

Abstract
Organic aerosols are ubiquitous in the atmosphere and play a central role in climate, air quality, and public health. The aerosol size distribution is key in determining its optical properties and cloud condensation nucleus activity. The dominant portion of organic aerosol is formed through gas-phase oxidation of volatile organic compounds, so-called secondary organic aerosols (SOAs). Typical experimental measurements of SOA formation include total SOA mass and atomic oxygen-to-carbon ratio. These measurements, alone, are generally insufficient to reveal the extent to which condensed-phase reactions occur in conjunction with the multigeneration gas-phase photooxidation. Combining laboratory chamber experiments and kinetic gas-particle modeling for the dodecane SOA system, here we show that the presence of particle-phase chemistry is reflected in the evolution of the SOA size distribution as well as its mass concentration. Particle-phase reactions are predicted to occur mainly at the particle surface, and the reaction products contribute more than half of the SOA mass. Chamber photooxidation with a midexperiment aldehyde injection confirms that heterogeneous reaction of aldehydes with organic hydroperoxides forming peroxyhemiacetals can lead to a large increase in SOA mass. Although experiments need to be conducted with other SOA precursor hydrocarbons, current results demonstrate coupling between particle-phase chemistry and size distribution dynamics in the formation of SOAs, thereby opening up an avenue for analysis of the SOA formation process.
Keywords: gas–particle interactions, heterogeneous chemistry, intermediate volatility organic compounds, alkane
Laboratory chamber experiments are the basis on which gas-phase photooxidation mechanisms and subsequent formation and evolution of secondary organic aerosols (SOAs) are evaluated. Despite major advances in understanding of the mechanisms of SOA formation, the long-standing question of the relative contributions of gas- vs. particle-phase routes to SOA formation and aging for the major classes of SOA-forming hydrocarbons is still unresolved (1). Photooxidation of parent hydrocarbons and subsequent multigeneration gas-phase chemistry produce an array of semivolatile organic compounds (SVOCs) that partition into the particle phase (2, 3). At the same time, particle-phase reactions involving these condensed SVOCs can lead to formation of low-volatility compounds, such as oligomers and other high molecular mass compounds (4–6). The extent to which particle-phase chemistry is important in SOA formation for each of the major classes of SOA precursors remains unclear. We show here that the evolution of the particle-size distribution, a measurement routinely made in chamber experiments of SOA formation, holds a key to evaluating the importance of particle-phase chemistry.
Current atmospheric aerosol models underpredict ambient SOA mass (1). The missing SOAs can be partly explained by intermediate volatility organic compounds (IVOCs) that may be emitted initially as primary organic aerosol but evaporate upon dilution and are subsequently oxidized in the gas phase, yielding substantial SOA mass (7, 8). Recent modeling studies have demonstrated that IVOCs are expected to be a major precursor of SOAs in megacity outflows (9). Long-chain alkanes, mainly emitted from incomplete fuel combustion (10), constitute a substantial fraction of IVOCs. Here we use dodecane (C12H26), a surrogate compound for IVOCs, to study the connection between the chemical mechanism of SOA formation and the evolution of the size distribution.
Results and Discussion
Dodecane Photooxidation Experiments and Modeling.
The photooxidation of dodecane and subsequent SOA formation is studied using the Caltech Environmental Chamber (11, 12). Dodecane at initial concentration of 34 parts per billion (ppb) in the presence of ammonium sulfate seed particles under dry conditions [relative humidity (RH) < 5%] was oxidized by OH radicals at low concentrations of NOx typical of nonurban conditions. The kinetic multilayer model of gas-particle interactions in aerosols and clouds (KM-GAP) (13) is used to simulate the evolution of SOA mass, the organic atomic oxygen-to-carbon (O:C) ratio, and particle-size distribution in the chamber experiments (Fig. 1). The model treats the following processes explicitly: gas-phase diffusion, reversible adsorption, bulk diffusion, and chemical reactions in the gas and particle phases. The model also accounts for the loss of gas-phase SVOCs to the chamber Teflon wall (14, 15) based on measurements for representative compounds in separate experiments (SI Materials and Methods, Gas-phase wall loss, Table S1 and S2). The physical state of the particle bulk is assumed to be semisolid with an average bulk diffusivity of 10−12 cm2⋅s−1 [a typical value for a semisolid state (16) (SI Materials and Methods)], consistent with observations that long-chain alkane-derived SOA particles bounce moderately on the smooth plates of an inertial impactor indicating behavior between that of liquid and glassy particles (17).
Fig. 1.
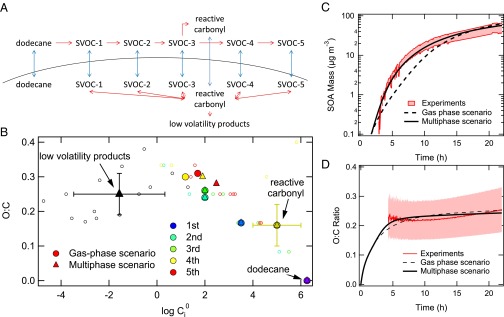
Modeling secondary organic aerosol formation from dodecane photooxidation. (A) Schematic of the mass transport (blue arrows) and chemical reactions (red arrows) in the dodecane SOA system. Multigeneration of SVOCs is considered in the gas phase. The multiphase scenario includes second-order reactions (red double arrows) between reactive carbonyl and SVOCs forming low-volatility products in the particle phase. (B) O:C ratios for products of dodecane oxidation vs. their predicted gas-phase saturation concentration (μg⋅m−3) over the pure (subcooled) liquids (Ci0). The smaller symbols indicate individual products predicted in the dodecane photooxidation chemical mechanism developed by Yee et al. (11). Color indicates the gas-phase generation in which compound is formed. These compounds include reactive carbonyl compounds (fourth generation). Black symbols indicate particle-phase products. The large solid circles and triangles indicate the surrogate SVOC compounds in the gas-phase and multiphase scenarios, respectively. Measured (red) and modeled SOA mass concentration (C) and O:C ratio (D) with the gas-phase (dashed line) and multiphase (solid line) scenarios, respectively. The upper and lower red lines in the C correspond to upper and lower bound wall loss corrections, respectively. The measured O:C ratio is subject to ±30% uncertainty (12).
In the gas phase, SVOCs resulting from up to five generations of OH oxidation are considered (Fig. 1A). Fig 1B shows the span of O:C ratio and gas-phase saturation concentrations for the surrogate SVOCs based on the dodecane oxidation mechanism of Yee et al. (11). Some of the fourth generation products have been established to be multifunctional carbonyl compounds, aldehydes, and ketones. The aldehydes can react with hydroperoxide, hydroxyl, and peroxycarboxylic acid groups, forming peroxyhemiacetal (PHA), hemiacetal, and acylperoxyhemiacetal, respectively, in the particle phase (2, 11, 18). Ketone functional groups can react with hydroxyl groups, catalyzed by acids, forming a variety of oligomers (4). These products have significantly lower volatility than their gas-phase precursors, as shown for PHA by the black triangles in Fig. 1B. We evaluate the potential effect of particle-phase chemistry on SOA formation by simulating two distinct scenarios. In the “multiphase” model scenario, the formation of low-volatility products arising from particle-phase chemistry is considered whereas, in the “gas-phase” scenario, only gas-phase reactions are accounted for. In the kinetic model simulations, the volatility and O:C ratio of each surrogate product are chosen to lie in the range of those of corresponding individual compounds, as shown by the large solid and triangle markers in Fig. 1B for the gas and multiphase scenarios, respectively. Note that the O:C ratios of later generation gas-phase and particle-phase products are both ∼0.25. Fig. 1C shows the experimental observations of SOA mass concentration for the cases of two limiting particle-wall loss corrections (19, 20): (i) wall-deposited particles are assumed to cease interaction with the gas phase (lower red line), and (ii) wall-deposited particles are assumed to continue interacting with the gas phase as if they were still suspended (upper red line). Fig. 1D shows the measured O:C ratio (12). Both scenarios, gas-phase (dashed lines) and multiphase (solid lines), are able to match the observed SOA mass concentration and O:C ratio within the experimental uncertainties, even though the gas-phase scenario initially underestimates SOA mass for times up to ∼10 h. This result exemplifies the ambiguity that arises in assessing the chemical routes to SOA formation based solely on SOA mass and O:C ratio.
Fig. 2A shows the measured evolution of the particle-size distribution. The mean particle diameter grows from ∼60 nm to ∼180 nm over ∼20 h. The simulated size distribution for the gas-phase scenario is shown in Fig. 2B. In contrast to the observed size distribution, the gas-phase scenario predicts a broad size distribution that is a characteristic also found for quasi-equilibrium growth mechanism (21), in which the condensed SVOCs equilibrate quickly with the gas phase. When SVOCs equilibrate with the particle population, they tend, on average, to partition to larger particles because of their greater absorption capacity and the reduction in equilibrium vapor pressure over the surface compared with that over smaller particles (the Kelvin effect). By contrast, the simulated number size distribution for the multiphase scenario captures the characteristic narrowing of the measured size distribution (Figs. 2 C and D, Fig. S1). The evolution of the volume size distribution is also well reproduced by the multiphase scenario (Fig. S2). Evaporation and recondensation of SVOCs tend to be inhibited in this case owing to the formation of low volatility products in the particle phase. The narrowing of the size distribution is characteristic of kinetically limited growth, in which the mass of condensing material is distributed proportional to the particle surface area (21, 22). The second-order reaction rate coefficient between reactive carbonyl and SVOCs (kBR), which is estimated to be 12 M−1⋅s−1 (2 × 10−20 cm3⋅s−1), is found to be the most sensitive parameter in controlling the evolution of the particle size distribution. The evolution of SOA mass and number size distribution obtained with an initial dodecane concentration of 8 ppb was also modeled well using the same parameters of the multiphase scenario (Fig. S3).
Fig. 2.

Evolution of particle number size distribution. Measured (with the upper-bound wall loss correction) (A) and modeled by KM-GAP with gas-phase (B) and multiphase (C) scenarios. (D) Initial number size distribution (black) and measured (red solid line) and modeled number size distribution after 20 h in the gas-phase (dotted line) and multiphase (dashed line) scenarios.
Fig. 3 shows the simulated evolution of the mass concentration ratio of oxidation products in the particle (Fig. 3A) and gas phase (Fig. 3B) in the multiphase scenario. During the first several hours, the particle phase is dominated by first- to third-generation SVOCs, which are progressively converted to higher generation SVOCs in the gas phase and low volatility products in the particle phase. The contribution of particle-phase products to the total SOA budget is predicted to exceed 60% after ∼5 h. The dominance of low-volatility particle-phase products is consistent with previous studies, in which peroxyhemiacetals were found to be major products in SOA derived from oxidation of alkenes (1-tetradecene) (23, 24), aromatic hydrocarbons (toluene) (25, 26), and monoterpenes (α- and β-pinene) (27).
Fig. 3.

Relative contribution and distribution of gas-phase and particle-phase reaction products. Mass concentration ratios of multigeneration dodecane gas-phase oxidation products (colored) and particle phase products (gray) in the particle phase (A) and the gas phase (B), modeled with the multiphase scenario. (C) Modeled radial profile of the production rate (cm−3⋅s−1) of particle-phase products for an initially 35-nm radius particle. The solid black line indicates the evolution of the particle radius. (D) Temporal evolution of a C6-carboxylic acid (tracer for aldehyde/reactive carbonyl) in the gas phase monitored by the CIMS. Two characteristic tracers of peroxyhemiacetal formation in the particle phase are measured by the AMS at m/z 183 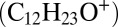 and m/z 215
and m/z 215 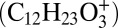 .
.
Fig. 3C shows the simulated radial profile of the production rate of low-volatility products along with the evolution of particle radius. The production rate becomes substantial, with a rate of >2 × 1017 cm−3⋅s−1 in the surface layer after ∼4 h when SOA mass starts growing (Fig. 1C). The particle-phase reactions are predicted to occur mainly at and near the surface, consistent with a product analysis study suggesting that PHA formation should occur mainly at the particle surface (24). Fig. 3D shows the measured evolution of gas-phase C6-carboxylic acid, which is a fourth-generation product and a proxy for the expected formation of aldehydes (11). Aerosol mass spectrometer (AMS) ions at mass-to-charge ratios (m/z) of 183 and 215 are also shown, which are representative fragments from the carbonyl hydroperoxide (CARBROOH; C12H24O3) and its derived PHA (11). The onset of gas-phase aldehyde, particle-phase PHA, SOA mass growth, and production rate of low-volatility products at ∼4 h suggests that the formation of PHA triggers initial SOA growth.
Tridecanal Injection Experiment.
To evaluate the hypothesis concerning the role of PHA production in SOA formation, we have conducted an additional chamber experiment with intentional injection of the aldehyde tridecanal (Fig. 4). Dodecane (initial concentration of 239 ppb) was photooxidized for 4 h under low-NOx and dry conditions in the presence of ammonium sulfate seed particles, at which time chamber UV lights were turned off to halt production of OH radicals, ceasing gas-phase chemistry and SOA growth. After 6 h, injection of 9.5 ppb tridecanal into the chamber led to a rapid increase in organic mass (measured by the AMS) of ∼45 μg⋅m−3 within 1 h. Simultaneously, the tracer ions for gas-phase CARBROOH decreased and that for particle-phase CARBROOH-derived PHA increased. The same trend was also observed for organic hydroperoxide and its derived PHA (Fig. S4). Direct analysis in real time mass spectrometry (DART-MS) spectra confirm the formation of high molecular mass compounds such as PHA and oligomers (Fig. S5 and Table S3). Note that the observed SOA growth cannot be explained by physical uptake of tridecanal, as its vapor pressure is too high (C0 = ∼105 μg⋅m−3).
Fig. 4.

Intentional aldehyde (tridecanal) injection experiments. SOA mass concentration and carbonyl hydroperoxide (CARBROOH; C12H24O3) signal monitored by CIMS at m/z 301 and CARBROOH-derived peroxyhemiacetals monitored by the AMS at m/z 183 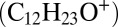 and m/z 215
and m/z 215 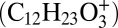 . UV lights are switched off after 4 h, and 9.5 ppb tridecanal is injected to the chamber at 6 h.
. UV lights are switched off after 4 h, and 9.5 ppb tridecanal is injected to the chamber at 6 h.
As the observed particle growth is predicted to be essentially a result of the particle-phase reaction, the second-order reaction rate between tridecanal and SVOCs can be estimated to lie in the range 0.3–12 M−1⋅s−1 (SI Materials and Methods, Estimation of the particle-phase reaction rate coefficient). This range is consistent with the estimated kBR of 12 M−1⋅s−1 derived from the KM-GAP modeling of the dodecane photooxidation experiments. Note that literature values of kBR for uncatalyzed reactions of various hydroperoxides and aldehydes in different solvents range from 10−4 to 0.06 M−1⋅s−1 (18, 28). The value of kBR inferred from the observations indicates that the reactions must be catalyzed (by two orders of magnitude) by the presence of acids (4, 18, 24) generated in the low-NOx dodecane mechanism (11). This magnitude of enhancement seems reasonable, as in the case of hemiacetal formation from acetaldehyde plus methanol, the presence of 1 M acetic acid increases the rate constants by a factor of ∼250 (18).
Summary and Implications.
The results of the current work have a number of implications for SOA models. Although the dynamics of an aerosol size distribution reflects the mechanism of growth (22, 29), we demonstrate here that it provides a key constraint in interpreting laboratory and ambient SOA formation. Aldehyde injection experiments suggest that peroxyhemiacetal formation by heterogeneous reactions between aldehydes and organic hydroperoxides can have a major impact on SOA formation. This work, although carried out specifically for the long chain alkane dodecane, is expected to be widely applicable to other major classes of SOA precursors. SOA consists of a myriad of organic compounds containing carbonyl, hydroxyl, and carboxyl groups (among other functional groups), which can generally undergo heterogeneous/multiphase reactions forming low-volatility products such as oligomers and other high molecular mass compounds (18). The importance of such a peroxyhemiacetal formation pathway depends on the reaction rate constants and concentrations of reactants as well as on particle acidity and hygroscopicity because the reactions can be acid-catalyzed (4, 30, 31) or hydration-based (32), depending on the environmental conditions and particle composition. Heterogeneous reactivity of SOA particles can be regulated by relative humidity and temperature, which will affect particle viscosity and bulk diffusivity (16, 33). If particle-phase chemistry is indeed central to SOA growth in general, the size-resolved SOA formation is better described in terms of kinetically limited condensational growth, rather than solely by thermodynamic equilibrium partitioning (22, 34). Analysis of field and laboratory data on size-distribution dynamics and heterogeneous chemistry for various SOA precursors and environmental conditions may lead to more robust predictions of chemical composition and particle-size distribution, with relevance to properties such as radiative forcing and cloud condensation nuclei activity.
Materials and Methods
Chamber Experiments.
Experiments were carried out in the Teflon reactors in the Caltech Environmental Chamber. Aqueous H2O2 solution was evaporated into the chamber, followed by the atomization of an aqueous ammonium sulfate solution generating seed particles, which were subsequently dried. Liquid dodecane was evaporated into the chamber to achieve 34 ppb gas-phase mixing ratio of dodecane. After an hour of mixing, the blacklights were turned on, initiating generation of the OH radical from H2O2 photolysis. The gas-phase composition of oxidation products was monitored using a chemical ionization mass spectrometer (CIMS) (11). The particle-phase chemical composition, including the atomic oxygen-to-carbon (O:C) ratio of SOA, was measured by an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) (12). In addition, particle-phase chemical composition was analyzed offline using direct analysis in real time mass spectrometry (DART-MS). Particle number size distribution was measured using a cylindrical differential mobility analyzer coupled to a condensation particle counter. More details are available in SI Materials and Methods.
Kinetic Modeling.
The kinetic multilayer model of gas-particle interactions in aerosols and clouds (KM-GAP) (13) is used for simulations. For size-resolved simulations, the bin method with full-moving size structure is used. KM-GAP consists of a number of compartments and layers in which semivolatile species can undergo mass transport and chemical reactions in the gas and particle phases. The required kinetic parameters for surrogate SVOCs include the gas-phase first-order reaction rate coefficients, the surface accommodation coefficient, the molecular desorption lifetime, gas and bulk diffusion coefficients, and the second-order bulk reaction rate coefficient for the reaction of reactive carbonyl with SVOCs (Table S1 and S2). The dynamics of compound concentrations in the gas and particle phases and of the aerosol size distribution were computed by solving the mass balance, transfer, and reaction rate equations. More details are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Xuan Zhang and Matt Coggon for assistance in the experiments. This work was supported by US Department of Energy Grant DE-SC0006626 and National Science Foundation Grant AGS-1057183. M.S. is supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307501110/-/DCSupplemental.
References
- 1.Hallquist M, et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos Chem Phys. 2009;9(14):5155–5235. [Google Scholar]
- 2.Kroll JH, Seinfeld JH. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos Environ. 2008;42(16):3593–3624. [Google Scholar]
- 3.Jimenez JL, et al. Evolution of organic aerosols in the atmosphere. Science. 2009;326(5959):1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- 4.Jang MS, Czoschke NM, Lee S, Kamens RM. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science. 2002;298(5594):814–817. doi: 10.1126/science.1075798. [DOI] [PubMed] [Google Scholar]
- 5.Kalberer M, et al. Identification of polymers as major components of atmospheric organic aerosols. Science. 2004;303(5664):1659–1662. doi: 10.1126/science.1092185. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, et al. Atmospheric nanoparticles formed from heterogeneous reactions of organics. Nat Geosci. 2010;3(4):238–242. [Google Scholar]
- 7.Robinson AL, et al. Rethinking organic aerosols: semivolatile emissions and photochemical aging. Science. 2007;315(5816):1259–1262. doi: 10.1126/science.1133061. [DOI] [PubMed] [Google Scholar]
- 8.Presto AA, et al. Intermediate-volatility organic compounds: A potential source of ambient oxidized organic aerosol. Environ Sci Technol. 2009;43(13):4744–4749. doi: 10.1021/es803219q. [DOI] [PubMed] [Google Scholar]
- 9.Lee-Taylor J, et al. Explicit modeling of organic chemistry and secondary organic aerosol partitioning for Mexico City and its outflow plume. Atmos Chem Phys. 2011;11(24):13219–13241. [Google Scholar]
- 10.Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT. Measurement of emissions from air pollution sources. 2. C1 through C30 organic compounds from medium duty diesel trucks. Environ Sci Technol. 1999;33(10):1578–1587. [Google Scholar]
- 11.Yee LD, et al. Secondary organic aerosol formation from low-NOx photooxidation of dodecane: Evolution of multigeneration gas-phase chemistry and aerosol composition. J Phys Chem A. 2012;116(24):6211–6230. doi: 10.1021/jp211531h. [DOI] [PubMed] [Google Scholar]
- 12.Craven JS, et al. Analysis of secondary organic aerosol formation and aging using positive matrix factorization of high-resolution aerosol mass spectra: Application to the dodecane low-NOx system. Atmos Chem Phys. 2012;12(24):11795–11817. [Google Scholar]
- 13.Shiraiwa M, Pfrang C, Koop T, Pöschl U. Kinetic multi-layer model of gas-particle interactions in aerosols and clouds (KM-GAP): Linking condensation, evaporation and chemical reactions of organics, oxidants and water. Atmos Chem Phys. 2012;12(5):2777–2794. [Google Scholar]
- 14.Matsunaga A, Ziemann PJ. Gas-wall partitioning of organic compounds in a Teflon film chamber and potential effects on reaction product and aerosol yield measurements. Aerosol Sci Technol. 2010;44(10):881–892. [Google Scholar]
- 15.Loza CL, et al. Characterization of vapor wall loss in laboratory chambers. Environ Sci Technol. 2010;44(13):5074–5078. doi: 10.1021/es100727v. [DOI] [PubMed] [Google Scholar]
- 16.Shiraiwa M, Ammann M, Koop T, Pöschl U. Gas uptake and chemical aging of semisolid organic aerosol particles. Proc Natl Acad Sci USA. 2011;108(27):11003–11008. doi: 10.1073/pnas.1103045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saukko E, et al. Humidity-dependent phase state of SOA particles from biogenic and anthropogenic precursors. Atmos Chem Phys. 2012;12(16):7517–7529. [Google Scholar]
- 18.Ziemann PJ, Atkinson R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem Soc Rev. 2012;41(19):6582–6605. doi: 10.1039/c2cs35122f. [DOI] [PubMed] [Google Scholar]
- 19.Loza CL, et al. Chemical aging of m-xylene secondary organic aerosol: Laboratory chamber study. Atmos Chem Phys. 2012;12(1):151–167. [Google Scholar]
- 20.Weitkamp EA, Sage AM, Pierce JR, Donahue NM, Robinson AL. Organic aerosol formation from photochemical oxidation of diesel exhaust in a smog chamber. Environ Sci Technol. 2007;41(20):6969–6975. doi: 10.1021/es070193r. [DOI] [PubMed] [Google Scholar]
- 21.Shiraiwa M, Seinfeld JH. Equilibration timescale of atmospheric secondary organic aerosol partitioning. Geophys Res Lett. 2012;39:L24801. [Google Scholar]
- 22.Riipinen I, et al. Organic condensation: A vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations. Atmos Chem Phys. 2011;11(8):3865–3878. [Google Scholar]
- 23.Tobias HJ, Docherty KS, Beving DE, Ziemann PJ. Effect of relative humidity on the chemical composition of secondary organic aerosol formed from reactions of 1-tetradecene and O3. Environ Sci Technol. 2000;34(11):2116–2125. [Google Scholar]
- 24.Tobias HJ, Ziemann PJ. Thermal desorption mass spectrometric analysis of organic aerosol formed from reactions of 1-tetradecene and O3 in the presence of alcohols and carboxylic acids. Environ Sci Technol. 2000;34(11):2105–2115. [Google Scholar]
- 25.Johnson D, Jenkin ME, Wirtz K, Martin-Reviejo M. Simulating the formation of secondary organic aerosol from the photooxidation of toluene. Environ Chem. 2004;1(3):150–165. [Google Scholar]
- 26.Sato K, Hatakeyama S, Imamura T. Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition. J Phys Chem A. 2007;111(39):9796–9808. doi: 10.1021/jp071419f. [DOI] [PubMed] [Google Scholar]
- 27.Docherty KS, Wu W, Lim YB, Ziemann PJ. Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O3. Environ Sci Technol. 2005;39(11):4049–4059. doi: 10.1021/es050228s. [DOI] [PubMed] [Google Scholar]
- 28.Capouet M, et al. (2008) Modeling aerosol formation in alpha-pinene photo-oxidation experiments. J Geophys Res Atmos 113(D2):D02308.
- 29.McMurry PH, Wilson JC. Growth laws for the formation of secondary ambient aerosols: Implications for chemical conversion mechanisms. Atmos Environ. 1982;16(1):121–134. [Google Scholar]
- 30.Zhao J, Levitt NP, Zhang R. (2005) Heterogeneous chemistry of octanal and 2,4-hexadienal with sulfuric acid. Geophys Res Lett 32(9):L09802.
- 31.Iinuma Y, Boge O, Gnauk T, Herrmann H. Aerosol-chamber study of the alpha-pinene/O3 reaction: Influence of particle acidity on aerosol yields and products. Atmos Environ. 2004;38(5):761–773. [Google Scholar]
- 32.Zhao J, Levitt NP, Zhang R, Chen J. Heterogeneous reactions of methylglyoxal in acidic media: Implications for secondary organic aerosol formation. Environ Sci Technol. 2006;40(24):7682–7687. doi: 10.1021/es060610k. [DOI] [PubMed] [Google Scholar]
- 33.Kuwata M, Martin ST. Phase of atmospheric secondary organic material affects its reactivity. Proc Natl Acad Sci USA. 2012;109(43):17354–17359. doi: 10.1073/pnas.1209071109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perraud V, et al. Nonequilibrium atmospheric secondary organic aerosol formation and growth. Proc Natl Acad Sci USA. 2012;109(8):2836–2841. doi: 10.1073/pnas.1119909109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.