

Significance
Voltage-gated sodium (Nav) channels contribute to physiological and pathophysiological electrical signaling in nerve and muscle cells. Because Nav channel isoforms exhibit tissue-specific expression, subtype selective modulation of this channel family provides important drug development opportunities. However, most available Nav channel modulators are unable to distinguish between Nav channel subtypes, which limits their therapeutic utility because of cardiac or nervous system toxicity. This study describes a new class of subtype selective Nav channel inhibitors that interact with a region of the channel that controls voltage sensitivity. This interaction site may enable development of selective therapeutic interventions with reduced potential for toxicity.
Abstract
Voltage-gated sodium (Nav) channels play a fundamental role in the generation and propagation of electrical impulses in excitable cells. Here we describe two unique structurally related nanomolar potent small molecule Nav channel inhibitors that exhibit up to 1,000-fold selectivity for human Nav1.3/Nav1.1 (ICA-121431, IC50, 19 nM) or Nav1.7 (PF-04856264, IC50, 28 nM) vs. other TTX-sensitive or resistant (i.e., Nav1.5) sodium channels. Using both chimeras and single point mutations, we demonstrate that this unique class of sodium channel inhibitor interacts with the S1–S4 voltage sensor segment of homologous Domain 4. Amino acid residues in the “extracellular” facing regions of the S2 and S3 transmembrane segments of Nav1.3 and Nav1.7 seem to be major determinants of Nav subtype selectivity and to confer differences in species sensitivity to these inhibitors. The unique interaction region on the Domain 4 voltage sensor segment is distinct from the structural domains forming the channel pore, as well as previously characterized interaction sites for other small molecule inhibitors, including local anesthetics and TTX. However, this interaction region does include at least one amino acid residue [E1559 (Nav1.3)/D1586 (Nav1.7)] that is important for Site 3 α-scorpion and anemone polypeptide toxin modulators of Nav channel inactivation. The present study provides a potential framework for identifying subtype selective small molecule sodium channel inhibitors targeting interaction sites away from the pore region.
Voltage-gated sodium (Nav) channels play an important role in the generation and propagation of electrical signals in excitable cells (1–3). Eukaryotic Nav channels are heteromeric membrane proteins composed of a pore-forming α-subunit and auxiliary β-subunits (3, 4). The mammalian genome encodes nine distinct α (Nav1.1–1.9) and four β subunits (3). The α-subunit comprises four homologous domains (D1–D4), each of which contains six transmembrane segments (S1–S6) (3–5). The S5 and S6 segments form the central pore separated by the SS1 and SS2 segments, which form the ion selectivity filter at its extracellular end. The S1 to S4 segments form the voltage sensor (3–5).
Both naturally occurring and synthetic pharmacological modulators of sodium channel have been identified (6–11), and for many, their site of interaction has been defined. For example, the marine toxin TTX inhibits several Nav subtypes by interacting with amino acid residues within the SS1–SS2 segments that define the outer pore of the channel (12, 13). In contrast, the polypeptide α- and β-scorpion venom toxins, which enhance sodium channel activation or delay inactivation, and spider venom toxin sodium channel inhibitors like Protx II interact with specific residues on the S1–S4 voltage sensor regions within homologous Domain 2 (i.e., β-scorpion toxins, Protx II) or Domain 4 (i.e., α-scorpion toxins, anemone toxins) of the channel (8, 14–18). Many synthetic small molecule inhibitors of Nav channels, including local anesthetic, antiepileptic, and antiarrhythmic agents, are believed to interact with amino acid residues within the S6 segment in Domain 4, which forms part of the pore lining and is structurally highly conserved across subclasses of mammalian Nav channels (11, 19–22). This structural homology probably accounts for many clinically used local anesthetics and related antiepileptic and antiarrhythmic inhibitors, exhibiting little or no selectivity across the nine subtypes of mammalian Nav channels (23). In the clinic this absence of subtype selectivity can result in toxicities associated with unwanted interactions with off-target Nav channels (e.g., cardiac toxicity due to inhibition of cardiac Nav1.5 channels) (24, 25). Therefore, because of their importance in normal physiology and pathophysiology, identification of selective pharmacological modulators of Nav channels is of considerable interest to the scientific and medical communities (9, 23, 26–29). For example, in addition to the therapeutic utility of sodium channel inhibitors described above, there has been recent interest in potentially targeting inhibition of specific Nav channel subtypes (i.e., Nav1.7, Nav1.8, and Nav1.3) for the treatment of pain (9, 30–32).
The present study describes the characterization of a class of subtype selective sodium channel inhibitor that interacts with a unique site on the voltage sensor region of homologous Domain 4. This inhibitory interaction site differs from previously reported inhibitor binding sites for TTX and local anesthetic-like modulators (11, 12).
Results
Characterization of a Subtype Selective Inhibitor of Human Nav1.3 Channels.
During a Nav channel inhibitor drug candidate discovery program, a previously uncharacterized agent, ICA-121431 [2,2-diphenyl-N-(4-(N-thiazol-2-ylsulfamoyl)phenyl) acetamide] was found to inhibit human TTX-sensitive Nav1.3 sodium channels stably expressed in HEK-293 cells (Fig. 1A). As with many known voltage-dependent sodium channel inhibitors, inhibition of human Nav1.3 by ICA-121431 was found to be dependent on channel gating state. When Nav1.3 currents were elicited by 20-ms depolarizing voltage steps from −120 mV, at which channels are predominantly in a resting closed state, ICA-121431 had a negligible effect on current amplitude at 1 µM (Fig. 1B). In contrast, when a test pulse was preceded by an 8-s conditioning voltage step to a membrane potential that resulted in approximately half of the channels being in an inactivated state, 1 µM ICA-121431 produced 92% ± 4% (n = 4) reduction in current amplitude (Fig. 1B). A preferential interaction with inactivated Nav1.3 channels was further supported by the finding that ICA-121431 elicited a concentration-dependent hyperpolarizing shift in the voltage dependence of inactivation (Fig. 1C).
Fig. 1.
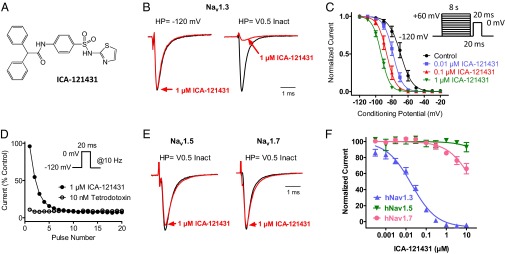
Properties of Nav channel inhibition by ICA-121431. (A) Structure of ICA-121431. (B) Current traces recorded on PatchXpress showing effect of 1 µM ICA-121431 on human Nav1.3 current amplitude elicited by a 20-ms voltage step to 0 mV from −120 mV or after an 8-s condition voltage step (HP) to −60 mV to inactivate approximately half of the available channels. Current traces are normalized so that control traces have same relative amplitude. (C) Voltage dependence of human Nav1.3 inactivation in the presence and absence of ICA-121431. Peak current amplitudes were measured during test pulses to 0 mV after an 8-s conditioning depolarization to the indicated potentials in the absence and presence of 0.01, 0.1, and 1µM ICA-121431. Values from individual cells were normalized to the largest amplitude current after a prepulse to the most hyperpolarized conditioning potential (−120 mV). Normalized inactivation curves were fit with a Boltzmann equation, whereby the half inactivation voltage in the presence of ICA-121431 was −78 ± 2 mV (0.01 µM), −86 ± 2 mV (0.1 µM), and −93 ± 2 mV (1 µM) compared with −67 ± 2 mV in the absence of inhibitor. (D) Use dependent inhibition of human Nav1.3 by 1 µM ICA-121431. Plot of current amplitude (normalized to control) during a train of 20-ms voltage steps from −120 mV to 0 mV given at 10 Hz in the presence of 1 µM ICA-121431 or 10 nM TTX. (E) PatchXpress recording showing effect of 1 µM ICA-121431 on human Nav1.5 and Nav1.7 currents elicited by a 20-ms voltage step to 0 mV after an 8-s condition voltage step to inactivate approximately half of the available channels. (F) Concentration dependence of human Nav1.3, Nav1.5, and Nav1.7 inhibition by ICA-121431 [IC50 18 ± 5 nM (n = 6) for Nav1.3 and >10 µM for Nav1.5 (n = 4) and Nav1.7 (n = 6), respectively]. Inhibition of sodium currents was recorded on IonWorks Quattro using a protocol comprising a 500-ms step from −120 mV to 0 mV to inactivate the channels, followed after a 100-ms recovery at −100 mV by a 20-ms test pulse to 0 mV to define inhibition.
Although ICA-121431 produced negligible inhibition when test pulses were applied from a −120 mV holding potential at a relatively low stimulation frequency (i.e., 0.05 Hz), cumulative (or use-dependent) inhibition became evident when stimulation frequency was increased to 10 Hz (Fig. 1D). This use-dependent inhibition of Nav1.3 by ICA-121431 clearly differs from the absence of frequency-dependent inhibition in the presence of TTX (Fig. 1D).
Although state- and/or frequency-dependent inhibition is a relatively common characteristic of small molecule voltage-gated sodium channel inhibitors (33), a more unique finding was the marked subtype selectivity exhibited by ICA-121431. Fig. 1E shows that in contrast to Nav1.3, 1 µM ICA-121431 produced little or no inhibition of human Nav1.7 or Nav1.5 currents. Selective inhibition of Nav1.3 by ICA-121431 is further illustrated in Fig. 1F, which shows the concentration dependence of inhibition of human Nav1.3, Nav1.5, and Nav1.7 measured on the IonWorks Quattro automated electrophysiology using a 500-ms conditioning pulse to 0 mV to promote availability of the inactivated state. Under these conditions, the IC50 for ICA-121431 inhibition of Nav1.3 was 19 ± 5 nM (n = 6), compared with >10 µM for both human Nav1.5 and Nav1.7. When additional Nav channel subtypes were examined, the selectivity profile of ICA-121431 was found to cover a spectrum of inhibitor potencies, with human Nav1.3 and Nav1.1 being the most sensitive to inhibition (IC50s <20 nM), Nav1.2 having an intermediate sensitivity (IC50 240 nM), whereas Nav1.4, Nav1.5, Nav1.6, Nav1.7, and Nav1.8 were relatively insensitive to inhibition (IC50s >10 µM) (Fig. S1 and Table S1).
ICA-121431 Interacts with Homologous Domain 4 of Nav1.3.
Given the potency and selectivity of ICA-121431, we were interested in further understanding its interaction with human Nav1.3 and the amino acid residues that may define selectivity for this channel over other related Nav channels, including Nav1.7 and Nav1.5. Our initial attempts focused on establishing whether inhibition resulted from interaction with a specific domain of the sodium channel. Restriction sites were introduced into the nucleotide sequences between the Domain 1–4 repeats of Nav1.3, Nav1.7, and Nav1.5 (details of sites in Fig. S2). Chimeras of Nav1.3 domains combined with varying homologous domains of either Nav1.7 or Nav1.5 were constructed and expressed in HEK-293 cells and then evaluated for inhibition by ICA-121431 using the 8-s half inactivation pulse protocol on conventional patch clamp described previously. Fig. 2A shows that replacement of Domain 3 and 4 of Nav1.3 with the equivalent domains from Nav1.7 resulted in a chimeric channel (Domains 1–4 = 3,3,7,7) that exhibited very weak sensitivity to inhibition by ICA-121431 (12% ± 4% at 3 µM, n = 3). This data suggested that the interaction site may lie somewhere within homologous Domains 3 and 4. Another chimera in which only Domain 4 of Nav1.3 was replaced with the equivalent domain from Nav1.5 (Domains 1–4 = 3,3,3,5) also lacked sensitivity to inhibition by ICA-121431 (19% ± 4% at 3 µM, n = 3). These findings suggest that Domain 4 may contain an interaction site for ICA-121431 inhibition. Further support of this hypothesis was demonstrated when a chimera comprising Domains 1–3 of Nav1.5 and Domain 4 of Nav1.3 (Domains 1–4 = 5,5,5,3) exhibited relatively potent inhibition by ICA-121431 (45% ± 2% at 0.1 µM and 90% ± 3% at 1 µM, n = 2) (Fig. 2A), which is ∼fivefold less potent than Nav1.3 but more than 500-fold more potent than inhibition of Nav1.5. It further suggests that the entire ICA-121431 interaction site may be contained within Domain 4 of Nav1.3.
Fig. 2.

(A) Defining interaction region on Nav1.3 required for inhibition by ICA-121431. Relative potency of ICA-121431 inhibition of chimeric sodium channels comprising various domains of Nav1.3 (blue) combined with domains from either Nav1.5 (green) or Nav1.7 (pink). Potent inhibition was only observed in chimeras containing a Nav1.3 Domain 4. (B) Concentration dependence of ICA-121431 inhibition of Nav1.3 sodium channel chimeras in which subregions of Domain 4 have been replaced with equivalent regions from Nav1.5 are compared with inhibition of unmodified Nav1.3 (blue) or Nav1.5 (green). (C) Concentration dependence of ICA-121431 inhibition of Nav1.3 sodium channel chimeras in which subregions of Domain 4 have been replaced with equivalent regions from Nav1.7 are compared with inhibition of unmodified Nav1.3 (blue) or Nav1.7 (pink). (D) IC50s for ICA-121431 inhibition of various Nav1.3-Nav1.5 or Nav1.3-Nav1.7 Domain 4 chimeras compared with unmodified Nav1.3, Nav1.5, and Nav1.7. Values are means ± SEM (n = 4–22). Inhibition of sodium currents in B, C, and D were recorded using conventional patch clamp and determined using an 8-s conditioning voltage step from −120 mV to a voltage producing half-maximal inactivation followed after a 20-ms recovery at −100 mV by a 20-ms test pulse to 0 mV.
It should be noted that the chimeras described above do exhibit significant differences in the voltage dependence of inactivation relative to unmodified Nav1.3 (midpoints potentials of −73 ± 1 mV for 3,3,7,7 chimera, −67 ± 1 mV for 3,3,3,5 chimera; −82 ± 2 mV for 5,5,5,3 chimera compared with −58 ±1 mV for wild-type Nav1.3). However, we believe that by using an 8-s conditioning pulse to the midpoint of inactivation protocol set appropriately for each of the chimeras being evaluated, we minimized the contribution of differences in voltage dependence of inactivation to apparent potency differences. Support for this comes from the finding that 1 µM amitriptyline [a nonselective Nav channel inhibitor that interacts preferentially with inactivated channels (34)] produced relatively similar inhibition of the chimeras compared with unmodified Nav1.3; 40% for 3,3,7,7 chimera, 60% for 3,3,3,5 chimera; 35% for 5,5,5,3 chimera compared with 51% for wild-type Nav1.3 (less than threefold differences in estimated IC50s).
ICA-121431 Interacts with the Voltage Sensor Region in Domain 4 on Nav1.3.
The apparent importance of Domain 4 for inhibition of Nav1.3 by ICA-121431 was further evaluated by constructing chimeras with homologous Domains 1–3 from Nav1.3 and various subregions of Domain 4 containing the equivalent amino acid residue sequences from either Nav1.5 (Fig. 2B) or Nav1.7 (Fig. 2C) (details of splice sites in Fig. S3). Replacement of Domain 4 S5 and S6 transmembrane regions of Nav1.3 with those from Nav1.5 or Nav1.7 (S5–S6) produced only small changes (two- to fivefold decrease) in ICA-121431 potency. In contrast, replacement of the Domain 4 voltage sensor region S1–S4 of Nav1.3 with the same region from Nav1.5 (S1–S4) resulted in a >500-fold loss potency for inhibition by ICA-121431 (IC50 10 ± 2 µM, n = 3) (Fig. 2 B–D), indicating that the main interaction site for ICA-121431 probably lies within the voltage sensor region.
To further delineate possible interaction sites within the Domain 4 S1–S4 voltage sensor region, three additional chimeras were constructed, in which either the S1, S2, or S3–S4 transmembrane segments or associated loops from Domain 4 of the Nav1.3 were replaced with the equivalent amino acid sequences of Nav1.5 or Nav1.7 (Fig. 2 B and C). Fig. 2D shows that whereas replacing S1 of Nav1.3 with S1 from Nav1.5 or Nav1.7 produced a relatively small (three- to eightfold) reduction in inhibition potency, replacement of either S2 or S3–S4 with the Nav1.5 or Nav1.7 sequence resulted in 20- to 100-fold loss in sensitivity to inhibition by ICA-121431 (IC50s for different chimeras are summarized in Table S2).
Three Amino Residues in the Voltage Sensor Region Contribute to Selectivity of ICA-121431.
To identify specific amino acid residues within the S2–S4 region of Domain 4 that contribute to ICA-121431 selective inhibition, we aligned human Nav channel amino acid sequences of Domain 4 S1–S4 and then grouped by relative sensitivity to inhibition. As noted earlier, the two most sensitive channels were Nav1.3 and Nav1.1. Nav1.2 exhibited an intermediate sensitivity (IC50 240 nM), whereas Nav1.4, Nav1.5, Nav1.6, Nav1.7, and Nav1.8 were relatively insensitive to inhibition (IC50s >10 µM) (Fig. 3A, Fig. S1, and Table S1).
Fig. 3.
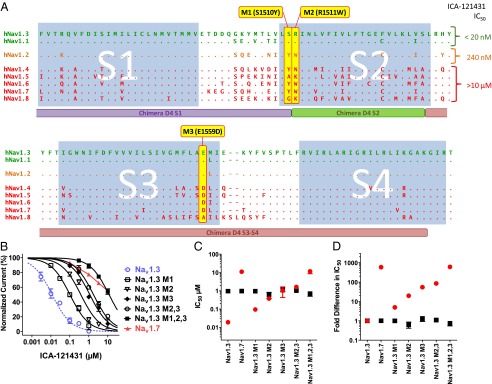
Defining amino acid residues important for inhibition of Nav1.3 by ICA-121431. (A) Comparison of amino acid sequence for S1–S4 voltage-sensor region of Domain 4 for different subtypes of human voltage dependent sodium channel. Residues that are different from Nav1.3 are shown (i.e., dots represent residues that are identical to Nav1.3). Sequence alignments for channels demonstrating high sensitivity to inhibition by ICA-121431 (IC50 <20 nM) are shown in green, intermediate sensitivity (IC50 ∼300 nM) are shown in orange, and insensitive channels (IC50 >10 µM) are shown in red. Residues that were present in ICA-121431 sensitive channels but absent in insensitive sodium channels (highlighted in yellow) were mutated, substituting in equivalent residues from Nav1.7 (termed M1, M2, and M3). The location of the chimera constructs D4 S1, D4 S2, and D4S3–S4 described in Fig. 2 are shown below the sequences for comparison. (B) Concentration dependence of ICA-121431 inhibition of human Nav1.3 mutated to contain single or combinations of human Nav1.7 residues at the three residue positions highlighted in A. Mutated Nav1.3 channels are compared with unmodified Nav1.3 (blue) and Nav1.7 (red). (C) Comparison of IC50 for ICA-121431 (red circles) and amitriptyline (black squares) inhibition of Nav1.3, Nav1.7, and various combinations of Nav1.3 M1 (S1510Y), M2 (R1511W), or M3 (E1559D) mutations. (D) Comparison of fold difference in IC50 for ICA-121431 (red circles) and amitriptyline (black squares) mediated inhibition of Nav1.7 and various combinations of Nav1.3 M1 (S1510Y), M2 (R1511W), or M3 (E1559D) mutations relative to wild-type human Nav1.3. Data were generated using conventional patch clamp using an 8-s conditioning voltage step from −120 mV to a voltage producing half-maximal inactivation followed after a 20-ms recovery at −100 mV by a 20-ms test pulse to 0 mV.
Focusing on the S2–S4 region of Domain 4, we looked for amino acid residues shared between Nav1.3 and Nav1.1 and not most other Nav channels. Three residues were identified: consecutive serine and arginine residues at positions 1510 and 1511 and a glutamate at position 1559 in S3 of Nav1.3 (Fig. 3A). S1510, R1511, and E1559 are predicted to be at the extracellular ends of the S2 and S3 transmembrane segments and were therefore initially considered to be promising candidates for potential ligand interaction. To examine their role, individually and together, these Nav1.3 residues were replaced with the equivalent residues of the ICA-121431–insensitive Nav1.7 channel. The effects of these mutations on inhibition by ICA-121431 are shown in Fig. 3B (also refer to Table S2). S1510Y (M1) resulted in a ninefold loss of potency of ICA-121431, whereas R1511W (M2) and E1559D (M3) produced 33-fold and 97-fold reductions in potency, respectively. The double mutation R1511W/E1559D (M2,3) produced a 152-fold loss of potency, whereas the triple mutation S1510Y/R1511W/E1559D (M1,2,3) resulted in a 600-fold loss of potency, exhibiting a lack of sensitivity to inhibition by ICA-121431 similar to Nav1.7. These results suggest that all three identified residues on Nav1.3 play a role in defining inhibition by ICA-121431. Although variations in the voltage dependence of inactivation for the various mutants were observed (Fig. S2), for the most part they were <7 mV and there was no obvious correlation with the widely varying potencies seen with ICA-121431. Moreover, all of the individual (M1, M2, M3) and multiple mutants (M2,3 and M1,2,3) of Nav1.3 exhibited relatively constant sensitivities to inhibition by amitriptyline (IC50 ∼1 µM) (Fig. 3 C and D), which as mentioned earlier would be expected to exhibit similar sensitivity to differences in the voltage dependence of inactivation as ICA-121431.
An obvious next question is whether the S1510/R1511/E1559 residues are sufficient to account for the selectivity of ICA-121431 for Nav1.3 over Nav1.7. To address this question, a triple mutation of Nav1.7 was made in which residues corresponding to M1, M2, and M3 were replaced by the equivalent Nav1.3 residues (Y1537S/W1538R/D1586E; Nav1.7 M1,2,3). Fig. 4A, Left compares ICA-121431 inhibition of Nav1.7 M1,2,3 with Nav1.3 M1,2,3, wild-type Nav1.3, and Nav1.7. Replacement of the three residues on Nav1.7 with those from Nav1.3 resulted in an 860-fold increase in potency for inhibition by ICA-121431; Nav1.7 M1,2,3 exhibited an IC50 of 33 nM, which is within twofold of the IC50 of 18 nM for unmodified Nav1.3. Furthermore, in contrast to the dramatic effects of Nav1.7 M1,2,3 and Nav1.3 M1,2,3 on ICA-121431 inhibition, these mutations had little or no effect on inhibition by the local anesthetic tetracaine (Fig. 4A, Right). Moreover, the M1,2,3 mutant forms of either Nav1.3 or Nav1.7 were associated with only minimal changes in biophysical properties, such as voltage dependence of activation (Fig. 4 B and C), inactivation (Fig. 4 D and E), and rate of recovery from inactivation (Fig. 4 F and G) (Table S3 lists biophysical parameters). These nominal biophysical differences are unlikely to account for the profound changes in pharmacological sensitivity seen in the mutated channels. Therefore, taken together, the evidence suggests that the S1510/R1511/E1559 residues in the Domain 4 voltage sensor region of Nav1.3 contribute to the potent and selective inhibition of Nav1.3 (and Nav1.1) by ICA-121431 by affecting the physical interaction of the molecule.
Fig. 4.

Pharmacological and biophysical properties of Nav1.3 and Nav1.7 M123 triple mutant. (A) Comparison of inhibition by ICA-121431(Left) or tetracaine (Right) after substituting the M1, M2, and M3 residues of Nav1.3 with residues from Nav1.7 (i.e., S1510Y/R1511W/E1559D) or vice versa in which the equivalent M1, M2, and M3 residues of Nav1.7 are substituted with equivalent residues from Nav1.3 (i.e., Y1537S/W1538R/D1586E). Inhibition was recorded using the IonWorks Quattro using the protocol described in Fig. 1F. Comparison of biophysical properties of human Nav1.3 and the Nav1.3 S1510Y/R1511W/E1559D triple mutant vs. human Nav1.7 and the Nav1.7 Y1537S/W1538R/D1586E triple mutant. (B and C) Current voltage relationships for wild-type and M123 mutant forms of Nav1.3 and Nav1.7. Peak current amplitudes were measured during 500-ms voltage steps to various membrane potentials from a holding potential of −120 mV. (D and E) Voltage dependence of inactivation for wild-type and M123 mutant forms of Nav1.3 and Nav1.7. Peak current amplitudes were measured during test pulses to −20 mV after 500-ms conditioning depolarizations to the indicated potentials. Individual normalized inactivation curves were fit with a Boltzmann equation, 1/1+exp[(Vh − V)/k], where Vh is the half-inactivation voltage, and k is the slope. Values for Vh and k are shown in Table S3. (F and G) Time course of recovery from inactivation for wild-type and M123 mutant forms of Nav1.3 and Nav1.7. Peak current amplitudes were measured during test pulses to −20 mV after a variable time period after a 500-ms conditioning depolarization to 0 mV. Individual normalized recovery curves were fit with a two phase exponential association equation, Y = FractionFast [1-exp(-t/tauFast)] + FractionSlow*[1-exp(-t/tauSlow)], where Y is the fraction of total current amplitude at time, t, and comprises fraction of current recovering with fast kinetics, FractionFast at time constant, tauFast or recovering with slow kinetics, FractionSlow at time constant, tauSlow. Values for time constants, tauFast, and tauSlow and fraction of current recovering with fast kinetics FractionFast are shown in Table S3. Data were generated using the PatchXpress automated electrophysiology platform.
The Same Three Amino Residues Can Contribute to Human Nav1.7 Inhibitor Selectivity.
The selectivity profile of ICA-121431, along with amino acid sequence diversity among Nav family members around the proposed interaction site (Fig. 3A), raises the question of whether this region of Nav channels provides opportunities for finding inhibitors with selectivity for other Nav channel subtypes. This question was addressed with another aryl sulphonamido-thiazole–containing molecule, PF-04856264 [3-cyano-4-(2-(1-methyl-1H-pyrazol-5-yl) phenoxy)-N-(thiazol-2-yl) benzenesulfonamide] (Fig. 5A), which was identified during a screen for Nav1.7 inhibitors. PF-04856264 was observed to interact preferentially with the inactivated state of Nav1.7. Fig. 5B shows that Nav1.7 currents elicited by 20-ms depolarizing voltage steps from −120 mV were insensitive to inhibition by 1 µM PF-04856264, whereas the same concentration produced 91% ± 4% inhibition when a test pulse was preceded by an 8-s conditioning voltage step to inactivate approximately half of the channels. Interestingly, using this latter protocol, human Nav1.3 and Nav1.5 exhibited little or no sensitivity to inhibition by 1 µM PF-04856264 (Fig. 5C). The subtype selectivity exhibited by PF-04856264 is further illustrated in the concentration response curves shown in Fig. 5D (generated using the same protocol used for ICA-121431 in Fig. 1D). The fitted IC50 for inhibition of human Nav1.7 was 28 ± 5 nM (n = 8), whereas little or no inhibition of human Nav1.3 or Nav1.5 currents were observed at concentrations up to 10 µM.
Fig. 5.
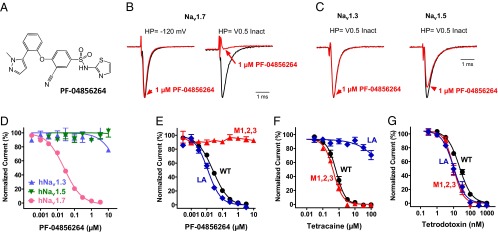
Selective Nav1.7 inhibitor also interacts with Domain 4 voltage sensor. (A) Structure of PF-04856264. (B) Current traces recorded on PatchXpress showing effect of 1 µM PF-04856264 on human Nav1.7 current amplitude elicited by a 20-ms voltage step to 0 mV from −120 mV or after an 8-s condition voltage step to −75 mV to inactivate approximately half of the available channels. Current traces are normalized so that control traces have same relative amplitude. (C) PatchXpress recording showing effect of 1 µM PF-04856264 on human Nav1.3 and Nav1.5 currents elicited by a 20-ms voltage step to 0 mV after an 8-s condition voltage step to inactivate approximately half of the available channels. (D) Concentration dependence of inhibition of human Nav1.3, Nav1.5, and Nav1.7 currents by PF-04856264 (IC50 is 28 ± 5 nM for Nav1.7 and >10 µM for Nav1.3 and Nav1.5). (E) PF-04856264 mediated inhibition of unmodified (WT) Nav1.7, Nav1.7 M1,2,3 (Y1537S/W1538R/D1586E), or the local anesthetic binding site mutant Nav1.7(F1737A/Y1744A) (LA) compared with inhibition by the local anesthetic tetracaine (F) or TTX (G). IC50s are provided in the text. Data for D–G were generated on the IonWorks Quattro using the protocol described in Fig. 1F.
Given the structural similarity (i.e., aryl sulphonamido-thiazole moiety) between PF-04856264 and ICA-121431, we evaluated the importance of the Domain 4 voltage sensor M1,2,3 residues of Nav1.7 for PF-04856264 inhibitor activity. Fig. 5E shows that when the M1,2,3 equivalent residues on Nav1.7 were substituted with residues found in Nav1.3 (i.e., Y1537S/W1538R/D1586E), inhibition by PF-04856264 was essentially abolished (IC50 >10 µM). The specificity of this modulatory effect is highlighted by the finding that inhibition by other established sodium channel blocking agents, tetracaine and TTX, which reportedly bind within or over the pore, was not changed by the Y1537S/W1538R/D1586E substitution (Fig. 5 F and G). Furthermore, Fig. 5 E and F provide supporting evidence that PF-04856264 and local anesthetic-like inhibitors interact with distinct binding sites, because mutation of two residues (F1737A/Y1744A) in S6 of Domain 4 of Nav1.7, which are reported to be involved in local anesthetic and antiepileptic drug binding to Nav channels (19, 20), resulted in a >100-fold loss of potency for inhibition by tetracaine (IC50 >100 µM compared with 0.9 µM for wild-type Nav1.7) but only a nominal change in the potency of PF-04856264 (IC50 = 12 nM compared with 28 nM for wild-type Nav1.7).
To further evaluate whether the M123 Y1537/W1538/D1586 amino acid motif was sufficient to account for the Nav1.7 vs. Nav1.3 selectivity observed with PF-04856264, a construct was generated in which the equivalent residues of human Nav1.3 were replaced with the Nav1.7 M123 residues (S1510Y/R1511W/E1559D). The potency for inhibition of Nav1.3 M123 (S1510Y/R1511W/E1559D) by PF-04856264 was found to be 7 µM. Although this IC50 is measurably lower than for Nav1.7 M123 (Y1537S/W1538R/D1586E), it is 250-fold greater than the IC50 for wild-type Nav1.7. These findings suggest that although the M123 residues are important for PF-04856264 inhibition of Nav1.7, they are not sufficient to account for all of the subtype selectivity observed with this particular inhibitor, indicating that additional residues in this region may play a role in determining subtype selective interactions.
Species Dependent Inhibition by ICA-121431 and PF-04856264 Is Determined by Amino Acid Sequence Differences in Domain 4 Voltage Sensor Region.
Although we have shown that ICA-121431 and PF-04856264 exhibit selectivity for human Nav1.3 or human Nav1.7, this profile was not observed with all mammalian species evaluated. For example, whereas cynomolgus monkey and dog Nav1.7 lacked sensitivity to inhibition by ICA-121431 (IC50s > 5,000 nM) similar to human Nav1.7, we found that rat Nav1.7 is potently inhibited by ICA-121431 with an IC50 of 19 nM, which is more comparable to its inhibition of human Nav1.3 (i.e., >1,000-fold more potent than human Nav1.7) (Fig. 6A). However, mouse Nav1.7 gave a more human-like profile, with an IC50 for inhibition by ICA-121431 of 8,700 nM. Intriguingly, an opposite differential rodent sensitivity to inhibition was observed with the human Nav1.7-selective inhibitor PF-04856264 (Fig. 6B). Whereas the IC50s for PF-04856264 inhibition of cynomolgus monkey (19 nM) or dog Nav1.7 (42 nM) were comparable to human Nav1.7, rat Nav1.7 was considerably less sensitive, exhibiting an IC50 of 4,200 nM. Again, in contrast to rat, mouse Nav1.7 exhibited a higher sensitivity to inhibition by PF-04856264 (IC50 131 nM).
Fig. 6.

Species orthologs of Nav1.7 exhibit different sensitivities to inhibition by ICA-121431 and PF-04856264. (A) Concentration dependent inhibition of rat Nav1.7 (blue squares), mouse Nav1.7 (red squares), and human Nav1.7 (black open circles) by ICA-121431. The fitted IC50s are 37 nM (rat), 8,800 nM (mouse), and >10,000 nM (human). (B) Concentration dependent inhibition of rat Nav1.7 (blue squares), mouse Nav1.7 (red circles), and human Nav1.7 (black open circles) by PF-04856264. The fitted IC50s are 4200 nM (rat), 131 nM (mouse), and 30 nM (human). (C) Amino acid sequence alignment of various species orthologs of Nav1.7 homologous Domain 4 transmembrane segments S1–S3 (light blue). Residues highlighted in yellow are the M1, M2, and M3 described in the text, which contribute to defining hNav1.3 vs. hNav1.7 selectivity by ICA-123431 and PF-04856264. Differences from human Nav1.7 sequence within the M1,2,3 are colored blue and underlined. Other sequence differences across species are colored green.
The amino acid sequence alignments of the Domain 4 S1–S4 transmembrane segments from Nav1.7 for different species shown in Fig. 6C provide a possible explanation for the variation in species sensitivities to inhibition by ICA-121431 or PF-04856264. Although there is a high degree of sequence homology across species in this region, there are notable variations including those in the M1, M2, or M3 residues (i.e., human Nav1.7-Y1537/W1538/D1586), which have been shown in previous sections to be important for defining human Nav1.3 and Nav1.7 selectivity. Specifically, species exhibiting the most human-like Nav1.7 inhibition profile (i.e., cynomolgus monkey, dog, and mouse Nav1.7) also exhibited the highest sequence homology over these three residues. In contrast, rat Nav1.7 differs at both the M1 and M3 residues.
Discussion
In the present study we characterize a unique class of nanomolar potent small molecule inhibitors of Nav channels that interact with and stabilize inactivated states of the channel. Furthermore, structurally related compounds within this class exhibit subtype selective inhibition of Nav channels, including human Nav1.3 and Nav1.7. For example, ICA-121431 exhibited a spectrum of inhibitory activity for Nav human channel subtypes; equipotent inhibition of Nav1.3 and Nav1.1, less potent inhibition of Nav1.2, and much weaker inhibition of Nav1.7, Nav1.6, Nav1.4, and the TTX-resistant human Nav1.5 and Nav1.8 channels (IC50s >10 µM). We exploited the subtype selectivity of ICA-121431 to investigate possible Nav interaction sites by constructing Nav1.3/Nav1.5 or Nav1.7 chimeras. Functional evaluation of these chimeras has provided evidence that the likely site of interaction for this class of inhibitor is at extracellular ends of the S2 and S3 transmembrane segments of Domain 4. By comparing the amino acid sequences of ICA-121431 sensitive vs. insensitive Nav channel subtypes over this region of the voltage sensor, we have identified three unique residues found only in channels sensitive to inhibition by ICA-121431[S1510 (M1), R1511 (M2), and E1559 (M3) of Nav1.3]. Their replacement with equivalent residues from the ICA-121431–insensitive Nav1.7 channel reduced potency for Nav1.3 inhibition by >500-fold. Further compelling evidence that the M1,2,3 residues are primary contributors to selective inhibition of Nav1.3 by ICA-121431 came from substituting the three M1,2,3 residues in human Nav1.7 with the Nav1.3 equivalent residues (Y1537S/W1538R/D1586E), which resulted in a striking gain in sensitivity to inhibition, comparable to unmodified Nav1.3. Although each of the M1, M2, or M3 residues contribute to inhibition, modification of the M2 and M3 residues alone and in combination had the largest impact on potency of ICA-121431. The M1,2,3 residues also seem to contribute to the inhibition of Nav1.7 by PF-04856264 given the substantial reduction of inhibition with replacement of the equivalent residues from human Nav1.3. Furthermore, the finding that both Nav1.3 and Nav1.7 selective inhibitors interact with similar but not entirely equivalent regions of the Domain 4 S2/S3 transmembrane segment indicates that additional interactions exist, possibly including residues that are conserved across Nav channel subtypes in this region of the voltage sensor domain.. An improved understanding of the functional importance of residues in and around the identified M1,2,3 residues may enable a fuller appreciation of the potential diversity of Nav subtype selective interactions that may be determined by this site.
As described, the dramatic changes in potency of ICA-121431 and PF-04856264 induced by modifications of the M1, M2, or M3 residues are unlikely to result from global structural rearrangements, because inhibition of individual or combined M1,2,3 Nav1.3 mutants (Nav1.3- S1510Y/R1511W/E1559D) or Nav1.7 M123 (Nav1.7-Y1537S/W1538R/D1586E) by amitriptyline, tetracaine, or TTX was comparable to wild-type channels. Furthermore, the absence of effect on local anesthetic-mediated inhibition suggests that Nav channel inactivation gating is not significantly affected by the M1,2,3 residue modifications. Indeed, an evaluation of the activation and inactivation gating properties of Nav1.3- S1510Y/R1511W/E1559D or Nav1.7-Y1537S/W1538R/D1586E are insufficient to account for the pharmacological changes observed.
Interestingly, observed species differences in sensitivity to inhibition by ICA-121431 or PF-04856264 can also be explained by differences in the M1,2,3 residue motif. The variability in species sensitivity to sodium channel inhibitors like the ones described in this study highlight the importance of gathering such information before evaluating compound efficacy and/or toxicity in animal tissues or in vivo models.
The present study provides compelling evidence that the unique subtype selective Nav channel inhibitors ICA-121431 and PF-04856264 interact with amino acid residues on an extracellular facing region of the homologous Domain 4 voltage sensor of Nav1.3 or Nav1.7, which is distinct from previously described interaction sites for TTX (12, 13) or local anesthetic-like (11, 20–22) Nav channel inhibitors (Figs. S4 and S5 show homology models illustrating relative locations of interaction sites). However, this small molecule interaction region on Domain 4 does overlap with the interaction sites for a number of polypeptide toxin modulators of sodium channel function (6, 14, 15, 18). For example, at least one residue, E1559 on Nav1.3 or D1586 on Nav1.7 (M3 residue), which contributes to the interaction of both ICA-121431 and PF-04856264, is also important for interaction of site 3 scorpion venom and anemone toxins that slow Nav channel inactivation (14, 15, 18, 35). The contribution of the M1 and M2 residues to peptide toxin binding is less well defined, although it has been reported that substitution of the tyrosine with alanine at position 1564 of Nav1.2 (equivalent to M1 residue position described in this study) has no effect on Lqh2 scorpion toxin-induced slowing of inactivation (18).
Inhibition of Nav channel function via interaction with the voltage sensor region has previously been reported for spider (e.g., Protx II, HWTX-IV) and marine snail (MuO conotoxins) peptide toxins (16, 36–38), although much of the published data suggest that they primarily interact with the voltage sensor of homologous Domain 2 (16, 17, 38–40). Several recent reports have also described possible interactions of Protx II with Domain 1, 3, and 4 voltage sensors (38–40). Interestingly, a residue equivalent to the Domain 4 M3 site on Nav1.3 and Nav1.7 has been reported to contribute to the interaction of Protx II, although this site may be more involved in a slowing of inactivation seen with the toxin rather than the more widely described inhibitory action (38). The mechanistic implications of preferential interactions with specific homologous domain voltage sensor regions by peptide vs. small molecule Nav channel inhibitors remain to be fully explored. However, these differences may underlie the contrasting ability of peptide toxins like Protx II to inhibit Nav channels by shifting the apparent voltage dependence of activation to depolarized potentials vs. apparent stabilization of one or more inactivated conformations by the small molecule inhibitors described in this study.
In conclusion, the present study highlights unique possibilities for identifying additional subtype selective small molecule sodium channel modulators by targeting allosteric interaction sites away from the pore region like those that contribute to voltage sensing and/or conformational changes associated with channel gating. Although the present studies have identified several residues on the Domain 4 voltage sensor region that are important for interaction and selectivity, further investigation is needed to elucidate the full extent of the interaction, perhaps by evaluating other residues reported to participate in peptide modulation of voltage sensor domains. Future studies will also be directed toward better understanding how pharmacological interactions with the voltage sensor in Domain 4 results in inhibition of conduction. Given the proposed involvement of Nav channels in pain, cardiovascular, and central nervous system disorders (9, 41–45), further exploration of subtype selective Nav channel inhibitors targeting this unique interaction site may provide opportunities to develop future voltage dependent Na channel directed therapeutics (27).
Methods
Reagents.
ICA-121431 and PF-04856264 were synthesized by the medicinal chemistry group at Icagen Inc. Tetracaine, amitriptyline, and TTX were obtained from Sigma Aldrich.
Generation of Chimeras and Mutants.
Chimeric channels comprising human Nav1.3, Nav1.5, and Nav1.7 domains and subdomains were assembled using unique restriction enzyme sites introduced by site-directed mutagenesis in the respective channel coding regions (QuikChange II Site-Directed Mutagenesis and QuikChange Multi Site-Directed Mutagenesis kits; Stratagene). Not I and Sal I restriction sites allowed transfer of full-length cDNA constructs into either pcDNA3.1 (Invitrogen) or pLNCX2 (BD Bioscience Clontech) mammalian expression vectors. All intermediate and final constructs were verified by sequencing and are based on nucleotide numbering from National Center for Biotechnology Information GenBank sequences NM_006922 for hNav1.3 and NM_198056 for hNav1.5 and NM_002977 for hNav1.7. Final constructs were either transiently expressed (domain swap chimeras) or stably expressed (sub-Domain 4 chimeras and all point mutations) in HEK-293 cells. Transient transfections were achieved using Fugene 6 (Roche Applied Science) according to the vendor’s protocol. Functional currents were recorded 48 h after transfections. Stable clonal cell lines were selected with G-418 (800 µg/mL) and prioritized by functional sodium current amplitude using conventional patch clamp or the IonWorks high-throughput electrophysiology platform. Controls for the wild-type human Nav1.3, Nav1.5, and Nav1.7 were stably expressed in HEK-293 cells using the same procedures described above.
Electrophysiology.
Functional characterization of voltage-gated sodium currents was accomplished using either conventional whole cell patch clamp or PatchXpress or Ionworks Quattro high-throughput electrophysiology platforms (Molecular Devices).
Conventional patch clamp.
Coverslips containing HEK-293 cells expressing unmodified and mutated Nav channels were placed in a recording chamber on the stage of an inverted microscope and perfused (∼1 mL/min) with an extracellular solution containing 138 mM NaCl, 2 mM CaCl2, 5.4 mM KCI, 1 mM MgCl2, 10 mM glucose, and 10 mM Hepes (pH 7.4), with NaOH. Recording patch pipettes were filled with an intracellular solution containing 135 mM CsF, 5 mM NaCl, 2 mM MgCl2, 10 mM EGTA, and 10 mM Hepes (pH 7.3) with NaOH and had a resistance of 1–2 MΩ. All recordings were made at room temperature (22–24 °C) using AXOPATCH 200B amplifiers and PCLAMP software (Molecular Devices). Nav currents were measured using the whole-cell configuration of the patch clamp technique. All compounds were dissolved in DMSO to make 10-mM stock solutions, which were then diluted into extracellular solution to attain the final concentrations desired. The final concentration of DMSO (<0.3%) was found to have no significant effect on sodium currents. Test compound effects were evaluated using a protocol in which cells depolarized from a holding potential of −120 mV (or −140 mV for Nav1.5) to a membrane potential that inactivated half of the available channels for 8 s (determined empirically for each channel type) followed after a 20-ms recovery at −120 mV by a test pulse to 0 mV to assess magnitude of inhibition. Concentration response data were analyzed using nonlinear least-squares fit of the Logistic Equation (GraphPad Prism 5) to provide IC50 determinations.
PatchXpress automated electrophysiological platform.
All assay buffers and solutions were identical to those used in conventional whole-cell voltage clamp experiments described above. Nav channel-expressing cells were grown as above to 50–80% confluency and harvested by trypsinization. Trypsinized cells were washed and resuspended in extracellular buffer at a concentration of 1 × 106 cells/mL. The onboard liquid handling facility of the PatchXpress was used for dispensing cells and application of test compounds. Test compound effects were evaluated and analyzed using the same protocols used for conventional patch clamp.
Ionworks Quattro automated electrophysiological platform.
Intracellular and extracellular solutions were as described above with the following changes: 100 µg/mL amphotericin was added to the intracellular solution to perforate the membrane and allow electrical access to the cells. Nav channel-expressing cells were grown and harvested as for PatchXpress, and cells were resuspended in extracellular solution at a concentration of 3–4 × 106 cells/mL. The onboard liquid handling of the Ionworks Quattro was used for dispensing cells and application of test compounds. Test compound effects were evaluated using a voltage protocol in which a 500-ms voltage step to 0 mV from −120 mV was applied to fully inactivate available sodium channels, followed after a 50 ms to 100 s hyperpolarized recovery period at −120 mV to allow partial recovery from inactivation, by a test voltage step to 0 mV to assess magnitude of inhibition. This protocol facilitates comparison of channel subtypes independent of their differences in midpoint potentials for voltage dependence of inactivation.
Supplementary Material
Acknowledgments
We thank Andrew Maynard, Aaron Gerlach, Sally Stoehr, Anrou Zuo, Theresa Mersch, Louise Heath, and Eva Prazak for additional experimental and cell reagent provision support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220844110/-/DCSupplemental.
References
- 1.Bezanilla F. The action potential: From voltage-gated conductances to molecular structures. Biol Res. 2006;39(3):425–435. doi: 10.4067/s0716-97602006000300005. [DOI] [PubMed] [Google Scholar]
- 2.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J Physiol. 2012;590(Pt 11):2577–2589. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol. 1998;508(Pt 3):647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Billen B, Bosmans F, Tytgat J. Animal peptides targeting voltage-activated sodium channels. Curr Pharm Des. 2008;14(24):2492–2502. doi: 10.2174/138161208785777423. [DOI] [PubMed] [Google Scholar]
- 7.Llewellyn LE. Sodium channel inhibiting marine toxins. Prog Mol Subcell Biol. 2009;46:67–97. doi: 10.1007/978-3-540-87895-7_3. [DOI] [PubMed] [Google Scholar]
- 8.Catterall WA, et al. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007;49(2):124–141. doi: 10.1016/j.toxicon.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 9.Wood JN, Boorman J. Voltage-gated sodium channel blockers; target validation and therapeutic potential. Curr Top Med Chem. 2005;5(6):529–537. doi: 10.2174/1568026054367584. [DOI] [PubMed] [Google Scholar]
- 10.Yanagidate F, Strichartz GR. Local anesthetics. Handb Exp Pharmacol. 2007;(177):95–127. doi: 10.1007/978-3-540-33823-9_4. [DOI] [PubMed] [Google Scholar]
- 11.Fozzard HA, Sheets MF, Hanck DA. The sodium channel as a target for local anesthetic drugs. Front Pharmacol. 2011;2:68. doi: 10.3389/fphar.2011.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fozzard HA, Lipkind GM. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar Drugs. 2010;8(2):219–234. doi: 10.3390/md8020219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noda M, Suzuki H, Numa S, Stühmer W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989;259(1):213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- 14.Leipold E, Lu S, Gordon D, Hansel A, Heinemann SH. Combinatorial interaction of scorpion toxins Lqh-2, Lqh-3, and LqhalphaIT with sodium channel receptor sites-3. Mol Pharmacol. 2004;65(3):685–691. doi: 10.1124/mol.65.3.685. [DOI] [PubMed] [Google Scholar]
- 15.Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271(27):15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 16.Schmalhofer WA, et al. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol Pharmacol. 2008;74(5):1476–1484. doi: 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- 17.Sokolov S, Kraus RL, Scheuer T, Catterall WA. Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol Pharmacol. 2008;73(3):1020–1028. doi: 10.1124/mol.107.041046. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, et al. Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci USA. 2011;108(37):15426–15431. doi: 10.1073/pnas.1112320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, et al. Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharmacology. 2003;44(3):413–422. doi: 10.1016/s0028-3908(02)00400-8. [DOI] [PubMed] [Google Scholar]
- 20.Panigel J, Cook SP. A point mutation at F1737 of the human Nav1.7 sodium channel decreases inhibition by local anesthetics. J Neurogenet. 2011;25(4):134–139. doi: 10.3109/01677063.2011.629702. [DOI] [PubMed] [Google Scholar]
- 21.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265(5179):1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 22.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA. 1996;93(17):9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.England S, de Groot MJ. Subtype-selective targeting of voltage-gated sodium channels. Br J Pharmacol. 2009;158(6):1413–1425. doi: 10.1111/j.1476-5381.2009.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolognesi R, Tsialtas D, Vasini P, Conti M, Manca C. Abnormal ventricular repolarization mimicking myocardial infarction after heterocyclic antidepressant overdose. Am J Cardiol. 1997;79(2):242–245. doi: 10.1016/s0002-9149(96)00727-8. [DOI] [PubMed] [Google Scholar]
- 25.Wolfe JW, Butterworth JF. Local anesthetic systemic toxicity: Update on mechanisms and treatment. Curr Opin Anaesthesiol. 2011;24(5):561–566. doi: 10.1097/ACO.0b013e32834a9394. [DOI] [PubMed] [Google Scholar]
- 26.Catterall WA. Ion channel voltage sensors: Structure, function, and pathophysiology. Neuron. 2010;67(6):915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.England S, Rawson D. Isoform-selective voltage-gated Na(+) channel modulators as next-generation analgesics. Future Med Chem. 2010;2(5):775–790. doi: 10.4155/fmc.10.26. [DOI] [PubMed] [Google Scholar]
- 28.Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9(4):413–424. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- 29.Tarnawa I, Bölcskei H, Kocsis P. Blockers of voltage-gated sodium channels for the treatment of central nervous system diseases. Recent Patents CNS Drug Discov. 2007;2(1):57–78. doi: 10.2174/157488907779561754. [DOI] [PubMed] [Google Scholar]
- 30.Clare JJ. Targeting voltage-gated sodium channels for pain therapy. Expert Opin Investig Drugs. 2010;19(1):45–62. doi: 10.1517/13543780903435340. [DOI] [PubMed] [Google Scholar]
- 31.Dib-Hajj SD, et al. Voltage-gated sodium channels in pain states: Role in pathophysiology and targets for treatment. Brain Res Brain Res Rev. 2009;60(1):65–83. doi: 10.1016/j.brainresrev.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Krafte DS, Bannon AW. Sodium channels and nociception: Recent concepts and therapeutic opportunities. Curr Opin Pharmacol. 2008;8(1):50–56. doi: 10.1016/j.coph.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Ragsdale DS, Scheuer T, Catterall WA. Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol Pharmacol. 1991;40(5):756–765. [PubMed] [Google Scholar]
- 34.Wang GK, Russell C, Wang SY. State-dependent block of voltage-gated Na+ channels by amitriptyline via the local anesthetic receptor and its implication for neuropathic pain. Pain. 2004;110(1-2):166–174. doi: 10.1016/j.pain.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 35.Gurevitz M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon. 2012;60(4):502–511. doi: 10.1016/j.toxicon.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 36.Bosmans F, Swartz KJ. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol Sci. 2010;31(4):175–182. doi: 10.1016/j.tips.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knapp O, McArthur JR, Adams DJ. Conotoxins targeting neuronal voltage-gated sodium channel subtypes: potential analgesics? Toxins (Basel) 2012;4(11):1236–1260. doi: 10.3390/toxins4111236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao Y, Blumenthal K, Jackson JO, 2nd, Liang S, Cummins TR. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol. 2010;78(6):1124–1134. doi: 10.1124/mol.110.066332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosmans F, Martin-Eauclaire MF, Swartz KJ. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature. 2008;456(7219):202–208. doi: 10.1038/nature07473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bosmans F, Puopolo M, Martin-Eauclaire MF, Bean BP, Swartz KJ. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J Gen Physiol. 2011;138(1):59–72. doi: 10.1085/jgp.201110614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox JJ, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10(7):1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- 43.Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hains BC, et al. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23(26):8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hains BC, Waxman SG. Sodium channel expression and the molecular pathophysiology of pain after SCI. Prog Brain Res. 2007;161:195–203. doi: 10.1016/S0079-6123(06)61013-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.