

Abstract
G protein-coupled receptors play a pivotal role in many physiological signaling pathways. Mounting evidence suggests that G protein-coupled receptors, including opioid receptors, form dimers, and dimerization is necessary for receptor maturation, signaling, and trafficking. However, the physiological role of dimerization in vivo has not been well-explored because of the lack of tools to study these dimers in endogenous systems. To address this problem, we previously generated antibodies to μ-δ opioid receptor (μOR-δOR) dimers and used them to study the pharmacology and signaling by this heteromer. We also showed that the heteromer exhibits restricted distribution in the brain and that its abundance is increased in response to chronic morphine administration. Thus, the μOR-δOR heteromer represents a potentially unique target for the development of therapeutics to treat pain. Here, we report the identification of compounds targeting μOR-δOR heteromers through high-throughput screening of a small-molecule library. These compounds exhibit activity in μOR-δOR cells but not μOR or δOR cells alone. Among them, CYM51010 was found to be a μOR-δOR–biased ligand, because its activity is blocked by the μOR-δOR heteromer antibody. Notably, systemic administration of CYM51010 induced antinociceptive activity similar to morphine, and chronic administration of CYM51010 resulted in lesser antinociceptive tolerance compared with morphine. Taken together, these results suggest that CYM51010, a μOR-δOR–biased ligand, could serve as a scaffold for the development of a unique type (heteromer-biased) of drug that is more potent and without the severe side effects associated with conventional clinical opioids.
Studies with mice lacking opioid receptors show that the antinociceptive actions of clinically administered opioids, such as morphine or fentanyl, involve the activation of μ-opioid receptors (μORs) (1). However, continued opioid use leads to undesired side effects, including respiratory depression, constipation, immunosuppression, and development of tolerance and addiction (2). In an effort to identify novel compounds that are as effective as morphine in the treatment of chronic pain but without the associated side effects, our group, among others, has investigated the modulation of μOR function by receptor heteromerization. We found that μOR can form interacting complexes with δ-opioid receptors (δORs), that both receptors are in close proximity to interact in live cells, and that, in heterologous systems, low nonsignaling doses of some δOR ligands can potentiate the binding and signaling of μOR agonists (3–5). The recently reported crystal structure of μOR (6), in which receptors were crystallized as parallel dimers, is consistent with the idea that μOR can associate in complexes.
We also generated mAbs selective to μOR-δOR heteromers; we showed that the latter can be detected in the brains of WT but not KO mice and that heteromer levels are increased in brain regions involved in pain processing after chronic morphine administration under a paradigm that leads to the development of tolerance (7). The idea that μOR-δOR heteromers may play a role in the development of tolerance to morphine is further supported by studies showing that genetic deletion of either δOR or β-arrestin or possible disruption of μOR-δOR heteromers leads to an enhancement of morphine-mediated antinociception and attenuation in the development of tolerance (8–10). Notably, we observed that a δOR antagonist, H-Tyr-Tic[CH2NH]-Phe-Phe-OH (TIPPψ), can potentiate morphine-mediated analgesia (4), and studies using bivalent ligands targeting μOR-δOR heteromers showed that these ligands induce antinociception with attenuated development of tolerance as well as conditioned place preference (11, 12). Taken together, these data suggest that occupancy of δOR by an antagonist could dissociate the antinociceptive effects of μOR agonists from the development of tolerance and addiction. Therefore, there is a need for ligands that selectively interact with μOR-δOR heteromers to understand their role in antinociception and development of tolerance to morphine.
In an attempt to identify μOR-δOR heteromer-selective agonists, we used a β-arrestin recruitment assay and screened small molecules available through the Molecular Libraries Probe Production Centers Network. This screen identified 94 compounds that were biased to μOR-δOR heteromers compared with μOR, δOR, or serotonin 5HT5A receptors. Among a dozen compounds that were repurchased and tested using secondary screens, one, which we named CYM51010 [PubChem compound identifier (CID)23723457; Probe Report ID ML335], exhibited a strong μOR-δOR–biased activity that was blocked by μOR-δOR heteromer-selective mAb (μ-δ mAb). Furthermore, systemic administration of CYM51010 led to antinociceptive activity similar to morphine but with a lower antinociceptive tolerance on chronic administration. Notably, although the intrathecal (i.t.) antinociceptive activity of CYM51010 could be significantly blocked by i.t. administration of μ-δ mAb, the i.t. antinociceptive activity of morphine was not. These results suggest that CYM51010 could serve as a scaffold for the development of unique therapeutics acting at the μOR-δOR heteromer for the effective management of pain.
Results and Discussion
To screen for μOR-δOR heteromer-biased ligands, we used a β-arrestin recruitment assay that is based on an enzyme fragment complementation technology. Specifically, receptor activation-mediated β-arrestin recruitment leads to reconstitution of β-gal activity (Fig. S1). This strategy was used to engineer cell lines stably expressing μβgalOR-δOR, δβgalOR, or μβgalOR (DiscoverX). Based on the finding that these cells bind radiolabeled μOR or δOR ligands with nanomolar affinity and exhibit heteromer-mediated increases in binding (potentiation of radiolabeled μOR binding by δOR antagonist and vice versa) and agonist-mediated increases in G-protein activity (Fig. S1), we proceeded to characterize their suitability to screen for μOR-δOR heteromer-biased ligands.
We found that treatment with a δOR-selective agonist deltorphin II (Delt II) leads to a dose-dependent increase in β-arrestin recruitment to μβgalOR-δOR with nanomolar affinity. Similar results were obtained with Flag μOR-δβgalOR cells (Fig. 1 A and B and Table S1). The Delt II-mediated β-arrestin recruitment exhibits a time-dependent increase that plateaus by about 40 min; this increase in β-arrestin recruitment is reduced by the μOR antagonist D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP). This effect of CTOP is selective for μOR-δOR heteromers, because it is not observed in cells expressing δβgalOR (Figs. S2 and S3 A–C and Table S1). Reciprocally, the μOR agonist, [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), increases β-arrestin recruitment, which is reduced by the δOR antagonist TIPPψ (Fig. S2 and Table S2). These results were surprising, because we had previously found that μOR-δOR heteromers constitutively recruit β-arrestin (13) and that activation of the heteromer causes a dissociation of associated β-arrestin (13). The fact that, in this study, we observe a time-dependent increase in β-arrestin recruitment suggests that the modified receptors and β-arrestin in μβgalOR-δOR cells behave differently from the native receptor system. Nonetheless, the observations that a combination of δOR agonist and μOR antagonist (or μOR agonist and δOR antagonist) causes a decrease in β-arrestin recruitment (a phenomenon similar to the phenomenon reported previously) (13) and that these effects are seen only in μβgalOR-δOR and not μβgalOR or δβgalOR cells suggests that the μβgalOR-δOR cell line would be suitable for screening μOR-δOR heteromer-selective ligands.
Fig. 1.

Recruitment of β-arrestin by the δOR agonist Delt II. Cells expressing (A) μβgalOR-δOR or (B) Flag μOR-δβgalOR (20,000 cells/well) were subjected to a β-arrestin recruitment assay with the δOR agonist Delt II (0–1 μM) as described in Materials and Methods. (C) Cells expressing μβgalOR-δOR were treated with Delt II (0–10 μM) in the absence or presence of μ-δ or CB1-AT1 mAb (1 μg/well), and β-arrestin recruitment was measured. (D) Cells expressing Flag μOR-δβgalOR were treated with DAMGO (0–1 μM) in the absence or presence of μ-δ, δ-CB1, μ-CB1, or CB1-AT1 mAb (1 μg/well), and β-arrestin recruitment was measured. Results are mean ± SE (n = 3–6). RLU, relative luminescence unit.
To directly test the extent of involvement of μOR-δOR heteromers in Delt II-mediated β-arrestin recruitment, we used a μ-δ mAb that was previously shown to selectively block μOR-δOR heteromer activity (7). Treatment with the μ-δ mAb leads to a substantial decrease in Delt II-mediated recruitment of β-arrestin in μβgalOR-δOR cells as well as Flag μOR-δβgalOR cells (Fig. 1C and Fig. S3D). Notably, μ-δ mAb-mediated decreases in recruitment are not seen with mAbs directed at other heteromers, such as CB1R-AT1R (CB1-AT1 mAb), μOR-CB1R (μ-CB1 mAb), or δOR-CB1R (δ-CB1 mAb) (Fig. 1 C and D), or in cells expressing δβgalOR-CB1R (Fig. S3E), suggesting that this effect is selective for μOR-δOR heteromers. These studies gave us confidence to use the β-arrestin recruitment assay for high-throughput screening aimed at the identification of small molecules targeting the μOR-δOR heteromer.
The set of ∼335,461 small molecules available through the Molecular Libraries Probe Production Centers Network was screened using cells expressing μβgalOR-δOR, μβgalOR, δβgalOR, or 5HT5Aβgal receptors. Primary screening carried out at a single concentration (9.3 μM) in cells expressing either μβgalOR-δOR or 5HT5Aβgal receptors led to the identification of 993 hits with μβgalOR-δOR and 2,039 hits with 5HT5Aβgal cells (Fig. 2A). Comparison of the hits between the two cell lines showed that 885 of 993 hits were unique to μβgalOR-δOR. Secondary screening (triplicate at a single concentration of 9.3 μM) led to the identification of 375 hits for μβgalOR-δOR, among which 346 hits were unique to μβgalOR-δOR (Fig. 2B). Tertiary screening (10-point dilution series in triplicate) of 229 of these compounds in cells expressing μβgalOR-δOR, 5HT5Aβgal receptors, μβgalOR, or δβgalOR led to the identification of 125 hits (Fig. 2C). We compared the dose–response curves obtained with cells expressing μβgalOR-δOR, μβgalOR, or δβgalOR and identified a number of potential μOR-δOR–biased ligands based on the criteria that they exhibit EC50 values of ≤10 μM with μβgalOR-δOR cells, a fivefold difference in EC50 between μβgalOR-δOR and either μβgalOR or δβgalOR cells. These criteria led to the selection of 94 compounds as potential μOR-δOR–biased ligands (Table S3). For validation of the identified μOR-δOR–biased ligands, we selected 14 compounds based on their potency, efficacy, and uniqueness of structure compared with other opioid ligands. Novelty of the chemical scaffolds with respect to 9,934 annotated opioid receptor ligands in the ChEMBL database (https://www.ebi.ac.uk/chembl/) was evaluated by way of Tanimoto coefficients (Tcs) to the nearest neighbors, excluding the aforementioned 94 compounds. Of 14 compounds selected for validation, 13 compounds exhibited Tc ≤ 0.3 to the closest annotated opioid ligand (Table S4); 14 compounds were repurchased and retested for their ability to recruit β-arrestin in cells expressing μβgalOR, δβgalOR, or μβgalOR-δOR. Among the compounds tested, six exhibited higher efficacy in μβgalOR-δOR cells compared with μβgalOR or δβgalOR cells alone (Fig. 2 D–I and Table S4). Among these six compounds, PubChem CID23723457 (CYM51010) exhibited a robust β-arrestin recruitment (Emax = 1,197 ± 31% basal) that was at least twofold higher than the activity obtained with μβgalOR or δβgalOR alone (Fig. 2E and Table S4); this compound was selected for additional characterization.
Fig. 2.

Screening for μOR-δOR–biased agonists. (A) Cells expressing μβgalOR-δOR or 5HT5Aβgal receptors (1,000 cells/well) were treated with different compounds at a single concentration of 9.3 μM in singlicate, and β-arrestin recruitment was measured as described in Materials and Methods. (B) Compounds (834) identified in A as unique for μβgalOR-δOR were tested in triplicate at a single concentration (9.3 μM) in cells expressing μβgalOR-δOR or 5HT5Aβgal receptors. (C) Compounds (229) identified in B as unique for μβgalOR-δOR were tested in a 10-point dilution series in triplicate (0–92.6 μM) in cells expressing μβgalOR-δOR, 5HT5Aβgal receptors, μβgalOR, or δβgalOR. No hits were observed with cells expressing 5HT5Aβgal receptors. (D–I) Validation of hits identified in C carried out with commercially available compounds (Tables S3 and S4) using cells expressing μβgalOR, δβgalOR, or μβgalOR-δOR (5,000 cells/well). Data with six compounds that exhibited higher efficacy or potency at μβgalOR-δOR cells are shown.
CYM51010-mediated β-arrestin recruitment could be significantly reduced by μ-δ mAb and to a lesser extent, μOR- or δOR-selective Ab but not anti-Flag Ab (Fig. 3A). We find that CYM51010 causes a robust increase in [35S]GTPγS binding in μβgalOR-δOR cell membranes compared with μβgalOR or δβgalOR cell membranes (Fig. 3B and Table 1). Moreover, pretreatment with a μOR antagonist, naltrexone (NTX), but not with a δOR antagonist, naltriben (NTB), significantly, albeit partially, decreases CYM51010-mediated increases in [35S]GTPγS binding (Fig. 3C). We also examined CYM51010-mediated increases in G-protein activity using spinal cord membranes from WT animals and compared them with membranes from mice lacking μOR or δOR. CYM51010 induces a robust dose-dependent increase in [35S]GTPγS binding in WT membranes (Fig. 3D and Table 1). In addition, pretreatment with a μOR antagonist NTX partially decreases this CYM51010-mediated increase in [35S]GTPγS binding (Fig. 3E). Interestingly, a combination of NTX with μ-δ mAb, but not CB1-δ mAb, completely blocks CYM51010-mediated increases in [35S]GTPγS binding (Fig. S3F), consistent with the idea that CYM51010 behaves as a μOR-δOR heteromer-biased ligand.
Fig. 3.

Validation of CYM51010 as a μOR-δOR–biased agonist. (A) Cells expressing μβgalOR-δOR (5,000 cells/well) were treated with CYM51010 (51010; 10 μM) in the absence or presence of μ-δ mAb, μ, δ, or Flag Ab, and β-arrestin recruitment was measured. (B) Membranes (20 μg) from cells expressing μβgalOR, δβgalOR, or μβgalOR-δOR were subjected to a [35S]GTPγS binding assay with CYM51010 (0–10 μM final concentration). (C) Membranes (20 μg) from cells expressing μβgalOR-δOR were subjected to a [35S]GTPγS binding assay with CYM51010 (1 μM) in the absence or presence of the μOR antagonist NTX (10 μM) or the δOR antagonist NTB (10 μM). (D) Spinal cord membranes (20 μg) from WT mice or mice lacking μOR (μ k/o) or δOR (δ k/o) were subjected to a [35S]GTPγS binding assay with CYM51010 (0–10 μM final concentration). (E) Spinal cord membranes (20 μg) from WT or δ k/o mice were subjected to a [35S]GTPγS binding assay with CYM51010 (1 μM) in the absence or presence of the μOR antagonist NTX (10 μM) or the δOR antagonist NTB (10 μM). Results represent mean ± SE (n = 3–6).
Table 1.
EC50 and Emax for CYM51010
| β-Arrestin recruitment | [35S]GTPγS | |||
| EC50 (M) | Emax (% basal) | EC50 (M) | Emax (% basal) | |
| µβgal OR | 1.8E-6 | 557 ± 11 | 2.1E-7 | 138 ± 6 |
| δβgal OR | 2.7E-6 | 423 ± 49 | 3.0E-7 | 113 ± 1 |
| µβgalOR-δOR | 8.3E-6 | 1,197 ± 31 | 5.4E-8 | 168 ± 3 |
| WT spinal cord | n.d | n.d | 7.1E-7 | 141 ± 2 |
| µ k/o spinal cord | n.d | n.d | 7.9E-6 | 106 ± 4 |
| δ k/o spinal cord | n.d | n.d | 1.8 E-7 | 117 ± 3 |
n.d., not determined.
Next, we examined if CYM51010 exhibits μOR-δOR heteromer-mediated activity in vivo using the tail-flick assay to measure antinociception, because previous studies have implied a role for μOR-δOR heteromer in spinal analgesia (4, 10–12); s.c. administration of CYM51010 leads to dose-dependent antinociception, which is similar to antinociception of morphine (Fig. 4 A and B and Fig. S4A). Notably, although the antinociceptive effects of morphine were completely blocked by NTX, the effects of CYM51010 were only partially blocked by the same dose of NTX (Fig. 4C and Fig. S4 B and C); NTB had no effect on either morphine- or CYM51010-mediated antinociception (Fig. 4C and Fig. S4 B and C). These results suggest that a component of the CYM51010-mediated antinociception is through μOR; the fact that only a portion of CYM51010-mediated antinociception was mediated by μOR suggests that CYM51010 is mediating its effects by engaging the μOR-δOR heteromer.
Fig. 4.
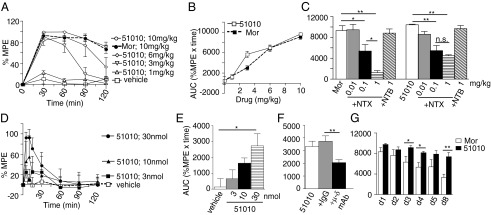
Antinociceptive activity of CYM51010. (A and B) Mice were s.c. administered with CYM51010 (51050) or morphine (Mor; 1, 3, 6, or 10 mg/kg body weight). (C) Mice were i.p. administered with vehicle, NTX, or NTB at the indicated doses 30 min before Mor (10 mg/kg s.c.) or CYM51010 (10 mg/kg s.c.) administration. (D) Mice were i.t. administered with CYM51010 (3, 10, or 30 nmol). E represents AUC calculated from data in D. (F) Mice were i.t. administered with vehicle, control IgG (anti-Flag IgG; 1 μg), or μ-δ mAb (1 μg) 30 min before CYM51010 (30 nmol i.t.) administration. (G) Mice were administered with either Mor or CYM51010 (10 mg/kg s.c.) one time daily for 8 d, and antinociception was measured daily. (A–G) Antinociceptive activity was measured as described in Materials and Methods. Results are mean ± SE (n = 3–18 mice per group). *P < 0.05; **P < 0.01 as determined by ANOVA followed by multiple comparison test (Student Newman–Keuls test) or unpaired t test. n.s., not significant.
The involvement of the μOR-δOR heteromer in CYM51010-mediated antinociception was tested by using μ-δ mAb. We find that coadministration of μ-δ mAb significantly blocks i.t. CYM51010-mediated antinociception (Fig. 4 D–F and Fig. S4F) but not the antinociception mediated by morphine (Fig. S4 D–H) or CID24891919 [area under the curve (AUC) [percentage of maximum possible effect (%MPE) × time] is 5,177 ± 844 in the absence and 4,998 ± 261 in the presence of μ-δ mAb], a nonselective opioid agonist (Table S3). Together, these results support the notion that CYM51010-mediated antinociception is, at least in part, mediated by μOR-δOR heteromers. Finally, we also examined the development of tolerance and physical dependence to CYM51010 and how it compared with morphine. We find that the development of antinociceptive tolerance to CYM51010 (10 mg/kg) is less than what is observed with morphine (Fig. 4G and Fig. S4 I–K). This observation is more apparent when a dose that induces ∼70% maximal antinociception (6 mg/kg) is used (Fig. S4 L–O). In naloxone precipitated withdrawal assay, chronic CYM51010 administration shows less severe signs of withdrawal for diarrhea and body weight loss compared with morphine (Fig. S5). Taken together, these results indicate that a major portion of the antinociception observed after i.t. administration of CYM51010 is mediated by μOR-δOR heteromers and that the chronic administration of the compound leads to less tolerance compared with morphine.
A major finding of this study is the identification of CYM51010 as a biased μOR-δOR agonist. Several recent reports have proposed classic μOR or δOR agonists to have μOR-δOR heteromer selectivity. For example, morphine and DAMGO have recently been reported to be more potent and efficacious at inducing Ca+2 mobilization in cells expressing μOR-δOR heteromers and more potent at eliciting [35S]GTPγS binding in these cells compared with cells expressing μOR (13, 14). Also, 4-[(R)-[(2S,5R)-4-allyl-2,5-dimethylpiperazin-1-yl](3-methoxyphenyl)methyl]-N,N-diethylbenzamide (SNC80), a δOR-selective agonist, was reported as a potent and efficacious μOR-δOR heteromer-selective agonist based on a Ca+2 mobilization assay and a decrease in antinociceptive activity in mice lacking μOR or δOR (15). These studies suggest that the ability of a compound to elicit a signaling response through μOR-δOR heteromers may depend on the signaling pathway being examined. However, these studies did not critically evaluate whether the μOR-δOR heteromer was the actual target of these agonists by either disrupting the heteromer or blocking the heteromer with selective Ab. In this study, we used the heteromer-selective mAb to infer that CYM51010 is a μOR-δOR heteromer-biased agonist. It is interesting to note that CYM51010 exhibits higher potency at μβgalOR-δOR compared with μβgalOR and δβgalOR alone for activating G proteins (∼50 vs. ∼300 nM) and lower potency for β-arrestin recruitment (∼8 vs. 3 μM) (Table 1). Improving the G-protein signaling bias in addition to increasing its μOR-δOR selectivity are likely to make CYM51010 an ideal tool to explore the in vitro and in vivo pharmacology of μOR-δOR heteromers.
Early studies showing that morphine-mediated antinociception could be potentiated by δOR antagonists (4, 16) suggested a functional interaction between μOR and δOR. More recent studies using bivalent ligands targeting the μOR-δOR heteromer (μOR agonist linked to δOR antagonist by a spacer) showed that they exhibit antinociceptive activity without the development of tolerance (11, 12). Recently, a compound named MuDelta, exhibiting μOR agonistic activity and δOR antagonistic activity, was identified using structure–activity relationship studies with the heterocyclic core of opioid receptor agonists (17, 18). This compound is currently under phase II clinical studies for the treatment of irritable bowel syndrome (17, 18). Given that μOR-δOR heteromers represent potential targets for the development of unique therapeutics to treat pain, the identification of CYM51010 as an antinociceptive μOR-δOR–biased agonist is exciting, because it provides a chemical scaffold for the development of therapeutics with reduced side effects commonly associated with chronic morphine use. In addition, compounds developed based on modifications of the CYM51010 structure (both agonists and antagonists) will help in the elucidation of the physiological role of μOR-δOR heteromers in vivo.
Materials and Methods
Cell Culture and Transfections.
μβgalOR–, δβgalOR–, or μβgalOR-δOR–expressing UO5S cells were a gift from DiscoverX. μβgalOR cells [expressing μOR tagged with a ProLink/β-gal donor (PK) fragment at the C-terminal region and β-arrestin tagged with a complementary β-gal activator (EA) fragment] and δβgalOR cells (expressing δOR tagged with the PK fragment at the C-terminal region and β-arrestin tagged with the EA fragment) were grown in MEMα containing 10% (vol/vol) FBS, streptomycin-penicillin, 500 μg/mL geneticin, and 250 μg/mL hygromycin. μβgalOR-δOR cells expressing WT δOR, μOR tagged with the PK fragment at the C-terminal region, and β-arrestin tagged with the EA fragment were grown in MEMα containing 10% (vol/vol) FBS, streptomycin-penicillin, 500 μg/mL geneticin, 250 μg/mL hygromycin, and 0.25 μg/mL puromycin.
δβgalOR cells were transfected with either Flag-tagged μOR or myc-tagged CB1R using Fugene 6 (Roche) as described in the manufacturer’s protocol.
Radioligand Binding Studies.
Saturation binding assays were carried out in cells (2 × 105) expressing μβgalOR, δβgalOR, or μβgalOR-δOR using either the radiolabeled δOR agonist ([3H]Delt II) or the radiolabeled μOR agonist ([3H]DAMGO; 0–10 nM final concentration) in 50 mM Tris⋅Cl buffer (pH 7.4) containing 0.32 M sucrose and protease inhibitor mixture (Sigma) as described in refs. 5 and 19. [3H]Delt II exhibits a Kd = 12.2 ± 3.8 nM and a Bmax = 3,723 ± 748 fmol/mg protein with cells expressing δβgalOR and a Kd = 12.7 ± 3.6 nM and a Bmax = 6,023 ± 1,099 fmol/mg protein with cells expressing μβgalOR-δOR (Fig. S1). [3H]DAMGO exhibits a Kd = 8.4 ± 2.3 nM and a Bmax = 670 ± 107 fmol/mg protein with cells expressing μβgalOR and a Kd = 2.5 ± 0.6 nM and Bmax = 370 ± 32 fmol/mg protein with cells expressing μβgalOR-δOR (Fig. S1).
Potentiation of [3H]Delt II binding by μOR antagonist CTOP (10 nM final concentration) or [3H]DAMGO binding by the δOR antagonist TIPPψ (10 nM final concentration) was examined in cells (2 × 105) expressing μβgalOR, δβgalOR, or μβgalOR-δOR in 50 mM Tris⋅Cl buffer (pH 7.4) containing 0.32 M sucrose and protease inhibitor mixture as described in refs. 4 and 5.
[35S]GTPγS Binding.
Membranes were prepared from cells expressing μβgalOR, δβgalOR, or μβgalOR-δOR and spinal cords of WT, μOR, or δOR KO mice as described previously (19). Membranes (20 μg) were subjected to a [35S]GTPγS binding assay using Delt II, DAMGO, or CYM51010 (0–10 μM final concentration) as described in ref. 4. In studies examining the effects of Ab, membranes were incubated with 1 μg indicated Ab for 30 min before the addition of agonists. In studies examining the effects of antagonists, membranes were incubated with 10 μM μOR antagonist NTX or δOR antagonist NTB for 30 min before addition of agonists.
β-Arrestin Recruitment.
Cells expressing μβgalOR, δβgalOR, or μβgalOR-δOR were plated in each well (20,000 cells) of a 96-well white clear bottom plate in 100 μL media. The next day, cells were treated with either Delt II or DAMGO (0–1 μM final concentration) for 60 min at 37 °C, and β-arrestin recruitment was measured using the PathHunter Chemiluminescence Detection Kit as described in the manufacturer’s protocol (DiscoverX). In studies examining the effects of μOR antagonist CTOP or δOR antagonist TIPPψ, cells were preincubated without or with the antagonists (10 μM final concentration) for 30 min before the addition of agonists. In studies examining the time course of β-arrestin, recruitment cells were treated with either 1 μM Delt II (±100 nM CTOP) or 1 μM DAMGO (±100 nM TIPPψ) for the indicated time periods (30 s and 1, 3, 5, 10, 30, and 60 min). In studies examining the effects of Ab, cells expressing μβgalOR-δOR, Flag μOR-δβgalOR, or δβgalOR-myc CB1R were incubated with 1 μg/well μ-δ, CB1 cannabinoid-AT1 angiotensin receptor (CB1-AT1), μ-CB1, or δ-CB1 heteromer-selective mAbs for 30 min before the addition of Delt II (0–1 μM). Validation studies were carried out using 5,000 cells/well and commercially available compounds identified as μOR-δOR–biased agonists in the high-throughput screen (0–100 μM final concentration). Differences observed in data obtained with the validated compounds and data from the high-throughput screening (described below) could be because of the differences in assay conditions.
High-Throughput Screening for μOR-δOR–Biased Agonists.
Cells expressing μβgalOR, δβgalOR, μβgalOR-δOR, or 5HT5Aβgal receptors were plated in each well (1,000 cells) of a 1,536-well white plate. Cells were incubated for 3 h at 37 °C with different compounds followed by 1 h of incubation with the PathHunter Chemiluminescence Detection Kit. In the primary screening, compounds were screened at a single concentration (9.3 μM) in singlicate in cells expressing either μβgalOR-δOR or 5HT5Aβgal receptors. In secondary screening assays, 834 compounds identified in primary screens as unique for μβgalOR-δOR were tested in triplicate at a single concentration (9.3 μM) in the same cell lines. This screen led to the identification of 375 hits with μβgalOR-δOR and 65 hits with 5HT5Aβgal cells. In tertiary assays, 229 compounds identified in secondary screens as unique for μβgalOR-δOR were tested in a 10-point dilution series in triplicate (0–92.6 μM) in cells expressing μβgalOR-δOR, 5HT5Aβgal receptors, μβgalOR, or δβgalOR. Detailed screening assay protocols as well as all screening results can be found in PubChem, a publically available database (pubchem.ncbi.nlm.nih.gov; BioAssay AID 504355) Hits identified as μOR-δOR–selective are summarized in Table S3.
Similarity Analysis.
Tc values were calculated with an in-house script using extended connectivity fingerprint maximum distance 4. They were generated with jCompoundMapper (20) after converting linear text SMILES formats for each molecule into corresponding SDF files with Corina (21). A Tc = 0 indicates maximally dissimilar compounds, whereas a Tc = 1 indicates maximally similar ones (22), with values below 0.40 suggesting reasonably unique compounds (23).
Animals.
Male C57BL/6 mice (25–35 g; 6–12 wk) were obtained from Jackson Laboratories. All mice were maintained on a 12-h light/dark cycle with rodent chow and water available ad libitum, and they were housed in groups of five until testing. Animal studies were carried out according to protocols approved by the Mount Sinai School of Medicine Animal Care and Use Committee.
Drug Administration.
Morphine sulfate (Sigma) and NTX (Tocris) were dissolved in saline. NTB (Tocris) was dissolved in 2% DMSO in saline. CYM51010 (ChemBridge) and CID24891919 (Life Chemicals) were dissolved in 6% DMSO and 5% Tween80 in saline. μ-δ mAb (7) and control IgG (anti-Flag Ab; 1 μg) were diluted in saline. Corresponding vehicle was used for the control group. Mice were administered drugs s.c. or i.p. for systemic treatment. For i.t. administration, the direct lumbar puncture method (24) was applied in awake, conscious mice. Mice were covered with a soft cloth over the head and upper body and gripped firmly by the pelvic girdle (iliac crest); 5 μL drug was administered with a 50-μL Hamilton syringe (Hamilton Co) attached to a 30-gauge, 1-in sterile disposable needle, which was inserted into the i.t. space at the cauda equine region according to the method described in ref. 24. Puncture of the dura was indicated by a flick of the tail. For measuring development of tolerance, mice were s.c. administered with 10 mg/kg morphine or CYM51010 one time per day for 8 d or 6 mg/kg daily for 14 d, and antinociception was measured daily from 0 to 120 min. Naloxone-precipitated withdrawal symptoms were measured in mice after administration of 10 mg/kg morphine or CYM51010 s.c. one time per day for 9 d. Withdrawal was induced by i.p. administration of naloxone (5 mg/kg) 2 h after the last drug administration, and withdrawal signs were recorded for 30 min as described (25). Body weight was measured before and 30 min after last naloxone injection.
Analgesia Assays.
Drug-induced antinociception was evaluated by using the tail-flick test (26). Using a tail-flick apparatus (IITC Life Science), the intensity of the heat source was set at 10, which resulted in the basal tail-flick latency occurring between 5 and 7 s for most of the animals. The tail-flick latency was recorded at the indicated time period (0–120 min) after drug administration; %MPE was calculated for each mouse at each time point according to the following formula: %MPE = [(latency after drug − baseline latency)/(20 − baseline latency)] × 100. Cutoff latency was selected at 20 s to minimize tissue damage. The area under the %MPE vs. time curves (AUCs) for each treatment condition is shown in Fig. 4 and Fig. S4.
Statistical Analyses.
The data were expressed as means ± SEMs. One-way ANOVA and multiple comparison tests (Student Newman–Keuls tests) were used to analyze the data. Tolerance and withdrawal data were analyzed by unpaired t tests. A difference was considered to be significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. John Pintar for the gift of brains from mice lacking μOR or δOR, DiscoverX for the μβgalOR, δβgalOR, and μβgalOR-δOR cells, and PathHunter Assay kits and Pierre Baillargeon, Lina DeLuca, and Louis Scampavia for compound management and analysis. These studies were supported by National Institutes of Health Molecular Library Probe Production Center Grant U54 MH084512 (to E.R. and P.H.) and National Institutes of Health Grants DA034049 (to M.F.), DA026434 (to M.F.), DA008863 (to L.A.D.), and DA019521 (to L.A.D.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222044110/-/DCSupplemental.
References
- 1.Matthes HW, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 2.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 3.Gomes I, et al. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J Neurosci. 2000;20(22):RC110. doi: 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes I, et al. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA. 2004;101(14):5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes I, Ijzerman AP, Ye K, Maillet EL, Devi LA. G protein-coupled receptor heteromerization: A role in allosteric modulation of ligand binding. Mol Pharmacol. 2011;79(6):1044–1052. doi: 10.1124/mol.110.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manglik A, et al. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta A, et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal. 2010;3(131):ra54. doi: 10.1126/scisignal.2000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y, et al. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24(1):243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 9.Bohn LM, et al. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286(5449):2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 10.He SQ, et al. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron. 2011;69(1):120–131. doi: 10.1016/j.neuron.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Daniels DJ, et al. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci USA. 2005;102(52):19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenard NR, Daniels DJ, Portoghese PS, Roerig SC. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur J Pharmacol. 2007;566(1–3):75–82. doi: 10.1016/j.ejphar.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 13.Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: Beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21(10):2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yekkirala AS, Kalyuzhny AE, Portoghese PS. Standard opioid agonists activate heteromeric opioid receptors: Evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective μ-δ agonists. ACS Chem Neurosci. 2010;1(2):146–154. doi: 10.1021/cn9000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metcalf MD, et al. The δ opioid receptor agonist SNC80 selectively activates heteromeric μ-δ opioid receptors. ACS Chem Neurosci. 2012;3(7):505–509. doi: 10.1021/cn3000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abul-Husn NS, Sutak M, Milne B, Jhamandas K. Augmentation of spinal morphine analgesia and inhibition of tolerance by low doses of mu- and delta-opioid receptor antagonists. Br J Pharmacol. 2007;151(6):877–887. doi: 10.1038/sj.bjp.0707277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wade PR, et al. Modulation of gastrointestinal function by MuDelta, a mixed µ opioid receptor agonist/ µ opioid receptor antagonist. Br J Pharmacol. 2012;167(5):1111–1125. doi: 10.1111/j.1476-5381.2012.02068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breslin HJ, et al. Identification of a dual δ OR antagonist/μ OR agonist as a potential therapeutic for diarrhea-predominant Irritable Bowel Syndrome (IBS-d) Bioorg Med Chem Lett. 2012;22(14):4869–4872. doi: 10.1016/j.bmcl.2012.05.042. [DOI] [PubMed] [Google Scholar]
- 19.Gomes I, Filipovska J, Devi LA. Opioid receptor oligomerization. Detection and functional characterization of interacting receptors. Methods Mol Med. 2003;84:157–183. doi: 10.1385/1-59259-379-8:157. [DOI] [PubMed] [Google Scholar]
- 20.Hinselmann G, Rosenbaum L, Jahn A, Fechner N, Zell A. jCompoundMapper: An open source Java library and command-line tool for chemical fingerprints. J Cheminform. 2011;3(1):3. doi: 10.1186/1758-2946-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasteiger JR, Rudolph C, Sadowski J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comp Method. 1990;3(6):537–547. [Google Scholar]
- 22.Rogers D, Brown RD, Hahn M. Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. J Biomol Screen. 2005;10(7):682–686. doi: 10.1177/1087057105281365. [DOI] [PubMed] [Google Scholar]
- 23.Wawer M, Bajorath J. Similarity-potency trees: A method to search for SAR information in compound data sets and derive SAR rules. J Chem Inf Model. 2010;50(8):1395–1409. doi: 10.1021/ci100197b. [DOI] [PubMed] [Google Scholar]
- 24.Hylden JL, Wilcox GL. Intrathecal morphine in mice: A new technique. Eur J Pharmacol. 1980;67(2–3):313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 25.Abul-Husn NS, et al. Chronic morphine alters the presynaptic protein profile: Identification of novel molecular targets using proteomics and network analysis. PLoS One. 2011;6(10):e25535. doi: 10.1371/journal.pone.0025535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72(1):74–79. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.