

Abstract
Genome-wide association studies have the potential to identify causal genetic factors underlying important phenotypes but have rarely been performed in bacteria. We present an association mapping method that takes into account the clonal population structure of bacteria and is applicable to both core and accessory genome variation. Campylobacter is a common cause of human gastroenteritis as a consequence of its proliferation in multiple farm animal species and its transmission via contaminated meat and poultry. We applied our association mapping method to identify the factors responsible for adaptation to cattle and chickens among 192 Campylobacter isolates from these and other host sources. Phylogenetic analysis implied frequent host switching but also showed that some lineages were strongly associated with particular hosts. A seven-gene region with a host association signal was found. Genes in this region were almost universally present in cattle but were frequently absent in isolates from chickens and wild birds. Three of the seven genes encoded vitamin B5 biosynthesis. We found that isolates from cattle were better able to grow in vitamin B5-depleted media and propose that this difference may be an adaptation to host diet.
Keywords: evolution, genomics, host adaptation, transmission ecology
Colonization of multiple host species increases the number of transmission opportunities for animal pathogens and symbionts but depends on making rapid adjustments to each new host (1). For organisms such as Campylobacter, relatively small genome size (1.6 Mb) limits the phenotypic flexibility of each bacterium. Single clones can multiply to large numbers within hosts, and genetic variation arising among these bacteria increases the range of available phenotypes. This might allow a bacterial lineage to passage successfully through multiple hosts by repeatedly evolving host adaptive traits.
Experimental work has shown that a large proportion of adaptations to new environments incur an equal or greater cost in other environments (2). This cost of adaptation might make a strategy of continuous evolution unstable by causing a progressive loss of fitness in the course of repeated host switching. Three factors that could reduce this cost of readaptation are canalization of genetic change via contingency loci (3, 4); coordinated genetic regulation of host-specific factors (5, 6); and import of DNA by recombination from other, already adapted, lineages in each new host species (7). The relative importance of these mechanisms for host specificity in Campylobacter remains unknown.
Campylobacter jejuni and Campylobacter coli are common components of the gut microbiota in numerous wild and domesticated animal species, as well as, together, being one of the most common causes of food poisoning in humans. The characterization of large numbers of C. jejuni and C. coli isolates from diverse sources and locations by multilocus sequence typing (MLST) has shown that there is genetic differentiation among sequence types (STs) associated with different hosts (8). Among wild birds, specific bird species most often harbor their own Campylobacter lineages (8, 9). However, in agricultural animals, although there are host-associated lineages that are largely restricted to chickens or to ruminants, some of the most abundant lineages are found at high frequencies in chickens, cattle, and humans with food poisoning (8). This multihost lifestyle is curious because of the challenges of colonizing organisms with such distinct gastrointestinal tracts, diets, immune systems, and body temperatures.
Here, we investigated the genetic basis of host specificity by analyzing the genome sequences of 192 isolates from cattle, chickens, clinical samples, and other sources. These included isolates from single-host lineages and multiple isolates from the host generalist ST-21 (C. jejuni), ST-45 (C. jejuni), and ST-828 (C. coli) clonal complexes (Table S1). We used phylogenetic analysis to investigate host switching and then sought to identify genetic elements that showed a stronger association with chicken or ruminants than expected based on the phylogeny. These elements represent candidate host adaptation elements. We identified one substantial cattle-associated region and demonstrate experimentally that it confers a phenotype (vitamin B5 biosynthesis) likely to aid in colonization of cattle.
Results
Clonal complexes, identified using MLST data, are groups of isolates that have identical sequences at most of or all the seven genotyped loci. Now that whole genomes are becoming available for large numbers of bacteria, it is possible to establish the accuracy of clonal complex designations in identifying clonal lineages. Phylogenetic analysis of C. jejuni genomes supports most of the clonal complex groupings identified by MLST and provides new insights into their relationships (Fig. 1A). For example, isolates from the chicken-associated ST-354, ST-443, ST-353, and ST-257 complexes (8) each form distinct clades in the tree and, together with a handful of other isolates, form a single chicken-associated supercomplex. Isolates from other chicken-associated clonal complexes, such as the ST-661 complex, branch elsewhere on the tree. There are also two cattle-associated lineages corresponding to the ST-61 and ST-42 complexes. In addition, there are lineages with levels of genetic variation comparable to those of the host-associated groups: specifically, the ST-21 and ST-45 complexes containing a mixture of isolates from cattle, chickens, and wild birds.
Fig. 1.
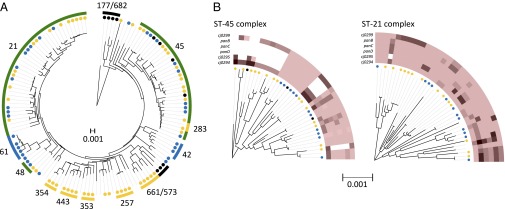
Genetic structure of C. jejuni isolates from different hosts. (A) Neighbor-joining tree of all isolates based on 1.53-Mb concatenated sequences (165,206 variable sites) of 1,623 loci in the NCTC11168 C. jejuni isolate genome. Host origin is indicated for chickens (yellow), cattle (blue), and wild birds and the environment (black). Other isolates are primarily from human infections. Clonal complex designations based on MLST are labeled around the tree. Host associations, based on the 2,764 isolates in the pubMLST database (pubMLST.org), are indicated in the halo using the same color scheme, with generalist lineages shown in green. (B) Tree of the ST-45 and ST-21 clonal complex isolates only, estimated using ClonalFrame. Allelic variation for six genes in the host-associated region is shown at the end of each branch. Each shade of red represents a unique allele at that locus. White denotes the absence or truncation of the gene.
To investigate relatedness within the multihost clonal complexes, we used ClonalFrame (10), which estimates clonal relationships allowing for the effect of recombination. Chicken and cattle isolates are found in multiple places on these trees (Fig. 1B), which implies relatively recent host switching, although there is one deeper branch of the ST-45 complex that may be chicken-associated.
To investigate the evolution of host specialization, we developed a genome-wide association mapping approach that identifies 30-bp DNA sequences (words) associated with colonization of particular species. The method identifies words that are more strongly associated with a particular host than would be expected based on neutral patterns of evolution, given the clonal relationships of the bacteria in the sample and their distribution among hosts. An attractive feature of this method is that it has the potential to identify host-adapting evolutionary events, including point mutation, homologous recombination, and lateral gene transfer.
In order for the method to have statistical power to detect associations, the dataset needs to contain multiple host switches within it. Furthermore, the presence of multiple isolates from the same host and clonal background (e.g., the chicken supercomplex) can reduce power because these isolates will share large amounts of DNA sequence due to clonal inheritance that do not confer host adaptive traits. We initially applied our method to 29 genome sequences from the ST-45 clonal complex, which are commonly found in wild and domesticated bird and mammal species. We included two genomes belonging to the ST-283 complex, which clusters within the ST-45 complex. We chose this lineage because it met the requirement of multiple host switches, and we reasoned that the low overall genetic variation within the lineage should enhance the power to detect adaptive events.
A total of 9,034 30-bp words were identified that were significantly associated with either chicken or cattle hosts. Of these, 8,999 mapped to 97 genes (Table S2) in the annotated genome of isolate NCTC11168 (11) in 10 genome locations (Fig. 2A). The association signal was replicated by determining the number of these words in the genomes of 161 C. jejuni and C. coli isolates from outside the ST-45 clonal complex. The pattern of host association varied among regions in the replication (Table S3). The most concordant signal, where the same pattern of host association occurred in the ST-45 complex isolates and in the remaining genomes, was among 7,307 cattle-associated words that came from 7 adjacent genes within a 5-Kb region (Table 1). We therefore focused on the evolutionary history of this region to investigate the events that generated the association signal.
Fig. 2.

Host-associated genomic regions. (A) Distribution of 10 locations containing 8,538 host-associated words for which homologs were found in reference genome NCTC11168, visualized using Artemis (12) and DNAPlotter (13). The red lines indicate host-associated words, the cyan rings show the protein coding sequences on the forward and reverse strands, and the green rings indicate other annotated features using default settings. (B) Schematic representation of seven adjacent genes within a 5-Kb region at location 3, to which 7,307 cattle-associated words mapped, and example flanking genes for comparison. Three regions (I–III) were identified based on host differences in word presence (Fig. S1). (C) Neighbor-joining trees of these seven genes in isolates from cattle (blue), chickens (yellow), and wild birds/environmental waters (green). The scale bars represent a genetic distance of 0.005.
Table 1.
Genes with cattle associated alleles
| Gene name* | Description |
| surE (Cj0293) | Stationary phase survival protein; catalyzes the conversion of a phosphate monoester to an alcohol and a phosphate |
| Cj0294 | Dinucleotide-using enzymes involved in molybdopterin and thiamine biosynthesis family 1 |
| Cj0295 | Putative acetyltransferase |
| panD (Cj0296c) | Aspartate α-decarboxylase; converts l-aspartate to β-alanine and provides the major route of β-alanine production in bacteria. β-alanine is essential for the biosynthesis of pantothenate (vitamin B5). |
| panC (Cj0297c) | Pantoate–β-alanine ligase; catalyzes the formation of (R)-pantothenate from pantoate and β-alanine |
| panB (Cj0298c) | 3-Methyl-2-oxobutanoate hydroxymethyltransferase; catalyzes the formation of tetrahydrofolate and 2-dehydropantoate from 5,10-methylenetetrahydrofolate and 3-methyl-2-oxobutanoate |
| Cj0299 | Putative periplasmic β-lactamase |
Gene names, numbering, and order are based on the C. jejuni strain 11168 annotation (11).
The pattern of gene presence varied for the six genes at the center of this region. The number of isolates from which they were completely absent varied from 3 isolates for surE to 78 and 98 isolates for Cj0299 and Cj0295, respectively. The panBCD genes were absent from between 34 and 39 isolates. Although the genes showed different patterns of presence and absence, they were all more common in cattle isolates than in chicken isolates. For example, in the ST-45 complex, the panBCD genes had been gained or lost three times (Fig. 1) and were not present in the two largest clades of the tree, where cattle isolates were absent. In contrast, the panBCD genes were present in all the ST-21 complex isolates examined.
The host association signal was the result of two forms of genetic variation. First, with the exception of surE, which had fewer host-associated words, all the genes in the major host-associated region were more likely to be absent in isolates from chickens. Second, even where genes were present, different sequences were found in cattle and chicken isolates. Alleles at these loci tended to show less homologous sequence variation in cattle isolates compared with chicken isolates, and as a result, cattle-associated alleles gave the strongest signals of host association. It is also notable that the frequency of both types of host-associated words was even lower in isolates from wild birds than in those from chickens (Fig. S1).
The majority of the cattle-associated words mapped to the three genes from the pantothenate (vitamin B5) biosynthesis pathway (panBCD). To test if cattle-associated isolates had a greater capacity to grow in a vitamin B5-depleted environment, we conducted in vitro growth experiments. This showed that isolates from cattle grew better, on average, in a low vitamin B5 environment than isolates from chickens (Fig. 3) and that this was due, at least in part, to the presence of the panBCD genes in a higher proportion of cattle isolates (Fig. S2).
Fig. 3.

Growth of C. jejuni in medium with and without pantothenic acid (vitamin B5). Curves in A and B represent growth levels in vitro (OD600) over a period of 18 h at 42 °C in microaerobic conditions in medium supplemented with vitamin B5 (solid lines) and lacking vitamin B5 (dashed lines). (A) Growth curves according to the host of origin, with cattle-associated isolates in blue (n = 29) and chicken-associated isolates in yellow (n = 22). (B) Growth curves according to the presence (n = 37, red lines) or absence (n = 20, black lines) of the panBCD operon in tested isolates.
The gene Cj0299 had exceptionally low sequence variation relative to other commonly present genes (Fig. S3), with only seven polymorphic sites among 774 nucleotides. This gene, which encodes a characterized enzyme called blaOXA-61 conferring resistance to β-lactam antibiotics (12), was found at the highest frequency in cattle isolates and was rarest in isolates from wild birds (Fig. S1).
Discussion
Our analysis provides insight into the tempo of adaptation of Campylobacter to its host. Host specificity can be a stable trait in some lineages. For example, specialization to chicken hosts presumably evolved in the common ancestor of the chicken supercomplex (Fig. 1A). There are also other less diverse chicken- and cattle-associated lineages. In addition, there are generalist lineages that are abundant in cattle, chickens, and other hosts. Within these lineages, there are sublineages with evidence of host association, suggesting that host specificity can also evolve on a much more rapid time scale.
Using the association mapping method described here, we have shown that one factor driving rapid host adaptation is gain and loss of the panBCD genes encoding the vitamin B5 biosynthesis pathway, which has already been highlighted as especially variable in a previous study (13). Vitamin B5 is abundant in cereals and grains but is found in a very low concentration in grasses (14), which, respectively, constitute the main diets of chicken and cattle. This makes it likely that Campylobacter needs to produce the vitamin itself to persist in cattle. We found that the genes are almost universally present in isolates from cattle.
Our methods for detecting association, like those used in humans (15, 16), are influenced by genetic linkage. Linkage affects association signals because the recombination events that lead to the gain or loss of host-adaptive elements can also affect the neighboring sequence. The situation is complicated by the possibility that gene order also evolves in response to selection for linkage (17), leading to elements that give advantages in the same or overlapping environments being inherited together, for example, in “pathogenicity islands” (18).
We found patterns of host association in the genes adjacent to panBCD similar to those in the genes themselves. Three upstream genes (surE, Cj0294, and Cj0295) are also involved in biosynthesis. Immediately downstream is an ampicillin resistance-associated gene, blaOXA-61 (12), which is also associated with cattle. The gene is notable for being the least diverse of the common genes in the Campylobacter genome (Fig. S3), implying that it has recently spread among the diverse C. coli and C. jejuni lineages in our sample. It is intriguing that these seven adjacent genes appear to code for two functions related to the challenges of the agricultural niche, namely, antibiotics and persistence in ruminants. More data will be needed to determine the extent to which selection is acting specifically on panBCD, on blaOXA-61, or on the combined effect of the genes together.
Our results suggest that host generalism in agricultural Campylobacter is possible because elements that are necessary for colonization of cattle (specifically, the genes responsible for vitamin B5 biosynthesis) persist in some isolates in chickens. However, many details remain to be investigated, and it is not clear whether generalism is a stable strategy taking advantage of the large number of transmission opportunities in agriculture or whether it reflects insufficient time for well-adapted host specialist lineages to have evolved in this recent manmade environment.
As well as being found in many chicken isolates from host generalist lineages, the panBCD genes are found in isolates from chicken-associated lineages. It is not known how large a fitness cost, if any, these genes incur in environments in which vitamin B5 is plentiful. Furthermore, part of the transmission ecology of chicken-associated Campylobacter lineages might depend on vitamin B5 biosynthesis, for example, if spread is dependent on transition via cattle or other host species. This could compensate for the metabolic cost of maintaining these genes in the primary host.
Our study illustrates the potential of genome-wide association methods for studying rapidly evolving traits in bacteria. The molecular Koch’s postulates, defined by Falkow (19), describe a set of experimental criteria that should be satisfied to show that a gene found in a pathogenic microorganism contributes to the disease caused by the pathogen. The postulates can also be applied to other microbial traits, such as host specificity. Genetic association study methods, such as those used here and elsewhere (20), will facilitate the identification of genetic elements likely to cause phenotypes of interest and provide targets for further laboratory investigation.
Materials and Methods
Isolates and Genome Sequencing.
Isolates were chosen from multilocus sequence-typed collections that, together, contain >10,000 isolates (www.pubmlst.org/campylobacter) (21) to represent common STs found in clinical samples (feces) and those associated with various agricultural animal and wild bird species. In addition, multiple isolates from the ST-21 (C. jejuni), ST-45 (C. jejuni), and ST-828 (C. coli) clonal complexes were chosen from different hosts to investigate the association of genomic elements with phenotype (host source). Details of all the isolates used, including genomes from published sources (22–26), such as the National Center for Biotechnology Information database, are provided in Table S1.
Campylobacter isolates were subcultured on Columbia Blood Agar, and plates were incubated in microaerobic conditions (5% CO2, 5% O2, 3% H2, and 87% N2) at 42 °C for 48 h. Single-colony cultures were harvested, cells were suspended in 125 μL of water in a 0.2-mL PCR tube, and extraction of genomic DNA was carried out using the QIAamp DNA Mini Kit (QIAGEN GmbH). The DNA was resuspended in 100–200 μL of the elution buffer supplied and stored at −20 °C.
Whole-genome sequencing was carried out using a multiplex sequencing approach on an Illumina Genome Analyzer using the standard indexing protocol. Fragmentation of 2 μg of genomic DNA was carried out by acoustic shearing using a Covaris E210 sonicator. Following enrichment for 200-bp fragments, DNA was end-repaired and cleaned using a 1:1 ratio of Ampure paramagnetic beads (Beckman Coulter, Inc.) to remove DNA <150 bp in length. A-tailing was carried out, and adapters were ligated, with each followed by additional DNA clean-up. An overlap extension PCR was carried out, using an Illumina 3 primer set, to introduce specific tag sequences between the sequencing and the flow-cell binding sites of an Illumina adapter. A final DNA clean-up was carried out before DNA quantification by quantitative PCR. Twelve separately tagged libraries were sequenced simultaneously in two lanes of an eight-channel Illumina Genome Analyzer II flow cell. The average overall output was 80 Mbp per isolate. High coverage short reads (25–50 bp) were assembled de novo using Velvet software (27) to produce contigs of up to 162 kb. The 80 genome sequences that are published in this paper are available in the Dryad repository (10.5061/dryad.28n35).
Background Population Genetic Structure.
The publically accessible Bacterial Isolate Genome Sequence Database (BIGSdb) (21) was used to store contiguous sequences and whole-genome data from the GenBank. The finished genome of isolate NCTC11168 (25, 28, 29) was used as a basis for defining locus designations and reference sequences for each of these that were stored in the database. The BLAST algorithm was used to scan other genomes for gene orthologs at these loci, defined as reciprocal best hits of a sequence with ≥70% nucleotide identity and a 50% difference in alignment length. MUSCLE software (30) was used to align gene orthologs on a gene-by-gene basis, and these data were concatenated into contiguous sequence for each isolate genome, including gaps for missing nucleotides (or entire genes). A whole-genome tree of alignments was reconstructed for the entire genus using MEGA (31) version 3.1 with the Kimura two-parameter model and neighbor-joining clustering. In addition, gene-by-gene alignments were extracted from the BIGSdb (21) for genes present in all three major multihost lineages (ST-828, ST-21, and ST-45 clonal complexes). Genealogies for these alignments were estimated using ClonalFrame, a model-based approach to determining microevolution in bacteria (10). This program differentiates mutation and recombination events on each branch of the tree based on the density of polymorphisms. Clusters of polymorphisms are likely to have arisen from recombination, and scattered polymorphisms are likely to have arisen from mutation. The program was run with 50,000 burn-in iterations, followed by 50,000 sampling iterations. The consensus tree represents combined data from three independent runs with 75% consensus required for inference of relatedness. Recombination events were defined as sequences with a length of >50 bp with a probability of recombination ≥75% over the length, reaching 95% in at least one site.
Association Mapping.
One key aim of this study was to search for the genomic variants underlying preferential host colonization in C. jejuni. Such genome-wide association mapping has been extensively performed in humans (15, 16) but not in bacteria (32), which meant that methodology had to be developed to account for the specificities of bacterial population genetics. For each genome, the presence or absence of each unique “word” of 30 bp on the forward or reverse strand of any contig was recorded. Such short words are also known as “features,” and they have been used previously to build alignment-free phylogenies (33, 34). Here, however, we wanted to measure the statistical association of the presence of a word with the host from which each genome was isolated. This approach presents the advantage of being scalable to large numbers of genomes, alignment-free, and able to encapsulate in the same framework any genomic variant, whether it is caused by point mutation, homologous recombination, or lateral gene transfer. One difficulty, however, is that naively measuring the association of word presence with the host (e.g, using a Fisher’s exact test) would produce many spurious association due to the confounding effect of population structure. This is a well-known problem when performing association mapping in humans (35), but it is likely to be even more problematic in bacteria due to their clonal mode of reproduction (32). To control for the population structure when measuring the association of a word with a host, we used a technique inspired from phylogenetic comparative methods through Monte Carlo simulations (36, 37). Based on the phylogeny reconstructed by ClonalFrame (10), words were simulated to evolve through a process of gain and loss along the branches of the phylogeny. The correlation of the real word was then compared with the distribution of the correlations of the simulated words to produce a phylogenetically correct P value (38). To account for multiple testing, only words with a P value below 10−4 were considered significant. The distribution of host-associated words for which homologs were found in reference genome NCTC11168 (11, 29) was visualized using Artemis (39), and DNAPlotter (40) was used to generate Fig. 2A. To test the sensitivity of the method to word size, we performed the same analysis with 20-bp and 40-bp words and obtained comparable results (Fig. S4).
Phenotypic Analyses.
The ability to grow in controlled rich medium in presence or absence of vitamin B5 was assessed in vitro. The synthetic medium used in this analysis was composed of 1× Earle’s balanced salt solution (EBSS), 1× MEM essential amino acid solution, 1× MEM nonessential amino acids (catalog no. 24010-043; Invitrogen), 1 mM sodium pyruvate, 20 μM FeSO4, and 2.1 μM d-pantothenic acid 1/2 Ca salts (catalog no. P2250; Sigma–Aldrich). The pantothenic acid was added (“EBSS + B5”) or not (“EBSS − B5”) depending on experimental context. Overnight cultures in Mueller–Hinton broth were normalized to an OD600 of 0.05 and were used to inoculate wells in 96-well plates (10 μL in 150 μL). Growth was examined in the synthetic EBSS-based medium after 24 h using an Omega Microplate Reader with an atmospheric control unit module (BMG Labtech) at 42 °C in a modified atmosphere composed of 10% CO2 and 5% O2.
Supplementary Material
Acknowledgments
We thank Dr. Bruce Pearson for helpful comments and advice. S.K.S. is a Wellcome Trust Fellow. This work was carried out with support from the Wellcome Trust and the Biotechnology and Biological Sciences Research Council.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Genome sequence data for isolates that were sequenced in this study have been deposited with Dryad, http://datadryad.org/. The data will be available in 48 h from the date of resubmission with the following doi:10.5061/dryad.28n35. Data will also be available via PubMLST, http://pubmlst.org/campylobacter/.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305559110/-/DCSupplemental.
References
- 1.Woolhouse ME, Taylor LH, Haydon DT. Population biology of multihost pathogens. Science. 2001;292(5519):1109–1112. doi: 10.1126/science.1059026. [DOI] [PubMed] [Google Scholar]
- 2.Giraud A, et al. Costs and benefits of high mutation rates: Adaptive evolution of bacteria in the mouse gut. Science. 2001;291(5513):2606–2608. doi: 10.1126/science.1056421. [DOI] [PubMed] [Google Scholar]
- 3.Moxon R, Bayliss C, Hood D. Bacterial contingency loci: The role of simple sequence DNA repeats in bacterial adaptation. Annu Rev Genet. 2006;40:307–333. doi: 10.1146/annurev.genet.40.110405.090442. [DOI] [PubMed] [Google Scholar]
- 4.Kim JS, et al. Passage of Campylobacter jejuni through the chicken reservoir or mice promotes phase variation in contingency genes Cj0045 and Cj0170 that strongly associates with colonization and disease in a mouse model. Microbiology. 2012;158(Pt 5):1304–1316. doi: 10.1099/mic.0.057158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Killiny N, Almeida RPP. Gene regulation mediates host speci. city of a bacterial pathogen. Environl Microbiol Rep. 2011;3(6):791–797. doi: 10.1111/j.1758-2229.2011.00288.x. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman S. Bacterial regulation: Global regulatory networks. Annu Rev Genet. 1984;18:415–441. doi: 10.1146/annurev.ge.18.120184.002215. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy ND, et al. Host-associated genetic import in Campylobacter jejuni. Emerg Infect Dis. 2007;13(2):267–272. doi: 10.3201/eid1302.060620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheppard SK, et al. Niche segregation and genetic structure of Campylobacter jejuni populations from wild and agricultural host species. Mol Ecol. 2011;20(16):3484–3490. doi: 10.1111/j.1365-294X.2011.05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griekspoor P, et al. Marked host specificity and lack of phylogeographic population structure of Campylobacter jejuni in wild birds. Mol Ecol. 2013;22(5):1463–1472. doi: 10.1111/mec.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175(3):1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkhill J, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403(6770):665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 12.Griggs DJ, et al. Beta-lactamase-mediated beta-lactam resistance in Campylobacter species: Prevalence of Cj0299 (bla OXA-61) and evidence for a novel beta-Lactamase in C. jejuni. Antimicrob Agents Chemother. 2009;53(8):3357–3364. doi: 10.1128/AAC.01655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearson BM, et al. Comparative genome analysis of Campylobacter jejuni using whole genome DNA microarrays. FEBS Lett. 2003;554(1-2):224–230. doi: 10.1016/s0014-5793(03)01164-5. [DOI] [PubMed] [Google Scholar]
- 14.McDonald P. Animal Nutrition. 7th Ed. Harlow, England: Prentice Hall/Pearson; 2011. [Google Scholar]
- 15.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wellcome Trust Case Control Consortium Craddock N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballouz S, Francis AR, Lan R, Tanaka MM. Conditions for the evolution of gene clusters in bacterial genomes. PLOS Comput Biol. 2010;6(2):e1000672. doi: 10.1371/journal.pcbi.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 19.Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–S276. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- 20.Beres SB, et al. Genome-wide molecular dissection of serotype M3 group A Streptococcus strains causing two epidemics of invasive infections. Proc Natl Acad Sci USA. 2004;101(32):11833–11838. doi: 10.1073/pnas.0404163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheppard SK, et al. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol Ecol. 2013;22(4):1051–1064. doi: 10.1111/mec.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson BM, et al. The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828) J Bacteriol. 2007;189(22):8402–8403. doi: 10.1128/JB.01404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fouts DE, et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol. 2005;3(1):e15. doi: 10.1371/journal.pbio.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkhill J, et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413(6858):848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 26.Lefébure T, Bitar PD, Suzuki H, Stanhope MJ. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol. 2010;2:646–655. doi: 10.1093/gbe/evq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabello H, et al. Bacterial colonization of distal airways in healthy subjects and chronic lung disease: A bronchoscopic study. Eur Respir J. 1997;10(5):1137–1144. doi: 10.1183/09031936.97.10051137. [DOI] [PubMed] [Google Scholar]
- 29.Gundogdu O, et al. Re-annotation and re-analysis of the Campylobacter jejuni NCTC11168 genome sequence. BMC Genomics. 2007;8:162. doi: 10.1186/1471-2164-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9(4):299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falush D, Bowden R. Genome-wide association mapping in bacteria? Trends Microbiol. 2006;14(8):353–355. doi: 10.1016/j.tim.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Sims GE, Jun SR, Wu GA, Kim SH. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc Natl Acad Sci USA. 2009;106(8):2677–2682. doi: 10.1073/pnas.0813249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sims GE, Kim SH. Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs) Proc Natl Acad Sci USA. 2011;108(20):8329–8334. doi: 10.1073/pnas.1105168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36(5):512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 36.Martins EP, Garland T. Phylogenetic analyses of the correlated evolution of continuous characters: A simulation study. Evolution. 1991;45(3):534–557. doi: 10.1111/j.1558-5646.1991.tb04328.x. [DOI] [PubMed] [Google Scholar]
- 37.Garland T, Jr, Bennett AF, Rezende EL. Phylogenetic approaches in comparative physiology. J Exp Biol. 2005;208(Pt 16):3015–3035. doi: 10.1242/jeb.01745. [DOI] [PubMed] [Google Scholar]
- 38.Garland T, Dickerman AW, Janis CM, Jones JA. Phylogenetic analysis of covariance by computer simulation. Syst Biol. 1993;42:265–292. [Google Scholar]
- 39.Rutherford K, et al. Artemis: Sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 40.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.