

Abstract
By covalently linking an azobenzene photoswitch across the binding groove of a PDZ domain, a conformational transition, similar to the one occurring upon ligand binding to the unmodified domain, can be initiated on a picosecond timescale by a laser pulse. The protein structures have been characterized in the two photoswitch states through NMR spectroscopy and the transition between them through ultrafast IR spectroscopy and molecular dynamics simulations. The binding groove opens on a 100-ns timescale in a highly nonexponential manner, and the molecular dynamics simulations suggest that the process is governed by the rearrangement of the water network on the protein surface. We propose this rearrangement of the water network to be another possible mechanism of allostery.
Subtle conformational transitions within the folded state of highly structured proteins are often an integral aspect in their functional mechanism. These conformational transitions can occur as a result of different events, such as ligand binding, covalent modification (e.g., phosphorylation), or proteolytic cleavage. When an event or perturbation at one site in a protein changes the enzymatic activity or the binding affinity to a ligand at another distant site, this process can be described as allostery. Hemoglobin has long served as the prototypical example to study this effect, where the binding of an oxygen in one subunit modifies the affinity of binding oxygen in another subunit (1, 2). The traditional models of allostery developed by Monod et al. (3) and Koshland et al. (4) attribute allosteric effects to conformational changes by which the allosteric binding site communicates with the distant active site. There is, however, increasing evidence that allostery can be mediated also without any conformational change, relying purely on changes in internal protein dynamics (5).
PDZ domains are an important class of protein interaction modules that have been studied extensively in the context of allostery. They are found in a large variety of proteins and generally bind the C termini of their targets (6–11). As scaffolding domains, they are molecular switches that play a central role in signal transduction. For several PDZ domain proteins, allosteric interactions are an important regulatory mechanism (10, 12–15).
NMR spectroscopy has been particularly useful to elucidate the equilibrium dynamics of proteins on various timescales. The notion of allostery mediated through a change in dynamic properties was corroborated by a study of the third PDZ domain from the PSD-95/SAP90 protein (16). This protein contains an additional C-terminal α-helix 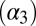 , which shows no direct interaction with the peptide ligand. Removal of
, which shows no direct interaction with the peptide ligand. Removal of 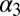 has a negligible effect on the structure of the PDZ core domain; however, it does lead to a large decrease in ligand binding affinity, which was shown to be entirely entropic in nature. Other studies have identified changes in (conformational) entropy of both backbone (17) and side-chain (18) dynamics in other systems to give rise to allosteric effects.
has a negligible effect on the structure of the PDZ core domain; however, it does lead to a large decrease in ligand binding affinity, which was shown to be entirely entropic in nature. Other studies have identified changes in (conformational) entropy of both backbone (17) and side-chain (18) dynamics in other systems to give rise to allosteric effects.
Here, we focus on the second PDZ (PDZ2) domain from human tyrosine-phosphatase 1E (hPTP1E), which has been demonstrated to possess allosteric properties (19). The PDZ domain is a small 96-residue protein with a binding groove between the 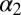 -helix and the
-helix and the 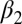 -strand (Fig. 1B). As mentioned previously, side-chain dynamics in contiguous sectors spanning the whole protein have been the proposed allosteric mechanism (19–21), but in this case, ligand binding also results in a small but significant structural change, albeit being quite small (22–24) (Fig. 1B and Table 1). Furthermore, a number of computational and experimental studies have addressed signal transduction pathways in the PDZ2 model system (25–34).
-strand (Fig. 1B). As mentioned previously, side-chain dynamics in contiguous sectors spanning the whole protein have been the proposed allosteric mechanism (19–21), but in this case, ligand binding also results in a small but significant structural change, albeit being quite small (22–24) (Fig. 1B and Table 1). Furthermore, a number of computational and experimental studies have addressed signal transduction pathways in the PDZ2 model system (25–34).
Fig. 1.

(A) Averaged NMR structures of the photoswitchable PDZ2 domain with the photoswitch (yellow) in the cis (Left, PDB ID 2M0Z) and trans (Right, PDB ID 2M10) conformations. (B) Overlays of the apo (blue) and holo (red) X-ray structures [3LNX and 3LNY (24)] together with the ligand (the Ras guanine nucleotide exchange factor 2 C-terminal peptide, in yellow) in the latter case. (C) The NMR structures with the photoswitch in cis (blue) and in trans (red) and (D) the averaged MD structures with the photoswitch in cis (blue) and 100 ns after switching into trans (red). For clarity, the photoswitch is not shown in C and D. The dotted lines in D indicate the Cα-Cα distances shown in Fig. 4B.
Table 1.
Structural comparison
| rmsd | X-ray (24): apo holo holo |
NMR: cis trans trans
|
MD: cis trans trans
|
| All secondary | 0.34 | 0.92 | 0.46 |
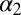 and and 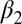
|
0.41 | 0.80 | 0.62 |
Structural difference of the apo vs. the holo form deduced from the X-ray structures (3LNX and 3LNY, ref. 24) or the cis vs. the trans conformer from the NMR structures and the MD simulations, respectively. The first row reports the rmsd (in angstroms) when considering all backbone atoms of regions with defined secondary structure and the second row that when considering only the 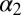 -helix and the
-helix and the 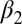 -strand.
-strand.
Allostery is the propagation of a signal between two sites of a protein. Most of the investigations so far have addressed the question of what that signal might be, e.g., a structural change vs. a change in dynamic properties. Even less is known of how such a signal propagates. Whereas NMR spectroscopy is extremely powerful in elucidating equilibrium dynamics on many timescales through relaxation experiments, its inherent time resolution is rather limited in studies of nonequilibrium processes, such as signal propagation. Transient IR spectroscopy, in contrast, provides essentially unlimited (i.e., picosecond) direct time resolution together with still significant chemical selectivity.
To make the best use of the high time resolution, it is crucial to be able to perturb the system locally and with a short laser pulse. Ideally, one would phototrigger the association or dissociation of a ligand. Here, we take an experimentally more feasible approach by covalently linking an azobenzene derivative across the binding groove, which can be switched between cis and trans isomers with a light of different wavelengths (Fig. 1A) (35–38). We carefully designed the system such that the structural perturbation upon isomerization of the photoswitch mimics that upon ligand binding, as is discussed in the next section. Subsequently, we use transient IR spectroscopy to investigate the nonequilbrium transition between both states and finally use nonequilibrium molecular dynamics (MD) simulations to complement the experimental results with atomistic detail.
Structural Characterization
In close analogy to ref. 39, we identified the surface-exposed amino acid pair Ser21 and Glu76 as anchor points for the photoswitch because their Cα-Cα distances in the apo and holo forms closely match those of the photoswitch length in its two configurations. Furthermore, these two residues face across the binding groove at the center. The residues were mutated to cysteines to which the photoswitch was covalently coupled (Fig. S1) (40). When searching for the anchor points, we used the NMR ensemble of structures of the PDZ2 domain [holo, 1D5G, ref. 23); apo, 3PDZ, ref. 22)], which reveals a rather large 1.5-Å change (on average) for the Cα(21)-Cα(76) distance. Recently, X-ray crystallography (3LNX and 3LNY, ref. 24) has shown, however, that this change is about a factor of 2 smaller (i.e., 0.8 Å). The fact that the linker could nevertheless be successfully coupled and resulted in a stable, well-folded protein indicates that the possible structural perturbation is well tolerated.
The two equilibrium structures of the PDZ2 domain with the photoswitch in the cis and trans configurations were determined by NMR spectroscopy [see SI Text for details, Protein Data Bank (PDB) IDs 2M0Z and 2M10]. Whereas the trans form of the azobenzene photoswitch is predominant (∼90%) after equilibration in the dark, the cis state was generated and maintained (>90%) by continuously illuminating the sample inside the spectrometer with a 370-nm laser coupled to a glass fiber leading into the NMR tube (for further information on the design see SI Text), similar to that described in previous work (41, 42). Both forms are stably folded and structurally similar to the corresponding X-ray structures (24), as can be seen in Fig. 1 B and C. More quantitatively, the rmsd of the NMR structure in cis compared with the X-ray structure in the apo form is 1.0 Å and that of trans to the holo form is 1.1 Å (considering all backbone heavy atoms of only the regions with defined secondary structure). These values include the uncertainties in the structure determination, the different environments (crystal vs. solvent), and the fact that the molecule is modified by the photoswitch. Also the adaptation of the structure upon isomerization of the photoswitch is comparable to that upon ligand binding. Table 1 lists the rmsds between the corresponding structures for all secondary structure elements and for residues of the 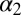 -helix and
-helix and 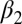 -strand only, with the latter constituting the binding groove. Our construct does slightly overemphasize the conformational perturbation, but the overall agreement is quite reasonable.
-strand only, with the latter constituting the binding groove. Our construct does slightly overemphasize the conformational perturbation, but the overall agreement is quite reasonable.
Transient IR Spectroscopy
Having established the equilibrium structures of the starting and final states by NMR spectroscopy, we now turn to IR spectroscopy. Fig. 2A displays stationary FTIR spectra in the region of the amide I band, which is a sensitive probe of the structure of the protein backbone. All IR experiments have been performed in a fully hydrated state [50 mM borate buffer (pH 8.5) and 150 mM NaCl, lyophilized and dissolved in D2O at 1.3 mM concentration].
Fig. 2.

(A) Absolute (photoswitch in trans, blue, downscaled by 50) and difference FTIR spectra (red and green) compared with the transient spectrum at 42 μs (black, upscaled by 7). (B) Transient difference spectra at −1 ns (yellow), 0 ps (light orange), and from 10 ps to 10 μs by decade (orange to black). (C) Contour plot of the IR response. Red indicates induced absorption, blue indicates a bleach, and contour lines are equally spaced.
A difference signal upon switching 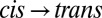 or
or 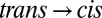 is clearly visible (Fig. 2A, red and green) with an intensity of about 1/50th that of the absolute amide I band (Fig. 2, blue), indicating that small changes in the protein backbone do indeed occur. The
is clearly visible (Fig. 2A, red and green) with an intensity of about 1/50th that of the absolute amide I band (Fig. 2, blue), indicating that small changes in the protein backbone do indeed occur. The 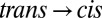 difference spectrum, induced by illuminating a dark-adapted sample at 370 nm, and the
difference spectrum, induced by illuminating a dark-adapted sample at 370 nm, and the 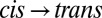 difference spectrum, after switching off the 370-nm light and subsequent relaxation back to trans in ∼30 min, are mirror images of each other (Fig. 2A, green and red lines), confirming that the molecule can be switched reversibly. In the next step, to observe the transition in time-resolved experiments, we prepared all samples in the cis configuration by continuously illuminating them at 370 nm with an excess amount of light and then initiated the
difference spectrum, after switching off the 370-nm light and subsequent relaxation back to trans in ∼30 min, are mirror images of each other (Fig. 2A, green and red lines), confirming that the molecule can be switched reversibly. In the next step, to observe the transition in time-resolved experiments, we prepared all samples in the cis configuration by continuously illuminating them at 370 nm with an excess amount of light and then initiated the 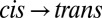 transition with a picosecond 420-nm pulse (Materials and Methods). At long times (42 μs), the transient IR response in the amide I region closely resembles the shape of the steady state (Fig. 2A, black vs. red line), indicating that the structural transition is essentially complete after this time. Its amplitude its about one-seventh of the latter, allowing one to estimate the combined excitation probability and isomerization yield.
transition with a picosecond 420-nm pulse (Materials and Methods). At long times (42 μs), the transient IR response in the amide I region closely resembles the shape of the steady state (Fig. 2A, black vs. red line), indicating that the structural transition is essentially complete after this time. Its amplitude its about one-seventh of the latter, allowing one to estimate the combined excitation probability and isomerization yield.
In reaching the final state, the data reveal a complex evolution over many orders of magnitude in time with bands that both change in intensity and shift in frequency (Fig. 2 B and C). No physically meaningful model with a limited number of discrete intermediate states could be identified to which we could globally fit the data. Similar observations have been made for downhill folding (43, 44), with different kinetic responses for different spectroscopic observables. This behavior is indicative of a continuous transition between initial and final states without any significant barriers on the pathway.
Three major phases of the overall process can nonetheless be identified. We illustrate these phases in Fig. 3 for the amide I band, choosing 1,640 cm−1 as probe wavelength (red circles), and a strong band at 1,491 cm−1 originating from the photoswitch linked to the PDZ2 domain (Fig. 3, green circles). This band is the amide II vibration of the amide unit of the linker connecting the azobenzene moiety to the protein (Fig. S1A). For comparison, the response of the same band of the unlinked photoswitch is shown as well (Fig. 3, blue circles; see Fig. S2, for the complete data). Phase I, clearly visible in the photoswitch bands in both the linked and unlinked form, is initiated by the absorption of a 420-nm photon and the subsequent ultrafast isomerization of the azobenzene moiety. This results in the deposition of a large amount of energy into the vibrational degrees of freedom of the molecule. The vibrational energy appears as heat and results in a broad IR signal, decaying within a few 10s of picoseconds as it quickly dissipates into the solvent (45). The heat signal happens to be zero for the amide I band at 1,640 cm−1, but it is clearly visible at other probe wavelengths (Fig. 2C).
Fig. 3.

Amide I response at a selected wavelength of 1,640 cm−1 (red, right scale) compared with that of a band at 1,491 cm−1 from the unlinked photoswitch (blue, left scale) and with that of the same band of the photoswitch linked to the PDZ2 domain (green, left scale). The signal of the photoswitchable PDZ2 domain (red and green) is fitted jointly to Eq. 1 (black).
Subsequently, in phase II, the band of the photoswitch linked to the PDZ2 domain evolves in a highly nonexponential manner until about 100 ns (Fig. 3, green circles), significantly slower than that of the photoswitch alone (Fig. 3, blue circles). Photoisomerization of the azobenzene moiety is an ultrafast picosecond process (46). The heat signal of the photoswitch linked to the protein appears on the same timescale as that of the unlinked photoswitch (Fig. S2); hence, in terms of crossing from the electronically excited back to the ground state, and thus in terms of the configuration of the central N = N bond, isomerization is equally fast in both cases. When bound to the protein, however, the photoswitch will find itself in a highly strained state after isomerization, because the protein cannot adapt instantaneously. This strain will affect also the vibrational states of the linker connecting the azobenzene moiety to the protein, i.e., the 1,491-cm−1 band. As the protein relaxes, the strain on the photoswitch is slowly released. Hence, the phase II signal of the photoswitch (Fig. 3, green circles) can be thought of as an indirect measure of the perturbation of the binding groove, as the binding groove is cross-linked by the photoswitch. Similar conclusions have been drawn for the electronic response of a similar azo-photoswitch in a smaller peptide (36). The perturbation of the binding groove is also reflected in the amide I band of the protein (Fig. 3, red circles).
Finally, in phase III, the photoswitch signal remains constant, because the binding groove has fully adapted to the perturbation. The amide I signal nevertheless continues to evolve in time. We assume that this signal is related to a slight rearrangement of the protein structure in a region different from the binding groove, for instance of some of the turn regions that are known to be quite flexible. This interpretation is corroborated by signal broadening of NMR resonances from the 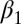 -
-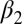 loop.
loop.
The two time traces from the photoswitchable PDZ2 domain at 1,640 cm−1 and at 1,491 cm−1 can be fitted jointly to a function (Fig. 3, black lines)
which is composed of a fast exponential contribution for the heat signal decay in phase I (τ1 = 15 ps), a stretched exponential contribution for the binding groove perturbation in phase II (τ2 = 7 ns, β = 0.49), and another exponential contribution (τ3 = 20 μs) for the final relaxation in the amide I band in phase III. In that fit, the time constants and the stretching factor were forced to be the same for both time traces, but the amplitudes were allowed to differ (Table S1). With a stretching factor of β = 0.49, the nonexponential time dependence of phase II is quite pronounced.
Nonequilibrium Molecular Dynamics
To facilitate the understanding of the transition on an atomistic level, we used nonequilibrium MD simulations in explicit water. Starting from an equilibrated cis ensemble (with an rmsd to the corresponding NMR structure of 1.4 Å), we launched very many nonequilibrium trajectories by instantaneously switching the potential energy function of the central N = N bond of the photoswitch from one that is stable in cis to one that is stable in trans (Materials and Methods) (36, 38, 47). As these simulations are limited to a maximum simulation time of 100 ns, we focus on phase II in the following discussion, i.e., the perturbation of the binding groove. The overall fold does not change during the first 100 ns after photoswitching (Fig. 1D), but the protein backbone is deformed slightly, as expressed by the rmsd that increases to 0.46 Å after 100 ns (Table 1 and Fig. 4A). The rmsd is larger when considering the 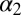 -helix and the
-helix and the 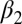 -strand only (0.62 Å), emphasizing that most of the structural changes occur at the binding groove on this timescale. As a function of time, the rmsd jumps relatively rapidly within the first 1 ps. It then continues to grow in a highly nonexponential manner, covering all orders of magnitude in time considered in this simulation, similar to the experimental observation (Fig. 3, green circles), and in fact it is not finished after 100 ns. We therefore did not attempt to fit the MD data, because a stretched exponential fit becomes robust only if data exist for times long enough so that the signal levels off. No quantitative agreement is expected for the time dependence because, for example, the self-diffusion coefficient of TIP3P (three-site transferrable intermolecular potential) water used in the simulation is more than a factor of 2 higher than the experimental value (48), and water plays an important part in determining the response of the protein (see discussion below). Nevertheless, qualitatively speaking, the response is similar to phase II in Fig. 3 (green circles) [note that phase I is not directly related to any structural process, but rather to a heat signal, which is based on the anharmonicity of the molecule’s potential (45) and as such is beyond the MD model]. Furthermore, the amount of structural change obtained from the MD simulation agrees reasonably well with what we find for the NMR structures (keeping in mind that the transition is not quite complete after 100 ns, Table 1).
-strand only (0.62 Å), emphasizing that most of the structural changes occur at the binding groove on this timescale. As a function of time, the rmsd jumps relatively rapidly within the first 1 ps. It then continues to grow in a highly nonexponential manner, covering all orders of magnitude in time considered in this simulation, similar to the experimental observation (Fig. 3, green circles), and in fact it is not finished after 100 ns. We therefore did not attempt to fit the MD data, because a stretched exponential fit becomes robust only if data exist for times long enough so that the signal levels off. No quantitative agreement is expected for the time dependence because, for example, the self-diffusion coefficient of TIP3P (three-site transferrable intermolecular potential) water used in the simulation is more than a factor of 2 higher than the experimental value (48), and water plays an important part in determining the response of the protein (see discussion below). Nevertheless, qualitatively speaking, the response is similar to phase II in Fig. 3 (green circles) [note that phase I is not directly related to any structural process, but rather to a heat signal, which is based on the anharmonicity of the molecule’s potential (45) and as such is beyond the MD model]. Furthermore, the amount of structural change obtained from the MD simulation agrees reasonably well with what we find for the NMR structures (keeping in mind that the transition is not quite complete after 100 ns, Table 1).
Fig. 4.

MD results. (A) Time evolution of the rmsd, averaged over an ensemble of nonequilibrium trajectories. The rmsd is relative to the averaged starting structure. (B) The same for the Cα-Cα distances across the binding groove, indicated as dotted lines in Fig. 1D. (C) Cα(21)-Cα(76) distance from four typical individual trajectories. A–C were all deduced from explicit water simulations. (D) Comparison of the Cα(21)-Cα(76) distance in simulations performed in explicit (red) vs. implicit (black) water.
As a more direct measure of the structural change of the binding groove, we also show in Fig. 4B the time evolution of various Cα-Cα distances across the binding groove. The Cα(21)-Cα(76) distance, corresponding to the sites that are directly linked by the photoswitch, is perturbed the most and jumps very quickly within the first 1 ps by a significant amount of ∼1 Å. We attribute this initial jump to the direct impact of the isomerization of the photoswitch on the protein backbone. Thereafter the Cα(21)-Cα(76) distance again increases steadily in a highly nonexponential manner. The neighboring pairs [Fig. 4B, Cα(19)-Cα(79) in green and Cα(23)-Cα(72) in blue] experience much less of the initial jump in change of distance, because they are not directly affected by the conformational change of the photoswitch, but the nonexponential response at later times is similar.
Nonexponential protein dynamics, modeled either as stretched exponentials or power laws, have been discussed extensively, for instance in the context of ligand (CO) dissociation and rebinding in hemoglobin or myoglobin (49–51) or the fluctuations of the pairwise distance between two sites in a protein (52–54). Two limiting scenarios of nonexponential relaxation kinetics can be distinguished (50): The parallel process is characterized by a distribution of exponential decay processes, originating from a distribution of barrier heights in an inhomogeneous ensemble of proteins. This mechanism applies in the limit when the timescale of the relaxation process is fast compared with the time the protein requires to sample its complete conformational space. In that case, individual single-molecule trajectories would still be a two-state system with either short or long Cα-Cα distances that are separated by one dominant barrier, so that one would observe essentially sudden jumps between these two states with a nonexponential distribution of jump times. Fig. 4C shows that we are in the opposite limit. That is, individual single-molecule trajectories essentially follow the averaged one (apart from statistical noise), without big jumps. In accordance with the conclusion already drawn from the experimental results, this observation implies that the transition is continuous without having to surmount any dominant barrier. Such a nonexponential response occurs when protein relaxation is the result of many small events, like defect diffusion that commonly leads to subdiffusive behavior (55).
Fig. 5 indicates what these many small events might be. Shown is a time series of the change of water density at the surface of the protein averaged over all nonequilibrium trajectories. As the photoswitch is isomerizing very quickly in the simulation (<1 ps), the water density changes immediately in the vicinity of the photoswitch (which is located on the right side in the structures in Fig. 5). As time proceeds, this perturbation of the water network travels around the protein and it in fact takes 100 ns until it reaches the back side. This relatively slow propagation of the perturbation of the water network needs to be put in contrast to the residence time of a given single water molecule at the protein surface, which was calculated to be 10–100 ps in the binding groove and 10–30 ps on the outside surface, respectively. Hence, water molecules need to exchange many times before a new equilibrium of the water network around the protein is established.
Fig. 5.
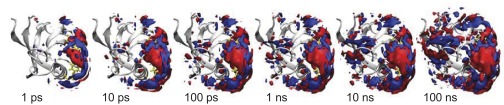
Change of water density as a function of simulation time, compared with that just before switching. Red depicts increased density and blue decreased density. The contour surfaces correspond to changes of ±0.01 water/Å3 (for comparison, the bulk water density is ∼0.033 water/Å3). The protein is shown as a gray ribbon and the photoswitch (visible only in part) is shown in yellow. See also Movie S1.
To gain further insights into the role of water during the structural transition, we repeated the switching simulations with implicit water (Fig. 4D, black line), which approximates water as a continuum of the correct dielectric constant, but without the internal degrees of freedom that would provide friction (no additional friction term was added in the Newtonian dynamics used in these simulations). Both states remain stably folded in the implicit water model, but the kinetics of the transition between them are very different from those of the explicit water model. That is, the average Cα(21)-Cα(76) distance completes a 1.4-Å jump within ∼1 ps in a ballistic fashion (a minor overshoot is observed) and then stays essentially constant out to 100 ns. Clearly, the implicit water simulation is artificial and not aimed to reveal results that are comparable to reality, but is used to reveal the consequences of the direct impact of water molecules on protein dynamics through the comparison with the explicit water MD simulation. This numerical experiment shows that on the length scale of this conformational transition, the intramolecular potential of the protein alone is not rugged at all and provides no internal friction. In other words, the process is entirely “slaved” by water (51, 56, 57), and water is an integral part of the observed protein dynamics.
Conclusion
In conclusion, we have shown in a closely linked experimental-simulation study that the perturbation of the binding groove of the PDZ2 domain evolves as a continuous subdiffusive process on a 100-ns timescale. The MD simulation (Fig. 4) can essentially quantitatively reproduce the experimentally observed kinetics (Fig. 3, green circles) both in terms of its timescale and in terms of its nonexponential character. This excellent agreement validates the MD simulation, from which atomistic details may then be extracted. We find that the ruggedness of the free energy landscape that governs the dynamics of the binding groove originates entirely from water, whereas the intramolecular potential of the protein is smooth for this small conformational transition. A similar conclusion was drawn for a significantly larger conformational change between a folding intermediate and the native state of a four-helix bundle protein (58). We find that in our system the overall protein response is dominated by the rearrangement of the water network on the protein surface. Interestingly, the perturbation of the water network propagates around the protein within the 100 ns (Fig. 5).
We propose this to be another possible mechanism of allostery, which addresses the question of how the ligand binding site communicates with remote parts of the protein. Although the photoswitch is a rather crude mimic of ligand binding, the peptide ligand will still introduce new partial charges in the binding pocket that will rearrange the water network in its vicinity and eventually also at larger distances. This mechanism would work without any significant structural change of the protein and might even unify the seemingly competing points of view, which explain allostery either as a structural or as a dynamical effect. That is, to the extent that the dynamics of a protein are slaved by water, a change in water structure can affect the dynamics of the protein. Independent from that, phase III in Fig. 3 also hints toward a conformational change of the protein backbone in a region different from the binding groove that also could be responsible for allosteric signaling in a more traditional sense (3, 4).
Materials and Methods
Protein Preparation.
The PDZ2 domain (S21C E76C) was expressed from Escherichia coli, using standard methods. The photoswitch 3,3′-bis(sulfonato)-4,4′- bis(chloroacetamido)azobenzene (BSBCA) was covalently linked to the two cysteines (40); see SI Text and Fig. S3 for details. We learned from mass spectrometry that the protein reacts photochemically with oxygen as well as with the initially used Tris buffer under the influence of the 420-nm laser pulses in the pump-probe experiment, where both presumably bind to the thioether groups of the cysteines linked to the photoswitch (Fig. S4). The experiments were therefore performed in 50 mM borate buffer (pH 8.5) and 150 mM NaCl, lyophilized and dissolved in D2O, and care was taken to maintain the sample oxygen-free.
Time-Resolved IR Spectroscopy.
Two synchronized 1-kHz Ti:sapphire oscillator/regenerative amplifier femtosecond laser systems (Spectra Physics) were used for pump-probe measurements (59). The jitter between both lasers (which effectively determines the time resolution of the experiment) was ∼10 ps and the delay could be adjusted up to 42 μs. The frequency-doubled pulses (420 nm, 3 μJ per pulse focused onto an ∼200-μm beam diameter in the sample and stretched to ∼1 ps to reduce sample deposition on the cuvette windows) of one laser system were used to excite the photoswitch. The IR probe pulses were obtained by sending the output of the second laser system through an optical parametric amplifier (100 fs, center wavenumber 1,635 cm−1 or 1,443 cm−1). The sample was circulated in a closed-cycle flow-cell system consisting of a reservoir followed by a CaF2 sample cell with 50 μm optical path length. The reservoir was continuously illuminated with a 150-mW, 370-nm continuous wave diode laser (CrystaLaser) so that all protein flowing into the sample cell was in the cis configuration.
NMR.
NMR spectra of PDZ2 with the photoswitch in trans were recorded in the dark after equilibration. For all measurements with the photoswitch in the cis configuration, the sample was continuously illuminated with the 370-nm cw laser specified above (Fig. S5) coupled into the NMR spectrometer through a custom-fabricated fiber terminated with an extended cylindrical diffuser (Molex). Spectrum assignment was achieved with a standard set of triple-resonance experiments. Structure calculation was performed from NOE distance restraints from 15N- and 13C-resolved NOESY spectra with 75-ms mixing times. NOE data were complemented by amide proton residual dipolar couplings (NH-RDCs) measured in Pf1 bacteriophage and n-dodecyl-penta(ethylene glycol)/n-hexanol liquid crystalline medium. The 20 conformers with lowest energy of both structures showed no NOE violations bigger than 0.5 Å and showed good Ramachandran plot statistics with only 0.3% of the residues in disallowed regions (for more detailed description of experiments, parameters, and structural statistics see SI Text and Tables S2 and S3).
Computational Methods.
MD simulations were performed with the Gromacs program package (60) and the Gromacs implementation of the Charmm27 force field (61, 62). The photoswitch was parameterized as in ref. 36. Details of the simulation protocol are given in SI Text.
Supplementary Material
Acknowledgments
We thank Ben Schuler and his group, in particular Hagen Hofmann, for tremendous help with the protein chemistry; and Andrew Woolley and Gerhard Stock for important discussions; and the Functional Genomics Center Zurich, especially Serge Chesnov, for help with mass spectrometry. This work has primarily been supported by an European Research Council (ERC) Advanced Investigator Grant (DYNALLO) and in part by the Swiss National Science Foundation through the National Center of Competence and Research (NCCR) MUST.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The NMR, atomic coordinates, chemical shifts, and restraints have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2M0Z and 2M10) and the BioMagResBank, www.bmrb.wisc.edu (accession nos. 18833 and 18834).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306323110/-/DCSupplemental.
References
- 1.Henry ER, Jones CM, Hofrichter J, Eaton WA. Can a two-state MWC allosteric model explain hemoglobin kinetics? Biochemistry. 1997;36(21):6511–6528. doi: 10.1021/bi9619177. [DOI] [PubMed] [Google Scholar]
- 2.Eaton WA, Henry ER, Hofrichter J, Mozzarelli A. Is cooperative oxygen binding by hemoglobin really understood? Nat Struct Biol. 1999;6(4):351–358. doi: 10.1038/7586. [DOI] [PubMed] [Google Scholar]
- 3.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions - a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 4.Koshland DE, Jr, Némethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5(1):365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 5.Cooper A, Dryden DTF. Allostery without conformational change. A plausible model. Eur Biophys J. 1984;11(2):103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 6.Songyang Z, et al. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275(5296):73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 7.Daniels DL, Cohen AR, Anderson JM, Brünger AT. Crystal structure of the hCASK PDZ domain reveals the structural basis of class II PDZ domain target recognition. Nat Struct Biol. 1998;5(4):317–325. doi: 10.1038/nsb0498-317. [DOI] [PubMed] [Google Scholar]
- 8.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114(Pt 18):3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 9.Jemth P, Gianni S. PDZ domains: Folding and binding. Biochemistry. 2007;46(30):8701–8708. doi: 10.1021/bi7008618. [DOI] [PubMed] [Google Scholar]
- 10.Lee H-J, Zheng JJ. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Ham M, Hendriks W. PDZ domains-glue and guide. Mol Biol Rep. 2003;30(2):69–82. doi: 10.1023/a:1023941703493. [DOI] [PubMed] [Google Scholar]
- 12.van den Berk LCJ, et al. An allosteric intramolecular PDZ-PDZ interaction modulates PTP-BL PDZ2 binding specificity. Biochemistry. 2007;46(47):13629–13637. doi: 10.1021/bi700954e. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Callaway DJE, Bu Z. Ezrin induces long-range interdomain allostery in the scaffolding protein NHERF1. J Mol Biol. 2009;392(1):166–180. doi: 10.1016/j.jmb.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T. Crystal structure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell. 2004;117(4):483–494. doi: 10.1016/s0092-8674(04)00454-4. [DOI] [PubMed] [Google Scholar]
- 15.Whitney DS, Peterson FC, Volkman BF. A conformational switch in the CRIB-PDZ module of Par-6. Structure. 2011;19(11):1711–1722. doi: 10.1016/j.str.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci USA. 2009;106(43):18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13(9):831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448(7151):325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J Mol Biol. 2004;335(4):1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes EJ, Gilmore SA, Mauldin RV, Lee AL. Evaluation of energetic and dynamic coupling networks in a PDZ domain protein. J Mol Biol. 2006;364(3):337–351. doi: 10.1016/j.jmb.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 21.Gianni S, et al. Demonstration of long-range interactions in a PDZ domain by NMR, kinetics, and protein engineering. Structure. 2006;14(12):1801–1809. doi: 10.1016/j.str.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Kozlov G, Gehring K, Ekiel I. Solution structure of the PDZ2 domain from human phosphatase hPTP1E and its interactions with C-terminal peptides from the Fas receptor. Biochemistry. 2000;39(10):2572–2580. doi: 10.1021/bi991913c. [DOI] [PubMed] [Google Scholar]
- 23.Kozlov G, Banville D, Gehring K, Ekiel I. Solution structure of the PDZ2 domain from cytosolic human phosphatase hPTP1E complexed with a peptide reveals contribution of the beta2-beta3 loop to PDZ domain-ligand interactions. J Mol Biol. 2002;320(4):813–820. doi: 10.1016/s0022-2836(02)00544-2. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, et al. Crystallographic and nuclear magnetic resonance evaluation of the impact of peptide binding to the second PDZ domain of protein tyrosine phosphatase 1E. Biochemistry. 2010;49(43):9280–9291. doi: 10.1021/bi101131f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ota N, Agard DA. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J Mol Biol. 2005;351(2):345–354. doi: 10.1016/j.jmb.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 26.De Los Rios P, et al. Functional dynamics of PDZ binding domains: A normal-mode analysis. Biophys J. 2005;89(1):14–21. doi: 10.1529/biophysj.104.055004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharp K, Skinner JJ. Pump-probe molecular dynamics as a tool for studying protein motion and long range coupling. Proteins. 2006;65(2):347–361. doi: 10.1002/prot.21146. [DOI] [PubMed] [Google Scholar]
- 28.Dhulesia A, Gsponer J, Vendruscolo M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J Am Chem Soc. 2008;130(28):8931–8939. doi: 10.1021/ja0752080. [DOI] [PubMed] [Google Scholar]
- 29.Kong Y, Karplus M. Signaling pathways of PDZ2 domain: A molecular dynamics interaction correlation analysis. Proteins. 2009;74(1):145–154. doi: 10.1002/prot.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerek ZN, Ozkan SB. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS Comput Biol. 2011;7(10):e1002154. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cilia E, Vuister GW, Lenaerts T. Accurate prediction of the dynamical changes within the second PDZ domain of PTP1e. PLoS Comput Biol. 2012;8(11):e1002794. doi: 10.1371/journal.pcbi.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 33.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10(1):59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 34.Chi CN, et al. Reassessing a sparse energetic network within a single protein domain. Proc Natl Acad Sci USA. 2008;105(12):4679–4684. doi: 10.1073/pnas.0711732105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumita JR, Smart OS, Woolley GA. Photo-control of helix content in a short peptide. Proc Natl Acad Sci USA. 2000;97(8):3803–3808. doi: 10.1073/pnas.97.8.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spörlein S, et al. Ultrafast spectroscopy reveals subnanosecond peptide conformational dynamics and validates molecular dynamics simulation. Proc Natl Acad Sci USA. 2002;99(12):7998–8002. doi: 10.1073/pnas.122238799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehm S, Lenz MO, Mensch S, Schwalbe H, Wachtveitl J. Ultrafast spectroscopy of a photoswitchable 30-amino acid de novo synthesized peptide. Chem Phys. 2005;323(1):28–35. [Google Scholar]
- 38.Ihalainen JA, et al. α-Helix folding in the presence of structural constraints. Proc Natl Acad Sci USA. 2008;105(28):9588–9593. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang F, et al. Structure-based approach to the photocontrol of protein folding. J Am Chem Soc. 2009;131(6):2283–2289. doi: 10.1021/ja807938v. [DOI] [PubMed] [Google Scholar]
- 40.Burns DC, Zhang F, Woolley GA. Synthesis of 3,3′-bis(sulfonato)-4,4′-bis(chloroacetamido)azobenzene and cysteine cross-linking for photo-control of protein conformation and activity. Nat Protoc. 2007;2(2):251–258. doi: 10.1038/nprot.2007.21. [DOI] [PubMed] [Google Scholar]
- 41.Rubinstenn G, et al. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat Struct Biol. 1998;5(7):568–570. doi: 10.1038/823. [DOI] [PubMed] [Google Scholar]
- 42.Kühn T, Schwalbe H. Monitoring the kinetics of ion-dependent protein folding by time-resolved nmr spectroscopy at atomic resolution. J Am Chem Soc. 2000;122:6169–6174. [Google Scholar]
- 43.Garcia-Mira MM, Sadqi M, Fischer N, Sanchez-Ruiz JM, Muñoz V. Experimental identification of downhill protein folding. Science. 2002;298(5601):2191–2195. doi: 10.1126/science.1077809. [DOI] [PubMed] [Google Scholar]
- 44.Ma H, Gruebele M. Kinetics are probe-dependent during downhill folding of an engineered lambda6-85 protein. Proc Natl Acad Sci USA. 2005;102(7):2283–2287. doi: 10.1073/pnas.0409270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamm P, Ohline SM, Zinth W. Vibrational cooling after ultrafast photoisomerization of azobenzene measured by femtosecond infrared spectroscopy. J Chem Phys. 1997;106:519–529. [Google Scholar]
- 46.Nägele T, Hoche R, Zinth W, Wachtveitl J. Femtosecond photoisomerization of cis-azobenzene. Chem Phys Lett. 1997;272:489–495. [Google Scholar]
- 47.Nguyen PH, Stock G. Nonequilibrium molecular dynamics simulation of a photoswitchable peptide. Chem Phys. 2006;323:36–44. [Google Scholar]
- 48.Mahoney MW, Jorgensen WL. Diffusion constant of the tip5p model of liquid water. J Chem Phys. 2001;114:363–366. [Google Scholar]
- 49.Lim M, Jackson TA, Anfinrud PA. Nonexponential protein relaxation: Dynamics of conformational change in myoglobin. Proc Natl Acad Sci USA. 1993;90(12):5801–5804. doi: 10.1073/pnas.90.12.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254(5038):1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 51.Frauenfelder H, et al. A unified model of protein dynamics. Proc Natl Acad Sci USA. 2009;106(13):5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Volk M, et al. Peptide conformational dynamics and vibrational Stark effects following photoinitiated disulfide cleavage. J Phys Chem B. 1997;101:8607–8616. [Google Scholar]
- 53.Yang H, et al. Protein conformational dynamics probed by single-molecule electron transfer. Science. 2003;302(5643):262–266. doi: 10.1126/science.1086911. [DOI] [PubMed] [Google Scholar]
- 54.Milanesi L, et al. Measurement of energy landscape roughness of folded and unfolded proteins. Proc Natl Acad Sci USA. 2012;109(48):19563–19568. doi: 10.1073/pnas.1211764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jäckle J. Models of the glass transition. Rep Prog Phys. 1986;49:171–231. [Google Scholar]
- 56.Fenimore PW, Frauenfelder H, McMahon BH, Parak FG. Slaving: Solvent fluctuations dominate protein dynamics and functions. Proc Natl Acad Sci USA. 2002;99(25):16047–16051. doi: 10.1073/pnas.212637899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vitkup D, Ringe D, Petsko GA, Karplus M. Solvent mobility and the protein ‘glass’ transition. Nat Struct Biol. 2000;7(1):34–38. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- 58.Sekhar A, Vallurupalli P, Kay LE. Folding of the four-helix bundle FF domain from a compact on-pathway intermediate state is governed predominantly by water motion. Proc Natl Acad Sci USA. 2012;109(47):19268–19273. doi: 10.1073/pnas.1212036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bredenbeck J, Helbing J, Hamm P. Continuous scanning from picoseconds to microseconds in time resolved linear and nonlinear spectroscopy. Rev Sci Instrum. 2004;75(11):4462. [Google Scholar]
- 60.Van Der Spoel D, et al. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 61.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 62.Mackerell AD, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.