested purified tagged CENP-A (Fig. S7C). Collisional activation of the unique dimer m/z value [2M + 3H]+3 (m/z = 1102.1549, −1.09 ppm) produced an MS2 spectrum corresponding to monomeric peptides of [M + 1H]+1 (m/z = 1652.7) and [M + 2H]+2 (m/z = 827.0). These results demonstrate that phosphorylated CENP-A N-terminal tails can form dimers.
Phospho-Mimetic CENP-A Nucleosome Arrays Resist Oligomerization.
Higher-order chromatin condensation is driven in part by histone tails. We have used in vitro-reconstituted nucleosome arrays to examine the behavior of CENP-A nucleosomes within chromatin. Here, polynucleosome arrays were assembled using 12 tandemly repeated 601-nucleosome positioning sequences and recombinant core histones, including either H3.1, CENP-A, or phospho-mimetic CENP-A S16D/S18D protein along with the other core histones (H4, H2A, and H2B). Assembled arrays were subjected to analytical ultracentrifugation (AUC) to assess the folding characteristics. In the absence of Mg2+, nucleosomes do not contact each other (i.e., beads-on-a-string conformation). Folding behavior upon Mg2+ addition can be observed at 50% boundary fraction as the average sedimentation value (Fig. 4F). H3-containing arrays become folded upon adding Mg2+, indicated by shifting 10S higher. As reported previously, CENP-A–containing arrays subjected to Mg2+ also display efficient folding at 50% boundary fraction (27). Additionally, above 50% boundary fraction, unmodified CENP-A arrays undergo extensive oligomerization, which is defined as sedimenting at >60 S (28). The sedimentation profile of the phospho-mimetic CENP-A arrays in the presence of Mg2+ lands between the unmodified H3 and CENP-A profiles. The extent of phospho-mimetic CENP-A array folding as well as oligomerization is more similar to the H3 array. We conclude that secondary structure generated by CENP-A phosphorylation greatly reduces the oligomeric state of the CENP-A arrays.
Phosphorylation of CENP-A Is Required for Proper Chromosome Segregation.
We expressed a CENP-A S16/S18A phospho-mutant in HCT116 cells to determine the effect of CENP-A Ser16/Ser18 phosphorylation on its function in vivo. Transiently transfected cells were synchronized in S-phase, released, and fixed after 10 h to examine the progression of cells through mitosis. Expression of CENP-A S16/S18A mutants caused an increase in the number of metaphase cells with unaligned chromosomes relative to expression of wild-type CENP-A (Fig. 5A) consistent with a defect in chromosome congression. We observed increases in lagging chromosomes as cells undergo chromosome segregation, suggesting that defects in centromere function persist throughout mitosis. The single mutation of either Ser16 or Ser18 shows limited effect. Only cells with CENP-A restricted to centromeres were analyzed for mitotic defects, suggesting that defects arise from filling centromere sites with an unphosphorylated CENP-A.
Fig. 5.
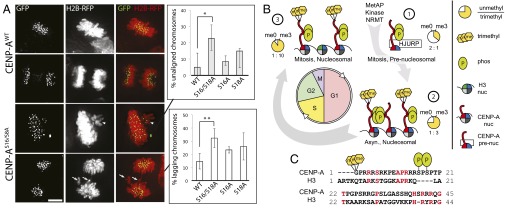
CENP-A Ser16/Ser18 phosphorylation is required for normal mitosis. (A) HCT116 cells transiently expressing histone H2B-mRFP and GFP-tagged wild-type or S16/S18A CENP-A mutants. The percentage of cells with unaligned metaphase (arrowhead) and lagging anaphase (arrow) chromosomes is higher in the S16/S18A mutant. *P = 0.018, **P = 0.002, Student’s t test, n = 3 independent experiments ± SD. (B) CENP-A PTM begins at translation and persists through the cell cycle. CENP-A α-N trimethylation occurs on a minority of the new prenucleosomal CENP-A and increases with cell cycle progression, constituting greater than 90% of the mitotic nucleosomes. (C) Human CENP-A PTM sites Gly1 α-N methylation and Ser16/Ser18 phosphorylation are especially divergent compared with H3.1. Conserved residues are in red.
Taken together, our data suggest a model where individual phosphorylated CENP-A tails are normally engaged in phospho-dependent intramolecular salt bridging that restricts the tendency to hypercondense chromatin arrays when unphosphorylated (Fig. 4F), and that weak oligomerization through CENP-A tails may occur when CENP-A nucleosomes are densely packed. The inability of these tails to undergo phosphorylation results in errors in chromosome segregation (Fig. 5B).
Discussion
The CENP-A N terminus is phosphorylated on Ser16 and Ser18 and α-N trimethylated on Gly1 following Metinit excision. Our in vitro experiments demonstrate that NRMT can efficiently catalyze the modification of the N terminus of CENP-A. The human CENP-A N-terminal Gly–Pro–Arg sequence is well conserved in mammals (Fig. 3C). In addition, Met–Pro–Arg, which is present in CENP-A of both birds and fish, was reported to be a human NRMT target in vitro (19). Both CENP-A N-terminal motifs are highly conserved in Tetrapoda, suggesting a conserved role for α-N methylation modification of CENP-A.
α-N methylation was shown to mediate stable chromatin localization of RCC1 (29). At centromeres, α-N–methylated CENP-A may play a similar role in DNA interactions. The addition of three methyl groups changes the N-terminal glycine so that it resembles a fixed positive charge buried within a hydrophobic shell. Such a structure might be capable of binding to nucleic acids through electrostatic interaction with negatively charged phosphates or polarizable π electrons on the aromatic nucleotide bases. α-N trimethylation could mediate an additional point of binding directly to underlying or neighboring DNA, perhaps contributing to the exceptionally high degree of phasing that CENP-A nucleosomes exhibit on centromeric α-satellite DNA (30).
We observed that CENP-A, which had been present for longer periods of time in the cell, showed higher degrees of α-N methylation; prenucleosomal CENP-A had the lowest degree of methylation whereas the CENP-A nucleosomes present in mitosis, which have been present in the chromatin for at least one cell cycle, were almost completely methylated. The increased proportion of CENP-A α-N methylation with cell cycle progression may be due to accessibility of CENP-A to NRMT activity or regulation of NRMT activity. The selective destabilization of unmethylated forms of CENP-A perhaps at particular points in the cell cycle such as DNA replication could also explain the observed changes in CENP-A methylation. Although it is possible that the unmodified form of CENP-A may represent protein that has undergone demethylation, no N-terminal demethylase has yet been identified.
Although the N-terminal tail of H3 is considered to be largely unstructured, amino acids 22–28 have been shown to form a short α-helix (31). Phosphorylation of human CENP-A Ser16 and Ser18 occurs in the context of an unusual proline-rich sequence. Moradi and colleagues calculated that Pro-X-Pro sequences (where X = Arg, Ser, or Thr) show high propensity to adopt beta sheet conformation, rather than polyproline or α-helix configuration (32). This short sequence of the CENP-A tail is completely divergent from H3 and appears to contain up to 4 additional residues (Fig. 5C). We find that salt-bridging generates a local secondary structure as well intermolecular CENP-A tail multimerization. We hypothesize that within highly condensed chromatin even low affinity interactions between CENP-A tails could influence chromatin organization and that CCAN proteins may further facilitate this process.
Materials and Methods
Stable Cell Line Generation.
CENP-A was C-terminally tagged with two tandem PreScission protease cleavage sites, a 6xHis, HA tag and GFP. Stable lines were made by retroviral infection as described (4). Polyclonal cell lines were FACS sorted for CENP-A expression. Spinner flask cultures were grown at a density of 3–8 × 105 cells per mL in Joklik media (Lonza) supplemented with 5% (vol/vol) newborn calf serum (HyClone), 1 mM Hepes, 1% penicillin streptomycin, 1% l-glutamine (Gibco, Life Technologies). Additional information is located in SI Materials and Methods.
Affinity Purification and Liquid Chromatography-MS Analysis.
HeLaS3 cells stably expressing tagged CENP-A were harvested and homogenized to generate a chromatin pellet for MNase digestion. CENP-A-LAP (Fig. S1) nucleosomes were recovered using αGFP beads and subsequently eluted by PreScission protease digestion. Eluted proteins were digested using trypsin, LysC, or GluC (Roche). Protein digests were HPLC-resolved and analyzed on-line using high-resolution MS. Spectra were analyzed as described in SI Materials and Methods.
Nucleosomal Array Reconstitution and Analytical Ultracentrifugation.
Nucleosomal arrays were reconstituted from the indicated histones and DNA using salt dialysis. The 601 12 × 200-bp DNA template was purified by anion exchange chromatography. The histones were expressed and purified as described (27). Reconstituted nucleosomal arrays were characterized by sedimentation velocity to obtain the integral distribution of sedimentation coefficients. Additional information is located in SI Materials and Methods.
Additional experimental details are available in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the Pacific Northwest National Laboratories for development of the Isotope Pattern Calculator software. We thank K. Luger (Colorado State), D. Cleveland (UCSD), and A. Straight (Stanford) for plasmids. This work was supported by National Institutes of Health (NIH) Grant T32 GM08715 (to A.O.B.), Robert A. Welch Foundation Grant A-1176 (to D.H.R.), the American Cancer Society (D.R.F.), NIH Grant R01 GM082989 (to B.E.B.), a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund (to B.E.B.), a Rita Allen Foundation Scholar Award (to B.E.B.), and NIH Grant GM037537 (to D.F.H.). T.P. acknowledges support from US National Institutes of Health Grant GM08275 (UPenn Structural Biology Training Grant).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300325110/-/DCSupplemental.
References
- 1.Stellfox ME, Bailey AO, Foltz DR. Putting CENP-A in its place. Cell Mol Life Sci. 2013;70(3):387–406. doi: 10.1007/s00018-012-1048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: From epigenetics to mitotic checkpoint signaling. Cell. 2003;112(4):407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 3.Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in flies and humans. Dev Cell. 2002;2(3):319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foltz DR, et al. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8(5):458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- 5.Okada M, et al. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat Cell Biol. 2006;8(5):446–457. doi: 10.1038/ncb1396. [DOI] [PubMed] [Google Scholar]
- 6.Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol. 2010;189(7):1143–1155. doi: 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol. 2007;176(6):795–805. doi: 10.1083/jcb.200701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foltz DR, et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137(3):472–484. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunleavy EM, et al. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137(3):485–497. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 10.Barnhart MC, et al. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol. 2011;194(2):229–243. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassett EA, et al. HJURP uses distinct CENP-A surfaces to recognize and to stabilize CENP-A/histone H4 for centromere assembly. Dev Cell. 2012;22(4):749–762. doi: 10.1016/j.devcel.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 13.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14(11):1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 14.Crosio C, et al. Mitotic phosphorylation of histone H3: Spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22(3):874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeitlin SG, Shelby RD, Sullivan KF. CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J Cell Biol. 2001;155(7):1147–1157. doi: 10.1083/jcb.200108125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunitoku N, et al. CENP-A phosphorylation by Aurora-A in prophase is required for enrichment of Aurora-B at inner centromeres and for kinetochore function. Dev Cell. 2003;5(6):853–864. doi: 10.1016/s1534-5807(03)00364-2. [DOI] [PubMed] [Google Scholar]
- 17.Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12(2):110–112. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- 18.Tooley CE, et al. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature. 2010;466(7310):1125–1128. doi: 10.1038/nature09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petkowski JJ, et al. Substrate specificity of mammalian N-terminal α-amino methyltransferase NRMT. Biochemistry. 2012;51(30):5942–5950. doi: 10.1021/bi300278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dephoure N, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Zubarev RA, Good DM, Savitski MM. Radical a-ions in electron capture dissociation: On the origin of species. J Am Soc Mass Spectrom. 2012;23(6):1015–1018. doi: 10.1007/s13361-012-0374-2. [DOI] [PubMed] [Google Scholar]
- 23.Keil B. Specificity of Proteolysis. Berlin: Springer; 1992. [Google Scholar]
- 24.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 25.Woods AS, Ferré S. Amazing stability of the arginine-phosphate electrostatic interaction. J Proteome Res. 2005;4(4):1397–1402. doi: 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruotolo BT, et al. Analysis of phosphorylated peptides by ion mobility-mass spectrometry. Anal Chem. 2004;76(22):6727–6733. doi: 10.1021/ac0498009. [DOI] [PubMed] [Google Scholar]
- 27.Panchenko T, et al. Replacement of histone H3 with CENP-A directs global nucleosome array condensation and loosening of nucleosome superhelical termini. Proc Natl Acad Sci USA. 2011;108(40):16588–16593. doi: 10.1073/pnas.1113621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarz PM, Hansen JC. Formation and stability of higher order chromatin structures. Contributions of the histone octamer. J Biol Chem. 1994;269(23):16284–16289. [PubMed] [Google Scholar]
- 29.Chen T, et al. N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol. 2007;9(5):596–603. doi: 10.1038/ncb1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasson D, et al. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat Struct Mol Biol. 2013;20(6):687–695. doi: 10.1038/nsmb.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen JC. Conformational dynamics of the chromatin fiber in solution: Determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 32.Moradi M, Babin V, Sagui C, Roland C. A statistical analysis of the PPII propensity of amino acid guests in proline-rich peptides. Biophys J. 2011;100(4):1083–1093. doi: 10.1016/j.bpj.2010.12.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.