

Abstract
Phagocytes clear the body of undesirable particles such as infectious agents and debris. To extend pseudopods over the surface of targeted particles during engulfment, cells must change shape through extensive membrane and cytoskeleton remodeling. We observed that pseudopod extension occurred in two phases. In the first phase, pseudopods extended rapidly, with actin polymerization pushing the plasma membrane forward. The second phase occurred once the membrane area from preexisting reservoirs was depleted, leading to increased membrane tension. Increased tension directly altered the small Rho GTPase Rac1, 3′-phosphoinositide, and cytoskeletal organization. Furthermore, it activated exocytosis of vesicles containing GPI-anchored proteins, increasing membrane area and phagocytosis efficiency for large particles. We thus propose that, during phagocytosis, membrane remodeling, cytoskeletal organization, and biochemical signaling are orchestrated by the mechanical signal of membrane tension. These results put a simple mechanical signal at the heart of understanding immunological responses.
Keywords: frustrated phagocytosis, immunology, phagocytic cell, Rho GTPases, traffic
Fcγ receptor (FcγR)-mediated phagocytosis is a vital component of innate immunity required for clearance of undesirable particles such as pathogenic bacteria or apoptotic cells (1–3). Following recognition of surface-bound IgG molecules, phagocytes dramatically change their shape, extending large pseudopodia around the target particle before engulfment. This change in shape is critical for ingestion of large particles (>3 µm) and is dependent upon membrane remodeling, cytoskeletal organization, and biochemical signaling. To promote efficient engulfment, phagocytic cells need to remodel their membrane and increase their surface area through exocytosis (4, 5). Actin cytoskeleton polymerization and reorganization in the pseudopod are directed by Rho GTPase (6) and phosphoinositide-mediated signaling pathways (7, 8). However, how these separate pathways operate in concert to achieve efficient phagocytosis of large particles is unclear.
Recently, plasma membrane area and tension (9, 10) have emerged as major players in determining cell shape, controlling the shape persistence of migrating keratocytes (11), and generating neutrophil polarity (12). Moreover, the mechanical feedback of plasma membrane tension informs spreading fibroblasts of their own physical limits (13), leading to exocytosis activation and cytoskeleton remodeling. Because the cell functions required for large particle phagocytosis are closely related to those under membrane tension control, we investigated the potential roles of membrane tension as a coordinating cue for phagocytosis.
Results and Discussion
To assess how membrane and cytoskeletal remodeling are linked to pseudopod extension, we used high-resolution microscopy of macrophages attempting to internalize an IgG-opsonized glass surface, a phenomenon termed “frustrated phagocytosis.” Although this system has a long history in the field (14–17), little attention has been given to dynamics at early times. Cells initially spread rapidly on the surface, displaying isotropic and homogenous pseudopod extension (Fig. 1A, Fig. S1A, and Movies S1 and S2), and then underwent an abrupt transition (denoted by “T”) in spreading rate such that the contact area progressively plateaued (Fig. S1 B and C). Contact area at the transition scaled proportionally with suspension cell size (i.e., larger cells spread to a bigger area before undergoing a transition), strongly indicating that the observed transition was linked to an intrinsic cell-size–related parameter (Fig. 1C), rather than a critical threshold of FcγR activation.
Fig. 1.

Macrophage pseudopod extension occurs in two phases during FcγR-mediated frustrated phagocytosis and correlates with membrane and cytoskeleton dynamics. (A) Spinning-disk confocal microscopy of a macrophage undergoing frustrated phagocytosis showing plasma membrane (PM-GFP, Upper) and F-actin (Ruby-Lifeact, Lower). A single confocal section at the glass interface is shown. Numbers indicate time in seconds (Movie S1). (Scale bar, 10 µm.) (B) Motility map of the cell in A showing protrusion velocity along the circumferential cell edge as a function of time. P1 and P2 indicate the motility phases before and following the transition, T, respectively. (C) Cell-contact area at the transition versus initial cell size on first contact with the opsonized surface. (D) Zoom from plasma membrane in A. Folds of plasma membrane (bright puncta visible in the first image) are exhausted before the transition. (E) Three-dimensional reconstruction of a spinning-disk microscopy time series during frustrated phagocytosis. Dorsal membrane folds (clearly visible at 0 s) are exhausted before the transition (at 120 s). Subsequently, ruffles and folds (indicated by the white arrow) recover on the cell surface.
Examination of the plasma membrane revealed that, initially, membrane folds were present on both the ventral (Fig. 1 A and D and Movie S2) and the dorsal cell surface (Fig. 1E and Movie S3). Isotropic pseudopod extension proceeded until all of the folds were depleted. By quantifying the amount of membrane contained in the folds in early first phase (P1) we estimated that cells kept at least 20–40% of membrane area in folds before the transition (Fig. S1D, 28 ± 0.04% SEM and ±13.4% SD of the total membrane area, n = 13 cells from 13 independent experiments). At the transition, the pseudopod edge drastically reduced its speed and lost homogeneity (Fig. 1B). Simultaneous examination of the actin cytoskeleton showed a stable organization with respect to the pseudopod edge during P1. At the transition, the actin presented a dramatic reorganization (Fig. 1A and Movie S2). At the beginning of the transition, a closer look at the pseudopod showed a clear reinforcement of the actin signal behind the leading edge (Fig. 1A: compare time 70 s with time 140 s; Movie S2), suggesting that the actin may react to a signal at the membrane–cytoskeleton interface. Once past the transition, in a second phase (P2), a global change in actin reorganization occurred with the apparition of large actin bundles in the central part of the cell (Fig. 1A, time 210 s; Movie S2), whereas the cell extended outward only slowly, with small and local cycles of protrusion–retraction, primarily restoring preexisting dorsal folds (Fig. 1E and Movie S3). This indicated that membrane and cytoskeleton remodeling are linked together through the transition.
But what signal controlled the transition? To investigate this further, we examined frustrated phagocytosis on 30-µm circles of IgG (18). Cells that landed on the circle edge extended pseudopods isotropically over both coated and noncoated areas alike, up to the transition (Fig. 2A and Movie S4). Only after the transition did cells acquire polarity, reducing their spread area on the uncoated substrate while continuing to expand and ruffling extensively on IgG (Fig. 2A and Movie S4). Thus, in contrast to the standard zipper-like model of phagocytic cup growth (19), -mediated adhesion is critical neither for early pseudopod extension (following initial activation) nor for the transition. By pulling membrane tethers with an optical trap during the first phase of pseudopod extension, we observed a clear peak in tether force as the cell passed through the transition (Fig. 2B and Movie S5). This is consistent with a peak in membrane tension occurring between the two steps of pseudopod extension [membrane tension is proportional to the square of tether force (20)], suggesting that an increase in plasma membrane tension is the signal for the transition. Moreover, by pulling membrane tethers during early P2, when local cycles of protrusion and retraction occur, we also observed a very clear fluctuation in tether force, with alternating increase (during protrusion) and decrease (during retraction) separated by a peak at the transition (47 pN during protrusion to 58 pN at the peak and then 45 pN during retraction, n = 12 cells; see Fig. S1E for quantification). Pseudopod extension occurs through actin polymerization pushing the membrane forward; therefore, a large proportion of the membrane tension observed could come from this pushing force. Using mild latrunculin A treatment (100 nM) to dampen but not totally inhibit actin polymerization (Fig. S2), we observed that the tether force was about three times lower in P1 than in controls (16.1 ± 2.2 pN SEM, n = 7 cells, Fig. 2B). In fact, pulling tether under mild latrunculin treatment was significantly easier than in control cases allowing for robust quantification. As we previously reported for lamellipodia extension during fibroblast spreading on fibronectin (21), radial pseudopod extension in P1 was also four times slower under latrunculin treatment (Fig. S2C), reinforcing the idea that actin polymerization is responsible in large part for tension generation by rapidly pushing the membrane forward. Moreover, at the P1–P2 transition, under latrunculin treatment, the characteristic temporary peak in membrane tension was stronger (in relative terms, 36.3 ± 6.2 pN, at the transition T, and then 16.6 ± 3.5 pN in early P2, SEM, n = 7 cells) and clearer (very strong and short lasting) than in the control case (Fig. 2B). Given that the contact area at the transition also scaled with cell size (Fig. 1C), our results indicated that the origin of the signal for the transition came only from the cell, reinforcing the notion that an increase in membrane tension is the key element.
Fig. 2.

An increase in membrane tension promotes the transition in pseudopod extension. (A) A RAW macrophage monitored by transmitted light-extending pseudopods on the edge of a circular pattern of IgG indicated by the red circle (Movie S4). (B) Membrane tension (indicated by tether force) dynamics at the transition in control (Left) or latrunculin A-treated (Right) cells. Blue dotted line in the center indicates approximate time of the transition, T. (Upper) Red dotted line shows the cell edge. (C) the small Rho GTPase Rac1 (CyPet-Rac, Top), active Rac (YPet-PBD, Middle), and activation ratio (YPet:CyPet ratio, Bottom) during frustrated phagocytosis (Movie S3). (D) Kymograph of Rac activation ratio shown in C. (E) Kymograph of 3′-phosphoinositide distribution (Akt-PH-GFP) through the transition. (F) Kymograph of YPet-PBD localization during early spreading when a hypotonic shock (0.5× Ringer’s) is applied. Recovery of tonicity (1.0× Ringer’s) results in restored protrusive activity.
We investigated how biochemical signaling could respond to the physical signal of membrane tension at the transition. The activity of the small GTPase Rac1 is known to relocate from the pseudopod to the cup base during engulfment of large particles (6) in a biphasic manner. By following Rac1 activity with fluorescent probes (22), we observed a clear redistribution of active Rac from the pseudopod to the cell center at the transition [Fig. 2 C and D and Movie S6, reduction in P-21 activated protein kinase binding domain (PBD):Rac1 ratio at the transition in the 5-μm region behind the pseudopodia edge: −48 ± 8%, SD, n = 11 cells]. Moreover, although the activity in the pseudopod appeared steady and stable during P1, the activity in the center of the cell during P2 seemed rather unstable and linked to a burst of local Rac1 activation. As Rac is also known to be activated by phosphatidylinositol trisphosphate (PIP3)-dependent guanine nucleotide exchange factors (GEFs), we examined the 3′-phosphoinositide distribution using the pleckstrin homologous domain of the protein Kinase AKT (GFP-Akt-PH) distribution (Fig. 2E and Movie S7). A significant decrease of fluorescence (reduction in Akt-PH domain level at the transition in the 5-μm region behind the pseudopodia edge: −37.7 ± 3.4%, SD, n = 10 cells) was apparent in the pseudopod at the transition, consistent with a deactivation of Rac-GEFs. Such a decrease was not observed in plasma membrane staining (Fig. 1A and Movie S2). The only method to date to artificially increase membrane tension is to subject cells to hypotonic stress (12, 13, 23, 24). Whereas it is not possible to totally exclude side effects on other signaling pathways, we did observe that replacing media with hypotonic solution at half tonicity clearly induced an increase in membrane tension in macrophages (Fig. S3C). This increased tension was correlated with a stalling of pseudopod extension concomitant with the immediate loss of Rac1 activity (Fig. 2F and Movie S8). A subsequent decrease in tension (by restoration of isotonic media) in the same cell relocated some Rac1 activity to the pseudopod, restoring protrusive activity (Fig. 2F and Movie S8). Reinforcing our observation, Rac1 activity has recently been shown to be modulated by membrane tension (12). Our results show that membrane tension not only induces the transition but also modulates the biochemical pathways responsible for pseudopod extension.
Macrophages have been described to increase their plasma membrane area during phagocytosis (4). This has been linked to activation of secretion pathways (25). However, both the spatiotemporal organization and the respective contribution of each pathway to the total area capacity are far from clear (17). Using a lipid-dye N-(3-triethylaminoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide (FM1-43)-based assay to monitor cell-surface area (26), no clear secretion was recorded before the transition (Fig. 3 A–C), confirming that preexisting membrane folds provide the extra membrane area required for initial pseudopod extension. Secretion was activated at the transition, associated with a global increase in plasma membrane area of 40 ± 7% (Fig. 3D). Moreover, to demonstrate that membrane tension can increase membrane area, we applied a hypotonic shock (9, 27) to macrophages cultured on glass. This resulted in rapid activation of exocytosis (Fig. S3A) and an increase in plasma membrane area of 30% (Fig. S3B), similar to that induced by frustrated phagocytosis. The two observed phases are thus linked to two membrane-remodeling mechanisms: (i) a rapid pseudopod extension while plasma membrane buffer (folds) is available and the membrane area stays constant and (ii) a much slower extension, after the increase in membrane tension, where secretion activation creates an extra membrane area. The increase in cell-surface area during frustrated phagocytosis is strikingly similar to the fibroblast capacity to increase membrane area during integrin-mediated spreading on fibronectin (26). This highlights how simple physical parameters (membrane tension) underlie shape changes, whereas chemical signaling and function may differ across cell type.
Fig. 3.

The transition promotes membrane remodeling. (A) FM 1–43 intensity and cell-contact area dynamics for a single cell during frustrated phagocytosis. Exocytosis (intensity increase) occurs following the transition, T. P1 and P2 indicate the phases before and after the transition, respectively. (B) FM 1–43 intensity versus contact area for the cell in A. An intensity increase occurs only once cells have passed through the transition. (C) Average rates of contact area and FM 1–43 intensity (exocytosis) increase during P1 and P2 (quantified from nine cells). Error bars show SEM. (D) Cell plasma membrane area (FM 1–43 intensity in arbitrary units) versus spread area (area of the cell in contact with the substrate) before (suspension cells just touching down on the substrate, blue dots) and after 30 min of frustrated phagocytosis (spread cells, red dots). Each point represents one cell. Mean and SD for each condition are shown. (E) Quantification of GPI exocytosis events and cell-contact area for a cell passing through the transition. (F) Comparison of exocytosis rates observed for four compartments (GPI, Golgi, LAMP, and ER) before (P1) and after (P2) transition and following a subsequent hypotonic shock (0.5 × R; R, ringer). (G) (Left) Overlay of PM-GFP and Ruby-Lifeact fluorescence in a macrophage spreading on IgG (arrow with dotted outline indicates vesicle before fusion). (Right) PM-GFP (Top), Ruby-Lifeact (Middle), and overlay of a cross-section through the cell in the Left panel containing a vesicle (Bottom). (H) PM-GFP (Top), Ruby-Lifeact (Middle), and overlay (Bottom, viewed from side) showing actin recruitment and subsequent shrinkage and fusion of the vesicle into the ventral surface (10 s per frame) (Movie S4).
In fibroblasts, the major compartment regulating membrane area during shape changes is a recycling pathway of membrane containing GPI-anchored proteins (26, 28). As the relative contribution of membrane from intracellular compartments to the phagocytic cup remains largely controversial (17), we investigated whether a GPI-linked compartment provides membrane area during frustrated phagocytosis. By quenching external GFP fluorescence on cells transfected with GFP–GPI, we observed a variety of differently sized unquenched vesicles apposed to the ventral surface (Fig. S4A). Fusion (Fig. S4B) was observed in the 3 min following the transition up to a peak of 30 events per minute (Fig. 3E and Movie S9). The contributions of Golgi (Fig. S5 A and B), endoplasmic reticulum (Fig. S5C), and lysozomes (Fig. S5D) were also addressed. Exocytosis of Golgi, previously implicated in cytokine secretion during phagocytosis (29), was observed but occurred at a relatively low rate (up to five events per minute in P2). No exocytosis of lysozomes or endoplasmic reticulum (ER) was recordable (Fig. 3F). Thus, we propose the GPI-anchored protein-containing compartment is a major contributor of the exocytosis occurring during phagocytosis to increase membrane area, strengthening its central role during shape changes (9, 30). Interestingly, following the transition, actin was transiently recruited to preexisting large vesicular structures [labeled with either plasma membrane GFP (PM-GFP) or GPI–GFP] at the back of the pseudopod (Fig. 3 G and H and Fig. S4C). These vesicles subsequently shrank, dissipating into the ventral membrane, whereas the actin coat initially concentrated before dissipating with similar kinetics (Fig. 3H, Fig. S6, and Movie S10). This strongly suggests that actin plays a dynamic role during exocytosis of this membrane-recycling pathway and is in agreement with our previous observations suggesting that this GPI-linked exocytic pathway was independent of microtubules (26).
To extend our observations, we addressed the role of membrane tension during the phagocytosis of beads, which, as opposed to frustrated phagocytosis, allowed macrophages to finish engulfment (Fig. 4A). As opposed to the isotropic geometry of frustrated phagocytosis, macrophage-ingesting beads are already attached to a substrate and very heterogenous in shape. This makes attempts to accurately measure the membrane tension fluctuations resulting from bead engulfment challenging. Thus, we compared the average membrane tension between populations of resting and phagocytosing cells. A 50% increase in tether force was observed for cells undergoing phagocytosis (extending pseudopodia around a bead), indicating that membrane tension was increased by more than twofold (Fig. 4B). Inhibition of PI3-kinase with wortmannin, which has been shown to block both pseudopod extension (7) and phagocytosis at an intermediate stage (31), prevented the increase in tension (Fig. 4B). This indicates that pseudopod extension leads to increased membrane tension. To mimic the spike in tension measured at the transition in frustrated phagocytosis (Fig. 2B), a hypotonic shock was applied following ligation of beads (Fig S3C). The increased membrane tension (Fig. S3C) resulted in a factor 4 increase in phagocytic efficiency after 5 min (Fig. 4C and Fig. S7A) compared with isotonic control. Remarkably, this effect partially overcame wortmannin treatment (Fig. 4C and Fig. S3C), indicating that, even if pseudopod extension is inhibited, restoring the signal of increased tension is enough to achieve engulfment.
Fig. 4.

Phagocytosis of particles is promoted by increased membrane tension. (A) Transmitted light (Top), Alexa-488–anti-IgG (Middle), and overlay of macrophages (Bottom) ingesting 8-µm IgG-opsonised latex beads, fixed after 20 min. (B) Comparison of tether force measured in either resting cells or macrophages ingesting 8-µm beads in the presence or absence of 100 nM wortmannin. “N” indicates the number of data points in each condition. (C) Phagocytic efficiency (relative to 20-min ingestion) for cells without shock, with isotonic shock, or with hypotonic shock with or without wortmannin (100 nM) or NEM (2 µM). (D) Macrophages were incubated with wortmannin (100 nM) and exposed to 8-µm IgG-opsonised latex beads, resulting in stalled phagocytosis. After 50 s, a hypotonic shock (0.5× Ringer’s) was applied, initiating particle ingestion. (E) Model for pseudopod extension. After bead engagement (i), PIP3-Rac1 signaling is activated and actin polymerization pushes the pseudopod membrane forward around the particle during the first phase (ii). Membrane tension is higher than prior engagement because of protrusion, leading to membrane buffer (folds) depletion. At the transition (iii), a spike in membrane tension due to competition between membrane area depletion and protrusive activity leads to PIP3-Rac1 deactivation and actin reorganization in the pseudopod as well as exocytosis activation. In the second phase of pseudopod extension (iv), the membrane area is increased by an actin-associated exocytosis of GPI-anchored protein-containing vesicles. Particle internalization is initiated, and pseudopodia extension progressively wraps the particle as it enters the cell body. After engulfment (v), maturation is initiated and subsequently involves lysosome fusion and, potentially, Golgi and ER compartments.
To determine how particles were ingested following the externally applied tension spike, we blocked pseudopod extension with wortmannin (to prevent a cell-mediated tension increase) and subsequently applied a hypotonic shock while observing F-actin and 3′-phosphoinositide distribution (Fig. 4D and Movie S11). Before the shock the beads were partially engulfed, but the nascent phagocytic cups were stable and F-actin had a uniform distribution, consistent with a lack of protrusion. On application of the tension spike the beads rapidly moved into the cell body, whereas pseudopods showed relatively little outward extension, as observed in the second phase of frustrated phagocytosis (Fig. 1 and Fig. S1). This indicated that the second phase, rather than promoting outward pseudopod protrusion, led to inward bead movement and ingestion, most probably through contraction and exocytosis activation. The tension-induced increase in efficiency was effectively prevented by inhibition of all secretion by N-ethylmaleimide (NEM) (32) (Fig. 4C), but was independent of myosin II-dependent contractility (Fig. S7B) or the Rho-signaling pathway (Fig. S7C). These results confirmed the role of membrane tension as an exocytosis activator and the requirement for exocytosis to complete phagocytosis.
A previous study reported a phagocytic-signaling transition following which there was a large increase in the density of PIP3 and actin in the phagocytic cup, described by a concentration threshold (33). The authors conclude that their data provide some support for the trigger model of phagocytosis, as opposed to the more conventionally favored zipper-like model. We observe that both models appear to be valid, but depending on the cell motile state and availability of membrane. The P1 phase is indicative of an initial FcγR-dependent trigger, and, following the tension-mediated transition, a global zippering on the IgG-coated surface occurs. Zhang et al. proposed that recruitment of the p85 subunits of the PI3 kinase may depend on surpassing a 3′-phosphoinositides threshold. Although this is possible, the leading-edge modifications that we observed at the transition and under osmotic shock are also consistent with a role for actin in directing lipid synthesis, as the physical force of tension acting on the cytoskeleton is concurrent with changes in Akt-PH distribution. Although lipid signaling has been shown to act upstream of cytoskeleton regulation (e.g., through RacGEFs), in this case we are of the view that the physical signal of tension most likely acts directly on the cytoskeleton. Therefore, we hypothesize that the cytoskeleton can in fact also direct lipid synthesis and that external physical stresses placed on the cytoskeleton could thus modify the distribution of phosphoinositides. We are currently investigating this possibility in further studies.
Conclusion
During phagocytosis, particularly of large particles, cortical tension (34), contractility (35), and phosphoinositide signaling (36) have been reported to display biphasic behavior. However, an integrated picture of the spatiotemporal organization of cell functions during these distinct phases has not been developed. We propose here that during phagocytosis the biphasic behavior of membrane remodeling, cytoskeletal organization, and biochemical signaling is coordinated by macrophages using the simple physical signal of the plasma membrane tension (see our working model in Fig. 4E). These results put membrane area and tension as master regulators of a fundamental immune response, and more generally outline, as previously suggested for phagocytosis (37), the increasing importance of mechanosensation as a means to orchestrate cell functions.
Experimental Procedures
Reagents, Solutions, and Plasmids.
Hypotonic medium (0.5× Ringer’s) is composed of 1× Ringer’s (150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM Hepes, and 2 g/L glucose, pH 7.4) mixed 1:1 with H2O, but maintaining 1 mM Ca2+ and 1 mM Mg2+. PM-GFP was described previously (38). Ruby-Lifeact was a kind gift from Roland Wedlich-Soldner (Max Planck Institute of Biochemistry, Martinsried, Germany) (39). Cyan fluorescent protein CyPet-Rac1 optimized for FRET with the yellow fluorescent protein YPet-PBD [the Rac1-GTP–binding domain of PAK, Addgene plasmids 22782 and 22781, from Klaus Hahn (University of North Carolina, Chapel Hill, NC) (22)] and GFP-Akt-PH [Addgene plasmid 21218 from Tobias Meyer (Stanford University, Stanford, CA) (40)] were from Addgene (www.addgene.org). GFP-GPI, YFP-Golgi, ER-dsRed (red fluorescent protein from Discosoma sp.), and GFP-lysosomal associated membrane protein 1 (LAMP1) were described previously (26). FM 1–43 was from Invitrogen. All other chemicals were from Sigma Aldrich.
Cell Culture.
Abelson murine leukemia virus transformed macrophages (RAW 264.7) were obtained from the American Type Culture Collection and grown in high-glucose DMEM supplemented with 10% heat-inactivated FBS (HI-FBS) and penicillin–streptomycin. Transfections were performed with a Neon electroporator (Invitrogen) as per manufacturer’s recommendations.
Frustrated Phagocytosis.
Cells cultured on cell culture plastic were lightly scraped and resuspended in RPMI supplemented with 20 mM Hepes and 10% HI-FBS, followed by rotation at 37 °C for 2–3 h. One hour before seeding back the cells, serum was removed. Cells were added to chambers containing 1× Ringer’s solution, and microscopy was performed on a heated stage at 37 °C.
Membrane Tension Measurements.
An optical tweezer (Elliot Scientific) was generated by focusing the output of a 5.0-W, 1,064-nm laser through a 100× (1.3 N.A.) objective (Nikon) mounted on a Nikon A1-R microscope. Tethers were pulled from cells using 1-µm polystyrene beads (Polysciences) coated in Con A. Bright-field images were acquired using an Andor camera. Trap strength was calibrated by observing Brownian motion of trapped beads with an exposure time of 0.6 ms to minimize motion blur [application of a previous method (41) led to corrections of the trap strength of less than 5%].
Quantification of Plasma Membrane Area.
This assay was described previously (26). In summary, frustrated phagocytosis was allowed to proceed for 30 min on a glass substrate coated in IgG. Thereafter, fresh suspension macrophages were added and allowed to attach to the substrate before media was replaced with ice cold 1× Ringer’s, and the sample was cooled to 4 °C to prevent further spreading and membrane turnover. Media was then replaced with ice-cold 1× Ringer’s containing 5 µg/mL FM 1–43. Differential interference contrast microscopy (DIC) and whole-cell fluorescence images were imaged through a 40× water objective on a microscope stage cooled with melting ice. Illumination was provided by a wide-field Hg source with a standard GFP filter set, and images were acquired using an Evolve EMCCD camera (Photometrics). Images were analyzed in ImageJ. Total FM 1–43 fluorescence intensity was calculated by subtracting background fluorescence from a region of interest enclosing individual whole cells. The amount of membrane present in folds was quantified by analyzing the density of fluorescence in the substrate-attached surface of cells expressing PM-GFP during frustrated phagocytosis (as presented in Fig. 1D). Early in the process, membrane is folded and thus bright puncta are visible; however, as the cell extends across the surface, the folds are lost. We considered the membrane at the transition to be smooth (and thus its fluorescence density is that of a smooth membrane) and assume that folds are distributed evenly over the entire cell surface. We thus estimate the amount of membrane in folds early in P1 by dividing the observed initial fluorescence density with that recorded at the point at which the cell passes through the transition.
FM 1–43 Fluorescence Dynamics.
Resuspended macrophages were mixed with an equal volume of 1× Ringer’s containing 10 µg/mL FM 1–43 and then added to a chamber containing 5 µg/mL FM 1–43 with an IgG-coated coverslip at its base. The chamber was mounted on a microscope at 37 °C, and DIC and whole-cell fluorescence images were acquired (as described in the preceding section) at 10-s intervals as the cells extended pseudopods on the surface. Intensities were extracted from images with ImageJ and background-corrected with Excel.
FM 1–43 is a soluble membrane impermeable dye that has a quantum yield 40× higher in lipid membranes. Thus, total FM 1–43 fluorescence emission from a cell at each time point is linearly related to the sum of the initial plasma membrane area (on addition of the dye assuming instantaneous attainment of loading equilibrium) and the total net membrane exocytosed (not recycled) up to that time point (equivalent to the instantaneous plasma membrane area plus the total membrane endocytosed up to that point); i.e., 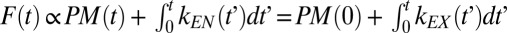 , where kEN(t) and kEX(t) are the time-dependent endocytic and exocytic rates, respectively. Thus, temporal variation of FM 1–43 reveals periods with high membrane turnover, visible as sharp increases in fluorescence.
, where kEN(t) and kEX(t) are the time-dependent endocytic and exocytic rates, respectively. Thus, temporal variation of FM 1–43 reveals periods with high membrane turnover, visible as sharp increases in fluorescence.
Microscopy.
Spinning disk confocal microscopy was performed with a Perkin-Elmer confocal spinning unit connected to an Olympus IX 81 microscope body. Images were acquired through a 100× oil immersion objective with a Hamamatsu C9100-13 EMCCD camera at a rate of 5 s per frame. Images of YPet-PBD and CyPet-Rac1 were background-corrected before calculation of their respective ratio. Total internal reflection fluorescence (TIRF) images of vesicle exocytosis were obtained through a 100× oil immersion objective with an iLas2 TIRF system connected to an Olympus IX 81 microscope body. Images were acquired in stream mode with 150 ms per frame.
Effect of Hypotonic Shock on Phagocytic Efficiency.
Macrophages were grown on glass coverslips overnight, placed in 1× Ringer’s solution, and allowed to engage beads on ice for 10 min to synchronize phagocytosis. Cells were then warmed to 37 °C for 2 min to initiate the phagocytic response, before no shock, an isotonic media replacement, or a hypotonic shock with 0.5× Ringer’s solution was applied. Cells were allowed to continue phagocytosis until fixation at the indicated time point (5, 10, or 20 min) and stained with Alexa-488–anti-IgG antibody to reveal uningested beads. Phagocytic efficiency was calculated as the number of beads eaten per total beads engaged, relative to the efficiency of the 20-min unshocked condition. Error bars were calculated from at least three experiments. A one-tailed Student t test was used to determine the significance level of differences between conditions.
Measurement of membrane area increase following hypotonic shock. Cells were cultured overnight on glass coverslips, placed in 1× Ringer’s solution, and imaged with DIC and Hg wide-field illumination with a 40× water immersion objective at 37 °C as described above. FM 1–43 was introduced into the imaging chamber and equilibrated. Subsequently, media was replaced with either 1× or 0.5× Ringer’s containing FM 1–43.
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1301766110/-/DCSupplemental.
References
- 1.Botelho RJ, Grinstein S. Phagocytosis. Curr Biol. 2011;21(14):R533–R538. doi: 10.1016/j.cub.2011.05.053. [DOI] [PubMed] [Google Scholar]
- 2.Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. 2008;9(8):639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 4.Holevinsky KO, Nelson DJ. Membrane capacitance changes associated with particle uptake during phagocytosis in macrophages. Biophys J. 1998;75(5):2577–2586. doi: 10.1016/S0006-3495(98)77703-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huynh KK, Kay JG, Stow JL, Grinstein S. Fusion, fission, and secretion during phagocytosis. Physiology (Bethesda) 2007;22(6):366–372. doi: 10.1152/physiol.00028.2007. [DOI] [PubMed] [Google Scholar]
- 6.Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15(8):3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem. 1999;274(3):1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- 8.Scott CC, et al. Phosphatidylinositol-4,5-bisphosphate hydrolysis directs actin remodeling during phagocytosis. J Cell Biol. 2005;169(1):139–149. doi: 10.1083/jcb.200412162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gauthier NC, Masters TA, Sheetz MP. Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 2012;22(10):527–535. doi: 10.1016/j.tcb.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Keren K. Cell motility: The integrating role of the plasma membrane. Eur Biophys J Biophy. 2011;40(9):1013–1027. doi: 10.1007/s00249-011-0741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keren K, et al. Mechanism of shape determination in motile cells. Nature. 2008;453(7194):475–480. doi: 10.1038/nature06952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houk AR, et al. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell. 2012;148(1–2):175–188. doi: 10.1016/j.cell.2011.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gauthier NC, Fardin MA, Roca-Cusachs P, Sheetz MP. Temporary increase in plasma membrane tension coordinates the activation of exocytosis and contraction during cell spreading. Proc Natl Acad Sci USA. 2011;108(35):14467–14472. doi: 10.1073/pnas.1105845108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michl J, Pieczonka MM, Unkeless JC, Silverstein SC. Effects of immobilized immune-complexes on Fc-receptor and complement-receptor function in resident and thioglycollate-elicited mouse peritoneal macrophages. J Exp Med. 1979;150(3):607–621. doi: 10.1084/jem.150.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright SD, Silverstein SC. Phagocytosing macrophages exclude proteins from the zones of contact with opsonized targets. Nature. 1984;309(5966):359–361. doi: 10.1038/309359a0. [DOI] [PubMed] [Google Scholar]
- 16.Cannon GJ, Swanson JA. The macrophage capacity for phagocytosis. J Cell Sci. 1992;101:907–913. doi: 10.1242/jcs.101.4.907. [DOI] [PubMed] [Google Scholar]
- 17.Touret N, et al. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123(1):157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Masters TA, et al. Easy fabrication of thin membranes with through holes: Application to protein patterning. PLoS ONE. 2012;7(8):e44261. doi: 10.1371/journal.pone.0044261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffin FM, Griffin JA, Leider JE, Silverstein SC. Studies on mechanism of phagocytosis. 1. Requirements for circumferential attachment of particle-bound ligands to specific receptors on macrophage plasma membrane. J Exp Med. 1975;142(5):1263–1282. doi: 10.1084/jem.142.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheetz MP. Cell control by membrane-cytoskeleton adhesion. Nat Rev Mol Cell Biol. 2001;2(5):392–396. doi: 10.1038/35073095. [DOI] [PubMed] [Google Scholar]
- 21.Fardin MA, et al. Cell spreading as a hydrodynamic process. Soft Matter. 2010;6(19):4788–4799. doi: 10.1039/c0sm00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machacek M, et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batchelder EL, et al. Membrane tension regulates motility by controlling lamellipodium organization. Proc Natl Acad Sci USA. 2011;108(28):11429–11434. doi: 10.1073/pnas.1010481108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha B, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144(3):402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bajno L, et al. Focal exocytosis of VAMP3-containing vesicles at sites of phagosome formation. J Cell Biol. 2000;149(3):697–705. doi: 10.1083/jcb.149.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gauthier NC, Rossier OM, Mathur A, Hone JC, Sheetz MP. Plasma membrane area increases with spread area by exocytosis of a GPI-anchored protein compartment. Mol Biol Cell. 2009;20(14):3261–3272. doi: 10.1091/mbc.E09-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai JW, Ting-Beall HP, Sheetz MP. The secretion-coupled endocytosis correlates with membrane tension changes in RBL 2H3 cells. J Gen Physiol. 1997;110(1):1–10. doi: 10.1085/jgp.110.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howes MT, et al. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol. 2010;190(4):675–691. doi: 10.1083/jcb.201002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray RZ, Kay JG, Sangermani DG, Stow JL. A role for the phagosome in cytokine secretion. Science. 2005;310(5753):1492–1495. doi: 10.1126/science.1120225. [DOI] [PubMed] [Google Scholar]
- 30.Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol. 2010;22(4):519–527. doi: 10.1016/j.ceb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Beemiller P, Hoppe AD, Swanson JA. A phosphatidylinositol-3-kinase-dependent signal transition regulates ARF1 and ARF6 during Fc gamma receptor-mediated phagocytosis. PLoS Biol. 2006;4(6):987–999. doi: 10.1371/journal.pbio.0040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci USA. 1988;85(21):7852–7856. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Hoppe AD, Swanson JA. Coordination of Fc receptor signaling regulates cellular commitment to phagocytosis. Proc Natl Acad Sci USA. 2010;107(45):19332–19337. doi: 10.1073/pnas.1008248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herant M, Heinrich V, Dembo M. Mechanics of neutrophil phagocytosis: Behavior of the cortical tension. J Cell Sci. 2005;118(9):1789–1797. doi: 10.1242/jcs.02275. [DOI] [PubMed] [Google Scholar]
- 35.Evans E, Leung A, Zhelev D. Synchrony of cell spreading and contraction force as phagocytes engulf large pathogens. J Cell Biol. 1993;122(6):1295–1300. doi: 10.1083/jcb.122.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Botelho RJ, et al. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151(7):1353–1367. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beningo KA, Wang YL. Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J Cell Sci. 2002;115(Pt 4):849–856. doi: 10.1242/jcs.115.4.849. [DOI] [PubMed] [Google Scholar]
- 38.Corbett-Nelson EF, Mason D, Marshall JG, Collette Y, Grinstein S. Signaling-dependent immobilization of acylated proteins in the inner monolayer of the plasma membrane. J Cell Biol. 2006;174(2):255–265. doi: 10.1083/jcb.200605044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riedl J, et al. Lifeact: A versatile marker to visualize F-actin. Nat Methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raucher D, et al. Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell. 2000;100(2):221–228. doi: 10.1016/s0092-8674(00)81560-3. [DOI] [PubMed] [Google Scholar]
- 41.Wong WP, Halvorsen K. The effect of integration time on fluctuation measurements: Calibrating an optical trap in the presence of motion blur. Opt Express. 2006;14(25):12517–12531. doi: 10.1364/oe.14.012517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.