

Abstract
Transcription factors (TFs) recognize short sequence motifs that are present in millions of copies in large eukaryotic genomes. TFsmust distinguish their target binding sites from a vast genomic excess of spurious motif occurrences; however, it is unclear whether functional sites are distinguished from nonfunctional motifs by local primary sequence features or by the larger genomic context in which motifs reside. We used a massively parallel enhancer assay in living mouse retinas to compare 1,300 sequences bound in the genome by the photoreceptor transcription factor Cone-rod homeobox (Crx), to 3,000 control sequences. We found that very short sequences bound in the genome by Crx activated transcription at high levels, whereas unbound genomic regions with equal numbers of Crx motifs did not activate above background levels, even when liberated from their larger genomic context. High local GC content strongly distinguishes bound motifs from unbound motifs across the entire genome. Our results show that the cis-regulatory potential of TF-bound DNA is determined largely by highly local sequence features and not by genomic context.
Keywords: gene regulation, transcription factor binding, systems biology
Detailed maps of transcription factor (TF)-bound genomic regions are being produced by consortium-driven efforts such as ENCODE (1), yet the sequence features that distinguish functional cis-regulatory sites from the millions of spurious motif occurrences in large eukaryotic genomes are poorly understood (2–6). Several models have been proposed to explain how TFs distinguish between functional cis-regulatory elements (CREs) and nonfunctional motif occurrences (3, 6, 7). In one model, large-scale chromatin context directs TF binding to target sites while limiting TF access to spurious motif occurrences (7–9). This model is supported by recent analyses of genomic DNaseI hypersensitivity, which show that only 1% of the genome typically resides in open chromatin in any given cell type (1, 10), suggesting that most spurious motif occurrences are inaccessible. A second model states that target sites are recognized through cooperative TF binding to highly specific combinations of sequence motifs, which are unlikely to occur by chance in nonregulatory regions of the genome (6, 11). This model is supported by evidence that the binding specificity of many TFs is affected by cooperative interactions with cofactors (12). A third model states that most TF binding is promiscuous, low occupancy, and nonfunctional, whereas functional CREs are characterized by high TF occupancy, achieved through either a permissive chromatin context or high affinity for TFs (3, 6). This model is motivated by recent genomewide binding studies demonstrating that binding locations of functionally diverse TFs overlap substantially (13, 14), a result that suggests binding is unlikely to be primarily determined by rare, specific combinations of cooperative interactions. Regardless of the mechanisms by which TFs select functional CREs, the distinction between functional and nonfunctional motif occurrences must ultimately depend on information encoded either locally or within the larger sequence context surrounding functional CREs.
To distinguish between these models, we used CRE-seq, a massively parallel reporter gene assay (15–17), to compare the cis-regulatory activity of Cone-rod homeobox (Crx)-bound DNA identified by ChIP-seq in murine photoreceptors (18), against the activity of unbound genomic regions with equivalent numbers of Crx motif occurrences. We assayed very short (84 bp) sequences centered on Crx ChIP-seq peaks (average length 267 bp); nonetheless, in our assay, we found major differences in activity between short genomic sequences with Crx motifs taken from ChIP-seq peaks and short sequences with equivalent numbers of Crx motifs taken from genomic regions that were unbound in ChIP-seq assays.
Results
We used high-throughput oligonucleotide synthesis (19) to create a library of 84-bp sequences, each fused to a 9-bp barcode. We included 1,298 of 5,595 ChIP-seq peaks [herein called Crx-bound regions (CBRs)] and 3,035 control sequences (Fig. 1A). Specifically, we included 865 CBRs with at least one Crx motif, and because we found that 35% of all CBRs lack a high-quality Crx motif, we included 433 CBRs that lack Crx motifs. As controls, we included 865 regions that contain Crx motifs, but which are not bound by Crx in the genome [unbound regions (UBRs)]. UBRs were specifically chosen to match the Crx motif content and chromosomal distribution of the CBRs with Crx motifs (Fig. S1). Additionally, we included mutant versions of CBRs in which each Crx motif was inactivated by a point mutation (20) and three sets of controls generated by scrambling the individual sequences of a subset of CBRs and UBRs while preserving all dinucleotide frequencies of the original sequences. Thus, the library included 4,333 sequences, each represented by three independent barcodes, resulting in a total library of 12,999 uniquely barcoded sequences.
Fig. 1.
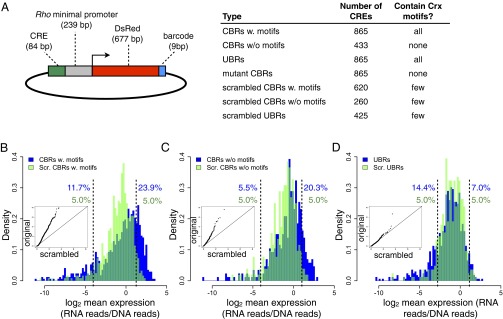
CBRs drive higher activation than scrambled DNA controls, whereas UBRs with Crx motifs do not. (A) Structure of CRE reporter genes and a list of number of sequences and Crx motif occurrences by CRE type. Some scrambled CREs contain motifs that were generated fortuitously by the scrambling algorithm. Distribution of CRE-seq log2 expression for (B) CBRs with Crx motifs, (C) CBRs lacking Crx motifs, and (D) UBRs with Crx motifs (blue) compared with the corresponding scrambled DNA distributions (green). Vertical dashed lines show 5th and 95th percentiles of scrambled distributions, and the percentage of the original (blue) and scrambled (green) distributions beyond the dashed lines are given. (Insets) Quantile-quantile plots, shown on a nonlogarithmic scale, comparing expression in scrambled and original distributions. Differences between distributions are indicated by the deviation of data points from the diagonal.
Between the library sequence and the barcode, we cloned the photoreceptor-specific Rhodopsin minimal promoter fused to a DsRed reporter gene, to create the final reporter gene library (Fig. 1A) (15). We electroporated the library into explanted newborn mouse retinas. After culturing, we measured reporter expression levels by extracting RNA and sequencing the barcodes in the RNA samples and the original plasmid DNA pool. To calculate expression for each library sequence, we averaged the RNA/DNA ratio across three barcodes from six replicate experiments. We observed a high correlation between CRE-seq expression of individual barcodes in different biological replicates (mean Pearson’s correlation coefficient = 0.95; Fig. S2).
Sequences from Crx ChIP-seq Peaks Drive Activation in CRE-seq, Whereas Unbound Sequences with Crx Motifs Do Not.
We found that many CBRs drove high levels of transcription, whereas UBRs with equivalent numbers of Crx motifs did not activate transcription above levels produced by their matched scrambled DNA controls, suggesting that unbound Crx motifs are intrinsically nonfunctional; 23.9% of the CBRs with Crx motifs drove reporter gene expression above the 95th percentile of the corresponding scrambled CBR sequences (Fig. 1B), and this was also true of 20.3% of CBRs lacking Crx motifs (Fig. 1C). In contrast with CBRs, only 7.0% of UBRs drove expression above the 95th percentile of scrambled UBRs (Fig. 1D), despite the fact that UBRs contained as many high-quality motifs as the CBRs and more Crx motifs than the scrambled sequences (Fig. S1). Overall, the distribution of CBR activity differed substantially from the corresponding scrambled sequence distributions at all expression levels, whereas this was not true of UBRs (Fig. 1 B–D, Insets). Pairwise comparisons between CBRs and their corresponding scrambled sequences showed that 77% of CBRs with motifs and 72% of CBRs lacking motifs showed activity that differed significantly from that of the scrambled version of the sequence; this was true for 64% of UBRs. Scrambled CBRs were more likely to lose activity relative to the original sequence, whereas scrambled UBRs were equally likely to gain or lose activity, further indicating that UBRs lack specific function (Fig. S3). Because the flanking sequence context in our plasmid-based assay was identical for CBRs and UBRs, our results demonstrate that much of the cis-regulatory potential of CREs is determined by sequence features that are independent of genomic context and highly localized within the central 84 bp of individual ChIP-seq peaks.
We found that both CBRs with Crx motifs and UBRs were more likely than the scrambled DNA controls to strongly repress transcription, whereas CBRs lacking Crx motifs were not; 11.7% of CBRs with motifs and 14.4% of UBRs drove reporter expression below the level of the fifth percentile of the corresponding scrambled distribution (Fig. 1, B and D), whereas only 5.5% of CBRs lacking motifs did so (Fig. 1C). Overall, our results show that both bound and unbound Crx motifs, removed from their genomic context, can produce repression, whereas only bound regions can strongly activate.
Although neither UBRs nor scrambled sequences drove high levels of transcription, nearly all of these sequences did exhibit some cis-regulatory potential in our assay. Individual CRE sequences produced distinct and reproducible levels of expression that differed from the distribution mean (Fig. S4). These results show that most sequences, including those generated by scrambling genomic sequences, can produce some cis-regulatory effects. They also explain why most UBR sequences alter their activity on scrambling, despite the overall resemblance between the UBR and scrambled distributions. This underscores the importance of comparing the results from ChIP-seq peaks to distributions of control sequences and not simply to a small set of representative controls.
Regulatory Potential of Crx ChIP-seq Peak Sequences Depends on Crx Motifs.
Because TFs can bind promiscuously, it is unclear what fraction of TF-bound regions are genuinely functional (2, 13, 14). We found that most CBRs exhibit activity that depends on the presence of Crx motifs, suggesting that most CBRs are potentially functional. We compared the cis-regulatory potential of WT CBRs with Crx motifs (WTCBRs) against the cis-regulatory potential of matching, mutated CBRs in which all Crx motifs were inactivated (mutCBRs); 59.5% of mutCBRs exhibited expression that differed significantly from the matching WTCBR (Fig. 2A), indicating that most CBRs have Crx-dependent regulatory potential. The strong correlation observed between the expression level of WTCBRs and the fold change in the mutCBRs shows that mutCBRs revert toward the mean scrambled DNA expression level. Thus, inactivating the Crx motifs caused mutCBRs to behave much like the scrambled DNA controls (Fig. 2B), which suggests that Crx motifs are necessary for nearly all of the activation and repression produced by WTCBRs.
Fig. 2.
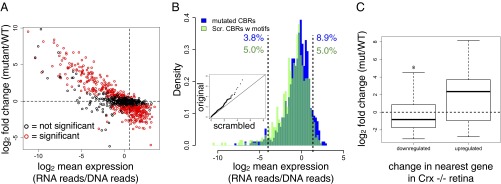
Activity of CBRs depends on Crx motifs. (A) Relationship between mean CRE log2 expression of WTCBRs and log2 fold-change in the matching mutCBRs. Red points indicate significantly different WT/mutant pairs (Benjamini-Hochberg adjusted P < 0.01, two-sided Welch’s t test, n = 12–18); black points indicate nonsignificantly changed pairs. The vertical dashed line indicates the distribution mean for WTCBR expression, and the horizontal dashed line indicates the position of no change in the mutant. (B) Distribution of CRE-seq expression for mutCBRs (blue) and scrambled CBRs with Crx motifs (green); compare with Fig. 1B. (C) Box and whisker plots showing the distribution of fold changes of significantly different WT/mutant pairs for CBRs near genes that are down-regulated (n = 42) or up-regulated (n = 14) in Crx−/− retina. Mutant versions of CBRs near up-regulated genes are more likely to show an increased fold change than mutant versions of CBRs near down-regulated genes (P = 0.047, one-sided Kolmogorov-Smirnov test). Distribution medians are indicated by bold horizontal lines, and the dashed horizontal line indicates the position of no change in the mutant.
Despite the short length of our library sequences and the plasmid context of our assay, we found that the effects of inactivating Crx sites often mirrored the expression changes of nearby genes previously observed to be up-regulated or down-regulated in Crx−/− retinas (21). Among the CBRs in our library whose expression changed significantly when Crx sites were inactivated, 14 are nearest up-regulated genes, and 42 are nearest down-regulated genes (18). Mutant versions of CBRs near genes up-regulated in Crx−/− retina were likely to increase expression in our assay (median fold change = 5.12), whereas mutant versions of CBRs near Crx−/− down-regulated genes were likely to decrease expression (median fold change = 0.56; Fig. 2C). These results provide additional support for the highly local and context-independent nature of the cis-regulatory potential of Crx-bound regions.
High Local GC Content Distinguishes Functional Crx Sites from Nonfunctional Crx Motif Occurrences in the Genome.
We sought to identify sequence features that might explain the differences in regulatory potential between CBRs and UBRs. Neither affinity for Crx (Fig. S5A) nor Crx occupancy previously measured by ChIP-seq (Fig. S5C) was quantitatively predictive of CRE expression level. We found no specific enrichment for additional TF motifs in either class of CBRs, including the photoreceptor TF Nrl (Fig. S6) (22).
High G and C nucleotide content was the sequence feature that most strongly differentiated CBRs from UBRs in our library. CBRs were previously shown to be GC rich relative to the genome average, including CBRs that do not reside in CpG islands (18). We found that this result also holds when CBRs are compared directly to unbound genomic regions with equal numbers of Crx motifs (Fig. 3A), although GC fraction does not quantitatively predict CRE expression (Fig. S5B). CBRs lacking Crx motifs are particularly GC rich, suggesting that these CBRs form a distinct class in which other sequence features compensate for the lack of Crx motifs. Consistent with this observation, CBRs lacking Crx motifs in our library are more likely to occur in CpG islands than CBRs with Crx motifs or unbound regions with Crx motifs (Fig. S7).
Fig. 3.

High GC nucleotide content distinguishes CBRs from UBRs with Crx motifs. (A) Box and whisker plots showing distributions of GC fraction in CRE sequences. Bold horizontal lines indicate distribution medians. All pairwise comparisons between distribution means are significant (P < 2.2 × 10−16, two-sided Welch’s t test). (B) Receiver operating characteristic curves showing discrimination between all bound (n = 1.4 × 104) and unbound (n = 6.7 × 106) Crx motifs in the mouse genome. Flanking %GC includes 40 bp on each side of the motif. (C) Distributions of CC/CG/GG/GC dinucleotides frequencies in CRE sequences. All pairwise comparisons between distribution means are significant (P < 2.2 × 10−16, two-sided Welch’s t test). (D) Total frequency of AAAA sequences per base pair across all sequences for different CRE categories. (E) Distributions of predicted minor groove width in CRE sequences, determined by ORChID2 (28). A larger score indicates a wider minor groove. All pairwise comparisons between distribution means are significant (P < 2.2 × 10−16, two-sided Welch’s t test). (F) Crx motifs in library CBRs (n = 1,762) exhibit wider average minor groove width than Crx motifs in UBRs (n = 1,515, P = 7.05 × 10−12, two-sided Welch’s t test).
We found that high local GC content identifies bound Crx motifs across the entire mouse genome. GC fraction in the flanking sequences around a Crx motif was substantially better than Crx motif quality as a discriminator between all bound Crx motifs (n = 1.4 × 104) and all unbound motifs (n = 6.7 × 106) in the mouse genome [Fig. 3B; area under the curve (AUC) for flanking GC = 0.79, AUC for Crx motif score = 0.60]. Remarkably, GC content of the 8-bp Crx motifs alone was equal to Crx motif quality as a discriminator between bound and unbound Crx motifs in the genome (Fig. 3B; AUC = 0.61), despite no correlation between the quality score and GC content of a motif (Pearson’s correlation coefficient = 0.01).
High GC content is associated with several distinct features that can affect the cis-regulatory potential of a sequence (4, 23), including high nucleosome occupancy (24–26) and a wide DNA minor groove (27, 28). In some cases, TF binding is promoted by high nucleosome occupancy (29), and minor groove width can affect the binding specificity of homeodomain TFs (12). Even considering only the 84-bp sequences included in our CRE library, CBRs were distinct from UBRs in sequence features related to nucleosome positioning and minor groove width. Compared with UBRs, CBRs were enriched in CC/CG/GG/GC dinucleotides (Fig. 3C) and depleted in poly-A sequences (Fig. 3D), suggesting that CBR sequences intrinsically promote higher nucleosome occupancy than UBR sequences. CBR sequences were also predicted to exhibit an overall wider minor groove width (Fig. 3E), which could affect the in vivo affinity of Crx for motif occurrences within CBRs relative to UBRs. We found that the sequences of the Crx motifs themselves were predicted to exhibit a wider minor groove in CBRs than in UBRs (Fig. 3F), despite comparable motif affinities, indicating that minor groove width could be a highly localized sequence feature that distinguishes functional from nonfunctional motifs.
Repressing ChIP-seq Peak Sequences Exhibit Low GC Content and High Affinity for Crx.
Crx is primarily known to function as a transcriptional activator (30); however, we observed that many CBRs produce repression that depends on the presence of a Crx motif (Fig. 2A). We discovered two sequence features that distinguish activating CBRs with Crx motifs from repressing CBRs with Crx motifs: (i) CBRs that repress contain more occurrences of Crx motifs than strongly activating CBRs (Fig. 4A; mean predicted Crx occupancy, determined by a binding model, of activating CBRs = 2.2, repressing CBRs = 3.7) and (ii) activating CBRs contain a higher GC fraction than repressive CBRs (Fig. 4B). The high GC content of activating CBRs relative to repressive CBRs parallels the high GC content of CBRs relative to UBRs (Fig. 3A), again indicating that functional Crx motifs that drive transcription depend on a high GC sequence context; in the absence of a high GC context, clusters of Crx motifs produce repression.
Fig. 4.

Predicted Crx occupancy as determined by a binding model (Materials and Methods) and GC content differentiate activating and repressing CBRs. (A) Box and whisker plots showing higher predicted Crx occupancy in repressing CBRs. Median Crx occupancy for each distribution is indicated by bold horizontal lines. (n per category = 49, P = 2.11 × 10−9, Welch’s t test). (B) Activating CBRs exhibit a higher GC fraction than repressing CBRs (n per category = 49, P = 4.92 × 10−7, Welch’s t test).
Discussion
By assaying the cis-regulatory activity of very short sequences, we demonstrated that highly local, context-independent sequence features distinguish the functional potential of TF-bound sequences from unbound sequences with Crx motifs. Because each CRE in our library was tested in the same plasmid context, any aspects of chromatin structure that distinguish the activity of CBRs from UBRs and from scrambled DNA controls must be locally encoded within the 84-bp sequences. Our results also indicate that most Crx motifs in the genome are likely to be intrinsically nonfunctional and not merely inaccessible within a broader repressive chromatin context. Our findings suggest that, despite evidence that TFs bind promiscuously, most TF-bound sequences have cis-regulatory potential, and this potential is encoded by a highly local regulatory grammar that could include specific arrangements of TF motifs, as well as more general sequence features such as minor groove width, which could potentially influence both TF affinity and local chromatin structure.
Our results demonstrate the importance of comparing the activity of candidate CREs against distributions of control sequences, as well as the value of using multiple approaches to assess the function of CREs. Although scrambled DNA elements are unlikely to drive very strong levels of activation or repression (Fig. 1 B–D), such sequences can produce distinct levels of enhancer activity within an intermediate range that overlaps with the activity of many functional sequences (Fig. S4). Thus, function cannot be assessed solely by applying a threshold level of activity; additional approaches to characterize function are necessary, such as mutagenesis of TF binding sites. Although it can be difficult to distinguish between the activity of individual functional CREs and random DNA, we have shown that our approach has the power to discover functional differences between classes of DNA elements, such as bound and unbound TF motifs, thereby providing a means to identify and test the role of sequence features that distinguish those classes from each other.
Comparisons between CBRs and the various controls in our experiment showed that a simple classifier based on the presence of a Crx motif embedded in a locally GC-rich sequence has remarkable power to distinguish functional from nonfunctional Crx motifs across the mouse genome (Fig. 3B). Our results suggest a model in which a combination of moderate affinity for Crx and high local GC content creates a favorable sequence context for activation, whereas repression is produced by the presence of multiple high-affinity Crx motifs and the absence of a favorable GC context. Although additional primary sequence features and the specific combinations of motifs that quantitatively account for the cis-regulatory activity of individual CBRs remain to be identified, our results show that such features are highly local and act independently of genomic context.
Materials and Methods
Design of CRE-seq Library Sequences.
A total of 1,298 CBRs were selected from 5,595 CBRs identified in two replicate ChIP-seq experiments (18). CBRs were chosen such that distributions of the number motifs and position weight matrix (PWM) scores of Crx motifs in our sample were representative of the total set of CBRs. CBRs in our sample were also selected to match the distribution of all CBRs among chromosomes. UBRs were selected by (i) dividing the genome into overlapping 84-bp windows with start positions spaced 10 bp apart, (ii) classifying each window by number and total PWM score of Crx motifs scoring better than a P < 0.001 threshold (see below), and (iii) for each chromosome, choosing every nth window that contained x Crx motifs (where x ranged from 1 to 6), such that the number of chosen windows with x Crx motifs matched the number of chosen CBRs with x Crx motifs from that chromosome. UBRs were resampled as necessary to ensure that (i) quality of the Crx motifs in the selected windows approximately matched that of CBRs and (ii) no window overlapped with any region identified as Crx bound. Sequences were taken from the July 2007 mouse genome assembly (NCBI37/mm9) (31). Mutant CBRs were designed by incorporating an adenine to cytosine point mutation at position 4 in each Crx motif (20). All mutant sequences were scanned to ensure that no new Crx sites were created by the A4C mutation. Scrambled sequences were designed by 100,000 iterations of the following procedure, which preserves all dinucleotide frequencies: (i) from the input sequence, randomly choose two nonoverlapping subsequences with identical flanking dinucleotides, and (ii) swap the positions of the two subsequences while retaining the flanking dinucleotides. Each CRE sequence was represented in the library three times linked to three unique 9-bp barcodes.
Identification and Scoring of Crx Motifs.
For library design, Crx motif occurrences were scored by the program FIMO (32), using the Crx PWM determined by Lee et al. (20). We used a threshold of P < 0.001, a conservative threshold that includes most, but not all, demonstrably functional low-affinity Crx motifs (20). For subsequent analyses (Fig. 3 B and F), we used a less conservative threshold of 5% affinity relative to the consensus sequence. As a separate, threshold-free measure of aggregate Crx affinity, we used a binding model to calculate predicted Crx occupancy. We used the binding model described in Eq. 1 of Zhao et al. (33), with energies E determined by the Crx PWM and the parameter µ = 9. Nrl motif occurrences were scored by FIMO using the previously described PWM (15). To perform de novo motif discovery and search for enrichment of known TF motifs in our library sequences, we used MEME-ChIP (34), DREME (35), and CentriMO (36).
CRE-seq Library Construction.
A library of 12,999 unique 150mer oligonucleotides (oligos) was ordered through a limited licensing agreement with Agilent Technologies. Each oligo was structured as follows: 5′ priming site (TAGCGTCTGTCCGT)/EcoRI site/84-bp CRE sequence/SpeI site/C nucleotide/SphI site/9-bp barcode/NotI site/3′ priming sequence (CAACTACTACTACAG). Library sequences are given in Dataset S1.
The library was amplified and cloned as previously described (15), except that the primers MO563 and MO564 were used (Table S1), and following four cycles of PCR, the library was purified by electrophoresis on a 17 × 16-cm 12% polyacrylamide gel in Tris-borate-EDTA (TBE) buffer run at 200 V. After staining with SYBR Gold (Invitrogen), gel bands were excised, and DNA was extracted by electrophoresis into 300 µL TBE in a 0.5-mL Slide-A-Lyzer Dialysis Cassette G2 2K MWCO (Thermo Scientific), followed by ethanol precipitation and resuspension in 10 mM Tris, pH 8, and 0.1 mM EDTA.
The library plasmid backbone, pJK03, was created from the plasmid Rho_basal-DsRed (18, 21) as described previously (15). Purified library amplicons were cloned into pJK03 using EcoRI and NotI (in the amplicon)/EagI (in the plasmid), and the Rho minimal promoter fused to DsRed was cloned between the CRE sequence and the barcode using SpeI (in the CRE sequence)/NheI (in the Rho promoter) and SphI, as previously described (15). CRE and barcode sequences for 90 constructs were verified by Sanger sequencing, of which 63 (70%) had no mutations, 19 (21%) had single base deletions, and 8 (9%) had larger deletions. Because each barcoded CRE was synthesized at a single array spot containing many molecules, the majority of sequences for any given CRE in the plasmid library will have no mutations.
CRE-seq Assay.
Retinal electroporations were performed as previously described (15, 21). Six replicate electroporations of the library were performed. cDNA synthesis and barcode amplification were performed as described (15), using PCR primers MO574 and MO575 (Table S1). Barcode amplicons were digested with SphI and MfeI and ligated to Illumina adapter sequences, followed by amplification as described (15). DNA from the plasmid library and cDNA samples were multiplexed and run on four lanes of an Illumina HiSeq machine, generating 48.9 million reads corresponding to the plasmid library DNA and an average of 49.4 million reads corresponding to each cDNA sample. Reads that did not perfectly match designed barcode sequences were discarded, and only barcodes with greater than one read per million in the DNA pool were used for analysis. Reads per million for each barcode from plasmid DNA and replicate cDNA samples are given in Dataset S1.
To account for differences in barcode representation in the original plasmid library, cDNA reads were normalized against DNA reads from the plasmid library. The average cDNA/DNA ratio and SEs for each CRE were calculated by error propagation across the six replicates and multiple barcodes as described (15). P values to determine significant differences in expression between WT and mutant CBRs were calculated using Welch’s t test (37), and P values were adjusted using the method of Benjamini and Hochberg (38). Mean normalized CRE-seq expression and SE for each sequence is given in Dataset S2 and for pairs of WT and mutant CBRs in Dataset S3.
Sequence Analyses.
CBR associations with Crx−/− up- or down-regulated genes were taken from Corbo et al. (18). Minor groove width was predicted at base pair resolution using the ORChID2 program (28), and ORChID2 scores were summed across all base pairs in each CRE or Crx motif to give a total sequence minor groove width score.
CpG island annotations were obtained from the UCSC Genome Browser at http://genome.ucsc.edu/index.html (39).
Statistical Tests.
Two-sample Welch’s t tests and Kolmogorov-Smirnov tests for differences in distribution means or cumulative distribution functions were calculated using the t.test and ks.test functions in the package stats in R (40). For t tests, approximate normality of the data wase evaluated using quantile-quantile plots. Receiver operating characteristic curves and AUC were calculated using the ROCR package in R (41).
Supplementary Material
Acknowledgments
We thank Jamie Kwasnieski, Ilaria Mogno, and Chris Fiore for advice on CRE-seq library construction and sequencing; Aaron Spivak for assistance with Crx motif analysis and the sequence scrambling algorithm; and members of the Cohen laboratory for advice on the manuscript. This work was supported by National Institutes of Health Grants GM092910 (to B.A.C.), EY018826 (to J.C.C.), and HG006346 (to B.A.C. and J.C.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307449110/-/DCSupplemental.
References
- 1.Dunham I, et al. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao Y, et al. Genome-wide MyoD binding in skeletal muscle cells: A potential for broad cellular reprogramming. Dev Cell. 2010;18(4):662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biggin MD. Animal transcription networks as highly connected, quantitative continua. Dev Cell. 2011;21(4):611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Landolin JM, et al. Sequence features that drive human promoter function and tissue specificity. Genome Res. 2010;20(7):890–898. doi: 10.1101/gr.100370.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitfield TW, et al. Functional analysis of transcription factor binding sites in human promoters. Genome Biol. 2012;13(9):R50. doi: 10.1186/gb-2012-13-9-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spitz FCO, Furlong EEM. Transcription factors: From enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 7.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43(3):264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 2006;16(12):1517–1528. doi: 10.1101/gr.5655606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guertin MJ, Lis JT. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 2010;6(9):e1001114. doi: 10.1371/journal.pgen.1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yáñez-Cuna JO, Dinh HQ, Kvon EZ, Shlyueva D, Stark A. Uncovering cis-regulatory sequence requirements for context-specific transcription factor binding. Genome Res. 2012;22(10):2018–2030. doi: 10.1101/gr.132811.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slattery M, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147(6):1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacArthur S, et al. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 2009;10(7):R80. doi: 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neph S, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489(7414):83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci USA. 2012;109(47):19498–19503. doi: 10.1073/pnas.1210678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melnikov A, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30(3):271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbo JC, et al. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res. 2010;20(11):1512–1525. doi: 10.1101/gr.109405.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeProust EM, et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 2010;38(8):2522–2540. doi: 10.1093/nar/gkq163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Myers CA, Williams N, Abdelaziz M, Corbo JC. Quantitative fine-tuning of photoreceptor cis-regulatory elements through affinity modulation of transcription factor binding sites. Gene Ther. 2010;17(11):1390–1399. doi: 10.1038/gt.2010.77. [DOI] [PubMed] [Google Scholar]
- 21.Hsiau TH-C, et al. The cis-regulatory logic of the mammalian photoreceptor transcriptional network. PLoS ONE. 2007;2(7):e643. doi: 10.1371/journal.pone.0000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rehemtulla A, et al. The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proc Natl Acad Sci USA. 1996;93(1):191–195. doi: 10.1073/pnas.93.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458(7236):362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tillo D, Hughes TR. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics. 2009;10:442. doi: 10.1186/1471-2105-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tillo D, et al. High nucleosome occupancy is encoded at human regulatory sequences. PLoS ONE. 2010;5(2):e9129. doi: 10.1371/journal.pone.0009129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohs R, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461(7268):1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop EP, et al. A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem Biol. 2011;6(12):1314–1320. doi: 10.1021/cb200155t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballaré C, et al. Nucleosome-driven transcription factor binding and gene regulation. Mol Cell. 2013;49(1):67–79. doi: 10.1016/j.molcel.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Chen S, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19(5):1017–1030. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- 31.Waterston RH, et al. Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 32.Grant CE, Bailey TL, Noble WS. FIMO: Scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Granas D, Stormo GD. Inferring binding energies from selected binding sites. PLOS Comput Biol. 2009;5(12):e1000590. doi: 10.1371/journal.pcbi.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Machanick P, Bailey TL. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics. 2011;27(12):1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40(17):e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sokal RR, Rohlf FJ. Biometry. 3rd Ed. New York: W. H. Freeman; 1994. pp. 404–405. [Google Scholar]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 39.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. R Core Team (2012) R: A Language and Environment for Statistical Computing (R Core Team, Vienna, Austria)
- 41.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: Visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.