

Abstract
Glioblastoma (World Health Organization grade IV) is an aggressive adult brain tumor that is inevitably fatal despite surgery, radiation, and chemotherapy. Treatment failures are attributed to combinations of cellular heterogeneity, including a subpopulation of often-resistant cancer stem cells, aberrant vasculature, and noteworthy immune suppression. Current preclinical models and treatment strategies do not incorporate or address all these features satisfactorily. Herein, we describe a murine glioblastoma stem cell (GSC) model that recapitulates tumor heterogeneity, invasiveness, vascularity, and immunosuppressive microenvironment in syngeneic immunocompetent mice and should prove useful for a range of therapeutic studies. Using this model, we tested a genetically engineered oncolytic herpes simplex virus that is armed with an immunomodulatory cytokine, interleukin 12 (G47∆-mIL12). G47Δ-mIL12 infects and replicates similarly to its unarmed oncolytic herpes simplex virus counterpart in mouse 005 GSCs in vitro, whereas in vivo, it significantly enhances survival in syngeneic mice bearing intracerebral 005 tumors. Mechanistically, G47∆-mIL12 targets not only GSCs but also increases IFN-γ release, inhibits angiogenesis, and reduces the number of regulatory T cells in the tumor. The increased efficacy is dependent upon T cells, but not natural killer cells. Taken together, our findings demonstrate that G47Δ-mIL12 provides a multifaceted approach to targeting GSCs, tumor microenvironment, and the immune system, with resultant therapeutic benefit in a stringent glioblastoma model.
Keywords: antiangiogenesis, immunotherapy, virotherapy
Glioblastoma (GBM), the most common primary brain tumor in adults, is invariably fatal with a median survival of 12–16 mo (1). It is characterized by rapid and invasive growth, heterogeneous morphology, and a complex microenvironment, including local immunosuppression and high degrees of neovascularization (1). Recently, GBM stem cells (GSCs) were isolated from patient tumors and characterized as cells that sustain themselves through self-renewal, differentiate into multiple more mature lineages, and efficiently initiate tumors in mice (2), making them an important and previously unrecognized target for therapy in GBM patients. GBM exhibits multiple mechanisms for evading the immune system that facilitate tumor progression, including systemic as well as local immunosuppression that has been attributed to a preponderance of regulatory T cells (Tregs) (3, 4). The standard of care for GBM (maximal surgical resection, temozolomide administration, irradiation, and sometimes bevacizumab) has only minimally increased survival for patients in the past decades. Therapy typically fails to eradicate GSCs, which contribute to infiltrating and treatment-resistant tumor cells, or induce immune responses. Newer molecular targeted therapies to single pathways are also unlikely to be sufficient for such a complex disease. Thus, novel combination strategies are needed to target not only the bulk tumor cells, but also GSCs and the tumor microenvironment.
It remains unknown whether GSCs can be successfully targeted in vivo by antitumor immune responses, partially due to the prior lack of an immunocompetent syngeneic GSC tumor model. In vitro analysis has demonstrated human GSC immunosuppressive activity, while suggesting susceptibility to T- and natural killer (NK)-cell killing (5–7). In turn, adoptive lymphocyte transfer studies showed that engineered antigen-specific T cells could eliminate human GSC-derived tumors in SCID mice (8, 9), but such immune-deficient tumor models have not been useful for examining the effects of host immune responses. Here, we use a mouse GSC-derived intracerebral tumor model in syngeneic immunocompetent mice. Mouse 005 GSCs were derived from gliomas arising after lentivirus transduction of activated Harvey-Ras (H-Ras) and protein kinase B (Akt) in tumor suppressor gene p53 (Tp53)+/− mice (10).
Oncolytic virotherapy is a distinct antitumor therapeutic modality where replication-competent viruses selectively kill cancer cells, amplifying and spreading throughout the tumor, as well as inducing antitumor immune responses (11). However, insufficient targeting of the complex GBM microenvironment limits its efficacy. In this study, we use a genetically engineered oncolytic herpes simplex virus (oHSV), G47Δ that is efficacious against human GSCs and has been safely administered to patients with recurrent malignant glioma (12, 13). To enhance oHSV efficacy, the virus can be “armed” to express therapeutic transgenes locally within the tumor. Given the need to target the tumor microenvironment, we generated G47∆-mIL12, expressing murine interleukin 12 (IL-12). IL-12 is a heterodimeric proinflammatory cytokine, which bridges innate and adaptive immunity (14). It strongly promotes the proliferation of activated T and NK cells, stimulates T-helper 1 (Th1) differentiation, and induces IFN (IFN)-γ production, which exerts pleiotropic effects, facilitating T-cell–mediated killing and antiangiogenesis (14). Owing to these diverse effects, IL-12 has been regarded as a master regulator of antitumor immunity.
Although we and others have previously tested IL-12-armed oHSV as a therapy in multiple tumor models (15–17), this study is unique in demonstrating the targeting of GSCs, as well as the complex tumor microenvironment in a stringent preclinical GBM model in immunocompetent mice. In this model, G47∆-mIL12 activity was multifaceted; direct oncolysis, reduction of local Tregs, stimulation of Th1-type immunity, T-cell–mediated survival advantage, and inhibition of tumor angiogenesis. G47∆-mIL12 thus represents a unique and potent therapy for GBM, with a broad antitumor repertoire.
Results
Mouse 005 GSCs Express Stem Cell Markers, Are Poorly Immunogenic, and Recapitulate the Histological Hallmarks of GBM in Immunocompetent Mice.
Most preclinical studies on GSCs have used human GSC-derived tumors in immunocompromised (nude or SCID) mice, which lack host adaptive immune responses and T-cell–mediated effects on the tumor and microenvironment. Mouse 005 GSCs (obtained from I. Verma, Salk Institute for Biological Studies, La Jolla, CA) were derived from GBM tumors induced by lentivirus transduction of Cre target sequence (loxP)-dependent activated H-Ras and Akt, and GFP into the stem cell niche of GFAP-Cre:Tp53+/− immunocompetent C57BL/6 mice (10). We confirmed that 005 GSCs form “neurospheres” in serum-free media containing EGF and FGF and express characteristic stem cell markers Nestin, m-Prominen (CD133 mouse homolog; 63% of cells from in vitro cultures) (Fig. 1A) and SRY-box containing gene 2 (Sox2), which is not expressed in the mouse glioma cell line CT2A (Fig. S1A). They also express proangiogenic VEGF (Fig. S1A), differentiate into multiple neural lineages that express glial GFAP or neuronal β-III tubulin when cultured in serum-containing media (Fig. 1A), and are temozolomide insensitive (Fig. S1B). The 005 GSCs were highly tumorigenic, forming hippocampal or striatal tumors after injection of only 20,000 cells into immunocompetent C57BL/6 mice (Fig. S1 C and D). These tumors are aggressive, morphologically heterogeneous with multinucleated giant cells, infiltrative along white matter tracts, and highly vascularized, as illustrated by CD31+ dilated blood vessels and VEGF expression, all hallmarks of GBM (Fig. 1B). The 005 GSCs maintain stem cell characteristics in vivo, as shown by immunohistochemical staining of Nestin and oligodendrocyte lineage transcription factor 2 (Olig2) and limited expression of GFAP (Fig. 1B).
Fig. 1.

In vitro and in vivo characterization of mouse 005 GSCs. (A) Neurospheres express the stem cell markers nestin (red; Left) and mProminin-1 (CD133 homolog; FACS, Center Left) and can be differentiated in serum-containing media into glial (GFAP; red, Center Right) and neuronal (βIII-tubulin; red, Right) phenotypes. (B) Murine 005 GSCs form characteristic gliomas in C57BL/6 mice. Pleomorphic, multinucleated giant cells seen by H&E staining (bold arrows and Inset; Upper Left) along with dense CD31-positive dilated vasculature (arrows) are observed within the tumors and not on the contralateral side of the brain. Murine 005 tumors also express VEGF and maintain stem cell characteristics, as seen by immunohistochemical staining (brown) of Olig2 and Nestin, and limited heterogeneous expression of GFAP (Lower Right). (C) In vitro, 005 GSCs only express MHC class I after treatment with IFN-γ (solid-Db and dotted-Kb red line), but strongly express NK ligand Rae-1 (Pan-Rae; solid green line). (D) Murine 005 GSCs lack expression of costimulatory signaling molecules CD40 and CD80 and contain only a minor population of CD86 (blue lines). Isotype controls, gray filled.
It is well documented that tumors can escape T-cell–mediated elimination by down-regulating molecules essential for immune recognition (18). We observed a lack of cell-surface expression of MHC I, CD40, CD80, and CD86 (Fig. 1 C and D) on 005 GSCs, as assessed by flow cytometry, demonstrating their nonimmunogenic phenotype and making them a stringent model for immunotherapy. MHC class I (Kb and Db) expression could be up-regulated by treatment with recombinant mouse IFN-γ for 24 h (Fig. 1C). The 005 GSCs do express high levels of NK-cell receptor ligands, retinoic acid early inducible-1 (Rae-1) (Fig. 1C) and CD155 (Fig. S1E). Thus, 005 GSCs, like human GBMs (4), inherently lack the capacity for antigen presentation and costimulation necessary to stimulate T-cell activation or proliferation.
G47Δ-mIL12 Can Replicate in and Kill Mouse 005 GSCs in Vitro While Expressing IL-12.
To ensure that G47∆ was capable of replicating and spreading in 005 GSCs, as mouse cells are typically not very permissive to oHSV replication (19), we constructed G47∆-mCherry, expressing the fluorescent reporter mCherry. G47∆-mCherry spread within and between 005 GSC spheres after low multiplicity of infection (MOI) (Fig. 2A). To assess therapeutic efficacy of oHSV armed with IL-12, we constructed G47Δ-mIL12 and control G47Δ-E, with no transgene (Fig. S2). G47Δ-E and G47Δ-mIL12 replicated similarly, but not well, in 005 GSCs (Fig. 2B). Both were equally cytotoxic to 005 GSCs in vitro (Fig. 2C), with a similar EC50 (∼0.1 MOI) as that reported for G47∆ in human GSCs (20). IL-12 was released from G47Δ-mIL12–infected 005 GSCs, with secretion increasing to about 60 ng/mL over 96 h, whereas cells infected with G47Δ-E did not produce any detectable IL-12 (Fig. 2D). Furthermore, recombinant IL-12 had no direct cytotoxic effect on 005 GSCs (Fig. S3A). Therefore, expression of IL-12 did not alter the oncolytic activity of G47∆ in vitro (Fig. 2C).
Fig. 2.

oHSV can replicate in and kill 005 GSCs in vitro while expressing IL-12. (A) Infection and spread of G47Δ-mCherry (MOI = 0.1; red) in 005 GSC spheres (phase contrast). (B) Replication kinetics of G47∆-E and G47∆-mIL12 in 005 GSCs after infection at MOI = 0.1. (C) Dose–response curves for G47Δ-E and G47Δ-mIL12 in 005 GSCs after 4 d, as measured by MTS assay; average of two experiments. (D) Secreted IL-12 (culture supernatants) at different times after infection (MOI = 0.1, same experiment as B), as measured by ELISA. No detectable IL-12 was measured after G47Δ-E infection. Error bars indicate SEM. Experiments were performed in triplicate.
G47Δ-mIL12 Treatment Inhibits Intracranial Tumor Growth and Extends Survival.
To assess the effects of G47∆-mIL12 on survival and tumor burden, mice bearing established intracranial 005 GSC-derived tumors were treated with intratumoral injections of PBS, G47Δ-E, or G47Δ-mIL12 on days 8 and 12 after tumor implantation. G47Δ-E had a modest effect (P < 0.05 versus PBS; Fig. 3A), whereas G47∆-mIL12 significantly enhanced survival of mice compared with either G47Δ-E (P < 0.005) or PBS (P < 0.001, >50% increase in median survival), with 10% mice surviving long term (Fig. 3A). Neither body weight loss nor other treatment-related toxicities were observed in any of the groups. To quantify tumor burden, cells were extracted from the right tumor-bearing quadrant of the brain 3 d after the second virus treatment (day 15) and GFP+ 005 cells were sorted by flow cytometry (Fig. 3B). Treatment with G47Δ-E did not alter the percentage of GFP+ cells (7.7% ± 1.9 vs. 8% ± 0.74 in PBS), whereas treatment with G47Δ-mIL12 significantly decreased the percentage obtained (3.3% ± 0.69; P < 0.05; Fig. 3B). After sorting, the GFP+ 005 cells were tested for their ability to form spheres in vitro (self-renewal). At limiting dilution, both G47Δ-E and G47Δ-mIL12 treatment significantly decreased the number of spheres per well (36% decrease) as well as the percentage of wells with spheres (25% decrease), indicating a similar effectiveness in reducing the stem cell subpopulation with self-renewal capability (Fig. 3C). Neither G47Δ-E nor G47Δ-mIL12 treatment changed the CD133+ fraction among GFP+ tumor cells compared with PBS, but G47∆-mIL12 significantly reduced the percentage compared with G47-E (Fig. 3D). The significant decrease in total GFP+ glioma cells with relative maintenance of the CD133+/CD133− ratio in tumors by G47Δ-mIL12 (Fig. 3 B and D) indicates that this treatment targets both the CD133-positive and -negative cell populations in vivo. This is important as recent publications have shown that tumorigenic and stem-cell–like cells exist in the CD133− subpopulation (21, 22). Because G47∆ did not replicate well in 005 cells in vitro, we examined whether virus replication was occurring in vivo using bioluminescent imaging and G47∆-Us11fluc (Fig. S2), with luciferase expression driven by the HSV-1 Us11 late promoter, so that bioluminescence is dependent upon virus replication and the intensity correlates with virus yield (23). Luciferase expression in 005 intracerebral tumors was elevated from days 1–3 and then decreased at day 6 (Fig. 3E), demonstrating virus replication in the established tumors.
Fig. 3.

Treatment of established intracranial 005 tumors in C57BL/6 mice. (A) Murine 005 GSCs (5 × 104 cells) were stereotactically implanted and mice treated with intratumoral injections of PBS, G47Δ-E, or G47Δ-mIL12 (n = 10) on days 8 and 12. Kaplan–Meier analysis shows a significant extension in survival with G47Δ-mIL12 treatment, with a median survival of 57 d compared with G47Δ-E (41 d) or PBS (37 d). (B) Representative flow cytometry results of GFP+ 005 cells (quadrant bordered in red; Left) extracted from brains on day 15. A significant decrease in tumor burden was observed after G47Δ-mIL12 treatment (*P < 0.05; Right graph). (C) GFP+ 005 GSCs were sorted from brain tumors and plated at 10–30 cells per well for self-renewal assays. Both G47Δ-E and G47Δ-mIL12 similarly (P = 0.3) decreased the ability of GSCs to form spheres, number per well, and percent sphere-positive wells versus PBS (*P < 0.05). (D) There is no difference when comparing treated and PBS in the percentage of CD133+ cells within the GFP+ population in tumors on day 15. The percent of CD133+ GFP+ cells after G47∆-mIL12 is significantly less than G47∆-E (*P < 0.05). (E) Bioluminescent imaging of virus replication in vivo. Established 005 tumors were injected with G47∆-Us11fluc on day 13 postimplantation of 105 cells. Left, images of four mice in the prone position on days 2 and 3 postinfection, with heat scale on Right (radiance; photons per s/cm2 per steradian). Right, mean luminescence (supine position) from four mice. Error bars indicate SEM.
Intratumoral Release of IL12 and IFN-γ in Vivo After G47Δ-mIL12 Treatment.
To understand the mechanisms by which G47Δ-mIL12 enhances survival, we first examined the local release of IL-12 by performing ELISAs on brain tumor homogenates (Fig. 4A). There was a significant increase in IL-12 on the day following the second injection of G47Δ-mIL12, although this was absent by day 6 posttreatment (Fig. 4B). Importantly, this IL-12 production was accompanied by marked release of downstream IFN-γ in the brain tumors on day 1 and, to a lesser extent, day 6 following treatment with G47Δ-mIL12 (Fig. 4C). No systemic IL-12 was detected in the serum, whereas systemic IFN-γ was detected at day 1 posttreatment only with G47Δ-mIL12 (Fig. 4D).
Fig. 4.
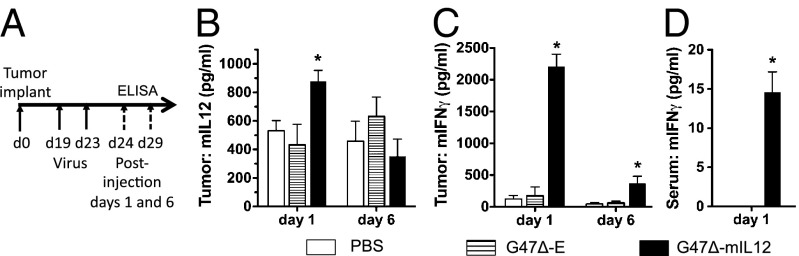
In vivo levels of IL-12 and IFN-γ after virus treatment. (A) Schema for experiment. Tumor-bearing mice were killed on days 1 and 6 posttreatment and tumor and serum isolated. (B) ELISA for mouse IL12p70 in tumor homogenates, showing a significant increase only after G47∆-mIL12 treatment (day 1; *P < 0.05 versus PBS or G47∆-E). (C) Tumor IFN-γ (ELISA) was significantly increased only after G47Δ-mIL12 treatment on days 1 and 6 (*P < 0.05 versus PBS). (D) Serum IFN-γ was increased on day 1 after G47Δ-mIL12 treatment (P < 0.05; paired t test). Error bars indicate SEM.
Antiangiogenic Effect of G47Δ-mIL12 in 005 GSC Brain Tumors.
To determine the vascularity of treated tumors, endothelial cells were stained with anti-CD31 antibody 6 d posttreatment (after the second injection). We observed a prominent reduction in CD31+ blood vessels only in brain tumors treated with G47Δ-mIL12 (Fig. 5A). The antiangiogenic activity of IL-12 has been attributed to IFN-inducible protein 10 [IP-10 or CXC chemokine ligand (CXCL)-10], an IFN-γ–activated chemokine that mediates chemotaxis of lymphocytes and angiostatic effects (14). Western blotting of protein lysates from brain tumor homogenates on day 6 posttreatment demonstrated a strong induction of IP-10 in all three tumors treated with G47Δ-mIL12 (Fig. 5B). We also observed a reduction in VEGF expression after G47Δ-mIL12 treatment, another likely contributor to decreased angiogenesis in the brain tumor, although one mouse in the G47∆-E group also had reduced expression (Fig. 5B). We previously reported that G47∆ inhibited VEGF secretion from U87 glioma cells to a limited extent in vitro (24). In vitro, supernatants from G47Δ-mIL12–infected 005 GSCs inhibited tube formation of human umbilical vein endothelial cells (HUVECs) (Fig. S3B), an indicator of antiangiogenic activity. Taken together, this suggests that arming G47Δ with IL-12 can elicit potent antiangiogenic effects.
Fig. 5.

Antiangiogenic effects of G47Δ-mIL12 in vivo. (A) Immunohistochemical staining of endothelial cells (anti-CD31 antibody) on brain tumor sections 6 d after treatment with PBS, G47Δ-E, and G47Δ-mIL12 (day 18). Right, histogram of CD31+ vessel area, as determined by ImageJ analysis (n = 3 per group). Only G47Δ-mIL12 treatment significantly decreased CD31+ microvessel density (**P < 0.005 versus G47Δ-E). Error bars indicated SEM. (B) Western blot of lysates from treated brain tumors on day 18 demonstrated an increase in IP-10 (Left) and a decrease in VEGF expression (Right) after G47Δ-mIL12 treatment. Each lane represents an individual mouse.
Survival Advantage of G47Δ-mIL12 Is T-Cell Mediated.
IL-12 acts as a “switch” that skews T cells toward Th1-type immune function, which is the desired mode for antitumor activity, and stimulates innate immunity, mediated by NK cells, which can also serve a role in antitumor immune responses. With regards to NK cells, the low expression of MHC I and high expression of NK ligand Rae-1 on 005 GSCs (Fig. 1C) makes them potentially susceptible to NK activity. We also observed an increase in NK tumor infiltration in G47Δ-mIL12–treated mice (Fig. S4). Given these observations, the enhanced efficacy conferred by G47Δ-mIL12 treatment and the strong immune effects of IL-12, we determined whether NK or T cells might be contributing to the G47Δ-mIL12–mediated survival advantage. For these purposes, C57BL/6 mice bearing 005 GSC intracranial tumors were depleted of NK cells using anti-NK1.1 antibody injected on day 6 after tumor implantation and every 3 d thereafter. Depletion of NK cells in vivo was confirmed by flow cytometry (Fig. S5). However, this had no effect on G47Δ-mIL12’s ability to enhance mouse survival, indicating that NK cells are not critical for the antitumor activity observed with G47Δ-mIL12 (Fig. 6A). Next, to investigate the role of T cells, 005 GSC were implanted in athymic/nude mice. In the absence of T cells, G47Δ-mIL12 was unable to significantly enhance survival over G47Δ-E (Fig. 6B), indicating a critical role of T cells in the IL-12–mediated antitumor activity.
Fig. 6.

Role of T cells in G47Δ-mIL12 treatment of intracranial 005 GSC tumors. (A) NK-cell depletion had no effect on the ability of G47Δ-mIL12 to enhance survival (P < 0.001; G47Δ-mIL12 with (G47∆-mIL12 + NKdepl) and without NK depletion (G47∆-mIL12) compared with PBS; P = 0.7, G47Δ-mIL12 with NK depletion versus without). (B) 005 GSCs were implanted in athymic mice, devoid of T cells, and treated with G47Δ-E or G47Δ-mIL12 on days 8 and 12. No significant difference was observed between these two treatments or compared with PBS. (C) FACS analysis of cells from treated 005 GSC tumors in C57BL/6 mice on day 6 posttreatment. Gates were drawn on live CD3+ cells, followed by CD4+ (Upper), and then the double-positive CD25+Foxp3+ cells were defined as Tregs. Representative flow cytometry plots of Tregs (black boxes, Lower) after PBS, G47Δ-E, and G47Δ-mIL12 treatment. (D) Significant decrease in Tregs (percent of CD4+) was observed with G47Δ-E versus PBS (**P < 0.005), and a further significant reduction with G47Δ-mIL12 versus G47Δ-E (**P < 0.005). Error bars indicate SEM.
G47Δ-mIL12 Decreases Tregs in 005 GSC Brain Tumors.
Given the demonstrated importance of T cells in mediating the survival advantage seen with G47Δ-mIL12, we further characterized the effects of G47Δ-mIL12 on T-cell responses, both locally and systemically. To quantitatively assess T-cell numbers, we performed multicolor flow cytometry on brain tissue, cervical draining lymph nodes, and spleens from mice 6 d posttreatment. No significant differences were observed between groups regarding numbers of CD3+ T cells, ratios of CD4+ and CD8+ T cells, or T-cell expression of the early activation marker CD69 (Table S1). Given the opposing roles of IL-12 and Tregs in fostering desired Th1 versus counterproductive Th2 polarization, respectively, we examined the effects of G47∆-mIL12 on local Tregs. There was a significant reduction in CD4+CD25+Foxp3+ Tregs in G47∆-E–treated brain tumors (Fig. 6C and D). These reductions were further significantly enhanced by IL-12 expression (Fig. 6C and D).
Discussion
The capabilities of GBM for GSC-mediated self-renewal, aggressive neovascularization, and marked local and systemic immunosuppression all contribute to current treatment inadequacy and tumor recurrence (25). GSCs, in addition to their key roles in GBM growth, heterogeneity, and resistance to therapy, also contribute directly to the tumor's immunosuppressive functions (5, 6, 26). Substantial evidence suggests that serum-cultured GBM cell lines do not recapitulate the genotype or phenotype of GBM and therefore have limitations in regards to translating therapies to the clinic (27, 28). Thus, the curative potential of anti-GBM therapies hinges on eradicating GSC in addition to countering beneficial features of the GBM microenvironment. Unfortunately, there has not been an immunocompetent GSC tumor model to evaluate the multitude of therapies being developed for GBM, especially those involving immunotherapy. Here we describe a GSC transplantable model in syngeneic mice. Murine 005 GSCs retain the important biological and pathophysiological characteristics of human GBM tumors; aggressive, infiltrative, angiogenic, heterogeneous, poorly immunogenic, and immunosuppressive. Therefore, this preclinical model provides a stringent test for investigating a given therapy's impact on the multiple features of GBM. Using this model, we demonstrate that oHSV G47∆ armed with mIL12 is highly efficacious in treating GBM, and its activity is multifaceted: (i) replicating and killing GSCs, (ii) inducing IFN-γ production, (iii) reducing abnormal vasculature, (iv) ameliorating the immunosuppressed microenvironment, and (v) prolonging survival that is T-cell mediated.
Oncolytic viruses have great potential for the treatment of tumors, using both direct cytotoxic and immune-stimulating mechanisms, and have progressed to phase III clinical trials in patients (11). A number of oHSVs have been developed that have proved safe for use in the brain, including G47∆ (19, 29). G47∆ has a number of advantages over other oHSVs examined in GBM clinical trials; it replicates in human GSCs (12) and lacks infected cell protein 47 (ICP47), which blocks MHC class I presentation in human cells (19). Thus, G47∆-infected tumor cells should be more visible to T cells, as we showed with human melanoma (19), particularly in the context of increased local IFN-γ production. It is also possible that this “heightened visibility” could decrease virus replication and efficacy, although OncoVexGM-CSF, with a similar ICP47 deletion, exhibited efficacy and induction of antitumor immune responses in patients (30). Arming oHSV with cytokines, including IL-12, has been shown to enhance their efficacy in mouse models (15–17, 31). However, none of these studies used cancer stem cells or mechanistically characterized the multiple contributions of IL-12.
IL-12 is a 70-kDa heterodimer composed of p35 and p40 subunits, and is one of the most effective immune-stimulatory cytokines, serving as a switch that initiates a path toward Th1-type immunity (14). IL-12 and its downstream Th1 mediator, IFN-γ, have multiple antitumor effects: unmasking the immunogenicity of cancer cells by up-regulating surface MHC class I, as illustrated for 005 GSCs (Fig. 1A); inducing Th1 differentiation; stimulating proliferation of NK, NKT, and T cells with induction of cytotoxic effectors; and antiangiogenesis (14). IFN-γ stimulates the production of angiostatic chemokines, such as MIG and IP-10, which have direct and CXC chemokine receptor (CXCR)3-independent effects on endothelial cells, in addition to their inflammatory effects on lymphocytes (32, 33). Systemic administration of IL-12, however, has been associated with serious toxicity as well as marginal clinical responses in most patients. Recent studies with engineered tumor-targeted T cells have highlighted the role of local IL-12 and consequent IFN-γ expression in modifying the tumor microenvironment and being key to mediating antitumor immune effects (34, 35).
Oncolytic HSVs are an excellent platform for localized IL-12 delivery, expression, and immunomodulatory activity, because they are tumor specific and activate a limited antitumor adaptive immune response that IL-12 augments (31). Typically mouse cells are much less permissive for oHSV replication and cytotoxicity than human cells (36), as we found for 005 GSCs; however, G47∆ replication was detected for at least 6 d in vivo. Whereas the in vitro replication and cytotoxicity of G47∆-mIL12 was similar to the nontransgene vector G47∆-E, IL-12 expression significantly improved in vivo efficacy in immunocompetent mice. In G47∆-mIL12–treated tumors, there was a strong induction of IP-10 and down-regulation of proangiogenic VEGF. Accordingly, the high microvessel density (CD31+ blood vessels) in the 005 tumors, part of the histological criteria of World Health Organization grades III and IV gliomas, was significantly reduced after treatment with G47∆-mIL12. Although oHSV has exhibited antiangiogenic effects in human U87 glioma models (37–40), G47∆-E did not reduce blood vessel density in the 005 tumor model. It is important to bear in mind that the mechanisms underlying the antitumor activity of antiangiogenic agents are multifaceted and not fully understood (41).
There is a dynamic cross-talk between tumors and the immune system that can regulate tumor growth, especially in GBM, which is highly immunosuppressive (4, 18). GBM employs multiple mechanisms to evade immune recognition, including impairment of MHC class I antigen presentation, down-regulation of costimulatory molecules (CD80 and CD86), and elaboration of immunosuppressive factors (i.e., TGF-β, IL-10, and STAT3) that recruit and expand Tregs, which in turn inhibit Th1 immunity and promote counterproductive Th2 responses (4, 18). Therefore, Tregs serve as an immune threshold that a successful antitumor immune response must overcome. These immunosuppressive features are present in the 005 GSC model. Interestingly, the percentage of Tregs among CD4+ T cells in patient GBMs is 25%–40% (42, 43), similar to what we detected in 005 tumors. In keeping with its role as a promoter of Th1 immunity, IL-12 was recently shown to inhibit Treg expansion, in a fashion dependent upon IL12Rβ2 and IFN-γ (44). In this study, G47∆-mIL12 injection resulted in a significant reduction in intratumoral Tregs, possibly via similar mechanisms given the local increases in IL-12 and IFN-γ. This presumably produced a more permissive environment for antitumor immunity, as evidenced by a T-cell–mediated survival advantage of G47∆-mIL12 treatment. G47∆-E also elicited a decline in Tregs (Fig. 6D), an effect seen in melanoma patients treated with oHSV expressing GM-CSF (30), but opposite that seen during peripheral HSV infection of mice, where Tregs were increased, activated, and protective (45). We did not observe any significant changes in T-cell numbers in the brains, spleens, or lymph nodes of mice after treatment, suggesting that the antitumor immune effects were local to the tumor.
The role of NK cells in oncolytic virotherapy is complex, with tumor cytotoxicity balanced by innate antiviral responses (46). Murine 005 GSCs expressed high levels of the NK group 2 member D (NKG2D) ligand Rae-1 and DNAX accessory molecule-1 (DNAM-1) ligand CD155, with no detectable MHC class I, which should make them sensitive to NK recognition and killing. Mouse breast cancer stem cells have also been shown to express Rae-1 and CD155 (47). Furthermore, G47∆-mIL12 treatment increased NK-cell tumor infiltration (Fig. S5). Therefore, we examined whether NK cell depletion would alter G47∆-mIL12–mediated efficacy. It did not, indicating that NK cells do not play a critical role in the ability of G47∆-mIL12 to enhance survival in this model. Rather, the IL-12–mediated benefit was dependent upon T-cell activity as it was completely abrogated in T-cell–deficient athymic mice. This is in contrast to the inhibitory effect of NK cells on oHSV rQNestin34.5 activity in a human glioma cell line xenograft model (48). The poor immunogenicity of 005 cells was likely countered by the observed increases in IFN-γ, which could up-regulate MHC I expression on 005 GSCs (Fig. 1A).
Cancer clinical trials using systemic delivery of IL-12 were suspended a decade ago because of dose- and regimen-related toxicities and limited clinical efficacy that was poorly understood (14). This study provides valuable information concerning the potential benefits and mechanisms when IL-12 is instead expressed locally in the context of oncolytic virus infection. In particular, it illustrates the multifaceted activities of G47∆-mIL12; antiangiogenesis, successful shifts toward Th1-type antitumor immunity, and viral oncolysis. These results are especially salient given the use of a stringent model that was representative of the diversity of subterfuges used by GBM. These promising data and the preclinical safety studies of another oHSV expressing IL-12 (M002) (49) provide a compelling rationale for the clinical translation of IL-12–armed oHSV for the treatment of GBM in patients.
Materials and Methods
A detailed description of the reagents and protocols used in this study is provided in SI Materials and Methods.
Cells and Viruses.
Mouse 005 GSCs, GFP-positive, p53+/−, with activated Ras and Akt (10), were cultured in neurosphere media with EGF and FGF2. Armed G47∆ vectors, G47∆-IL12, G47∆-E, G47∆-mCherry, and G47∆-Us11fluc (Fig. S2) were constructed using the flip-flop HSV BAC system (50).
Cell Viability and Virus Replication Assays.
Cells were treated as described previously (20). More details are provided in SI Materials and Methods.
Self-Renewal Neurosphere Formation Assay.
Three days after second virus infection, brain tumor quadrants were harvested and sorted for GFP+ cells by FacsAria. Viable cell (trypan blue excluding) counts were determined and cells resuspended in serum-free media and seeded into 96-well plates at 10 or 30 cells per well. Fifteen days later, the number of neurospheres (diameter, >60 μm) and wells containing neurospheres were counted. Six mice per group and counts were performed in a blinded manner.
In Vivo Studies.
Mice bearing intracerebral tumors were intratumorally treated with virus (∼5 × 105 pfu/3 μL). In some cases, brain tumor quadrants were excised and cells isolated or tissue was homogenized. NK-cell depletion was performed with anti-NK1.1 antibody (BioXcell). All mouse procedures were approved by the subcommittee on research animal care at Massachusetts General Hospital.
ELISA, Western Blots, Immunohistochemistry, and FACS.
Mouse IL-12 and IFN-γ were quantified using the Quantikine IL-12 and IFN-γ ELISA kits (R&D Systems). Western blots were performed as previously described (20) and immunohistochemistry using standard procedures. Antibody stained cells were sorted by flow cytometry.
Supplementary Material
Acknowledgments
We thank Drs. Inder Verma and Yasushi Soda for the 005 GSC cells; David Dombowski for sorting of GFP-positive cells; Kat Folz-Donahue and Laura Prickett-Rice from the Harvard Stem Cell Institute Flow Cytometry Core facility for assistance with flow cytometry analysis; John Chen and Derrick Jeon at the Massachusetts General Hospital Center for Systems Biology for assistance with the bioluminescence imaging; and members of the Molecular Neurosurgery Laboratory, especially Cecile Zaupa, Anoop Patel, Melissa Humphreys, and Howie Latimer for technical assistance and support. These studies were supported in part by National Institutes of Health Grants R01NS032677 (to R.L.M.), R01CA160762 (to S.D.R.), and R25NS065743 (to P.E.F.); a Joelle Syverson American Brain Tumor Association Research Fellowship (to T.A.C.); and the Rendina Family Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 11672.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307935110/-/DCSupplemental.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Dirks PB. Brain tumor stem cells: The cancer stem cell hypothesis writ large. Mol Oncol. 2010;4(5):420–430. doi: 10.1016/j.molonc.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fecci PE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 4.Rolle CE, Sengupta S, Lesniak MS. Mechanisms of immune evasion by gliomas. Adv Exp Med Biol. 2012;746:53–76. doi: 10.1007/978-1-4614-3146-6_5. [DOI] [PubMed] [Google Scholar]
- 5.Wei J, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res. 2010;16(2):461–473. doi: 10.1158/1078-0432.CCR-09-1983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Di Tomaso T, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16(3):800–813. doi: 10.1158/1078-0432.CCR-09-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castriconi R, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182(6):3530–3539. doi: 10.4049/jimmunol.0802845. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed N, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown CE, et al. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T cells. Clin Cancer Res. 2012;18(8):2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marumoto T, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15(1):110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakimoto H, et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ino Y. (2009) A clinical study of a replication-competent, recombinant herpes simplex virus type 1 (G47delta) in patients with progressive glioblastoma. World Health Organization International Clinical Trials Registry. Available at http://apps.who.int/trialsearch/trial.aspx?trialid=JPRN-UMIN000002661.
- 14.Del Vecchio M, et al. Interleukin-12: Biological properties and clinical application. Clin Cancer Res. 2007;13(16):4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- 15.Parker JN, et al. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA. 2000;97(5):2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong RJ, et al. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther. 2001;12(3):253–265. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- 17.Varghese S, et al. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006;13(3):253–265. doi: 10.1038/sj.cgt.7700900. [DOI] [PubMed] [Google Scholar]
- 18.Dunn GP, Fecci PE, Curry WT. Cancer immunoediting in malignant glioma. Neurosurgery. 2012;71(2):201–222, discussion 222–223. doi: 10.1227/NEU.0b013e31824f840d. [DOI] [PubMed] [Google Scholar]
- 19.Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA. 2001;98(11):6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanai R, et al. Oncolytic virus-mediated manipulation of DNA damage responses: Synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prestegarden L, et al. Glioma cell populations grouped by different cell type markers drive brain tumor growth. Cancer Res. 2010;70(11):4274–4279. doi: 10.1158/0008-5472.CAN-09-3904. [DOI] [PubMed] [Google Scholar]
- 22.Beier CP, Beier D. CD133 negative cancer stem cells in glioblastoma. Front Biosci (Elite Ed) 2011;3:701–710. doi: 10.2741/e280. [DOI] [PubMed] [Google Scholar]
- 23.Sgubin D, Wakimoto H, Kanai R, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cells. Stem Cells Transl Med. 2012;1(4):322–332. doi: 10.5966/sctm.2011-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu TC, Castelo-Branco P, Rabkin SD, Martuza RL. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol Ther. 2008;16(6):1041–1047. doi: 10.1038/mt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 26.Wu A, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-oncol. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 28.Li A, et al. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res. 2008;6(1):21–30. doi: 10.1158/1541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
- 29.Aghi M, Rabkin S. Viral vectors as therapeutic agents for glioblastoma. Curr Opin Mol Ther. 2005;7(5):419–430. [PubMed] [Google Scholar]
- 30.Kaufman HL, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 31.Todo T. Active immunotherapy: Oncolytic virus therapy using HSV-1. Adv Exp Med Biol. 2012;746:178–186. doi: 10.1007/978-1-4614-3146-6_14. [DOI] [PubMed] [Google Scholar]
- 32.Sgadari C, Angiolillo AL, Tosato G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood. 1996;87(9):3877–3882. [PubMed] [Google Scholar]
- 33.Campanella GS, Colvin RA, Luster AD. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PLoS ONE. 2010;5(9):e12700. doi: 10.1371/journal.pone.0012700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerkar SP, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121(12):4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Todo T, Ebright MI, Fong Y, Rabkin SD. In: Oncolytic Herpes Simplex Virus (G207) Therapy: from Basic to Clinical. Tumor-Suppressing Viruses, Genes, and Drugs. Maruta H, editor. San Diego: Academic; 2002. pp. 45–75. [Google Scholar]
- 37.Liu TC, et al. Dominant-negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res. 2006;12(22):6791–6799. doi: 10.1158/1078-0432.CCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 38.Aghi M, Rabkin SD, Martuza RL. Angiogenic response caused by oncolytic herpes simplex virus-induced reduced thrombospondin expression can be prevented by specific viral mutations or by administering a thrombospondin-derived peptide. Cancer Res. 2007;67(2):440–444. doi: 10.1158/0008-5472.CAN-06-3145. [DOI] [PubMed] [Google Scholar]
- 39.Hardcastle J, et al. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther. 2010;18(2):285–294. doi: 10.1038/mt.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, et al. Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther. 2012;20(1):37–45. doi: 10.1038/mt.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellis LM, Hicklin DJ. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 42.El Andaloussi A, Lesniak MS. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J Neurooncol. 2007;83(2):145–152. doi: 10.1007/s11060-006-9314-y. [DOI] [PubMed] [Google Scholar]
- 43.Crane CA, Ahn BJ, Han SJ, Parsa AT. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro-oncol. 2012;14(5):584–595. doi: 10.1093/neuonc/nos014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao X, et al. Interleukin 12 stimulates IFN-gamma-mediated inhibition of tumor-induced regulatory T-cell proliferation and enhances tumor clearance. Cancer Res. 2009;69(22):8700–8709. doi: 10.1158/0008-5472.CAN-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320(5880):1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez-Breckenridge CA, Yu J, Kaur B, Caligiuri MA, Chiocca EA. Deciphering the multifaceted relationship between oncolytic viruses and natural killer cells. Adv Virol. 2012;2012:702839. doi: 10.1155/2012/702839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M, Knight DA, Smyth MJ, Stewart TJ. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol Immunother. 2012;61(8):1255–1268. doi: 10.1007/s00262-012-1200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez-Breckenridge CA, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18(12):1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Markert JM, et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J Virol. 2012;86(9):5304–5313. doi: 10.1128/JVI.06998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuroda T, Martuza RL, Todo T, Rabkin SD. Flip-Flop HSV-BAC: Bacterial artificial chromosome based system for rapid generation of recombinant herpes simplex virus vectors using two independent site-specific recombinases. BMC Biotechnol. 2006;6:40. doi: 10.1186/1472-6750-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.