

Abstract
Phosphorodiamidate morpholino oligonucleotide (PMO)-mediated control of the alternative splicing of the chloride channel 1 (CLCN1) gene is a promising treatment for myotonic dystrophy type 1 (DM1) because the abnormal splicing of this gene causes myotonia in patients with DM1. In this study, we optimised a PMO sequence to correct Clcn1 alternative splicing and successfully remedied the myotonic phenotype of a DM1 mouse model, the HSALR mouse. To enhance the efficiency of delivery of PMO into HSALR mouse muscles, Bubble liposomes, which have been used as a gene delivery tool, were applied with ultrasound exposure. Effective delivery of PMO led to increased expression of Clcn1 protein in skeletal muscle and the amelioration of myotonia. Thus, PMO-mediated control of the alternative splicing of the Clcn1 gene must be important target of antisense therapy of DM1.
Myotonic dystrophy type 1 (DM1) is caused by expansion of the CTG repeat in the 3′ untranslated region (UTR) of the DMPK gene1,2,3,4. Patients with DM1 show multi-systemic symptoms, including muscle wasting, muscle weakness, myotonia, cardiac conduction defect, cataracts, mental retardation and insulin resistance5. A patient, however, does not always present with all of these symptoms and the severity of the disease varies among individuals. Among the symptoms, myotonia is the most prominent and common phenotype of DM1: most patients feel muscle stiffness and difficulty in relaxing muscles soon after developing the disease.
The characteristic feature of the pathology of DM1 is the aberrant regulation of dozens of alternative splicing events, and some of the abnormal splicing events have been suggested to be involved in some of the symptoms6,7,8,9,10. Myotonic discharge is thought to be caused by the aberrant alternative splicing of the chloride channel 1 (CLCN1) gene11,12. In patients with DM1, extra exons from intron 6 are spliced into the CLCN1 mRNA, leading to the appearance of a premature termination codon in the subsequent exon, degradation of the mRNA by nonsense-mediated decay and decreased expression of CLCN1 protein11. The idea that abnormal splicing of the CLCN1 gene causes myotonia is strongly supported by the fact that the CLCN1 gene is the only gene responsible for congenital myotonia, and the identification of multiple mutations in patients with the disease and their families13. Wheeler and his colleagues corrected the abnormal splicing of the Clcn1 gene in a DM1 mouse model, the HSALR mouse, by using an antisense oligonucleotide (AON) and successfully alleviated the myotonic phenotype14. Thus, correction of the abnormally regulated splicing is a promising treatment for DM1.
An AON is a short, synthetic nucleic acid molecule with a sequence complementary to a target transcript. It can be used to manipulate an alternative splicing event: the AON that binds to the region around the target exon, specifically splice sites or splicing enhancer domains, physically blocks assembly of the spliceosome on the exon and induces exon skipping15. The efficacy of an AON is dependent on its half-life, affinity for its target RNA and in vivo kinetics. A variety of AON molecules with 2′-O modifications and/or unnatural backbones have been developed to improve nuclease resistance and affinity for RNA15. Among them, Morpholino (also referred as phosphorodiamidate morpholino oligonucleotide [PMO]) is one of the most hopeful AONs. PMO has morpholine rings linked with phosphorodiamidate linkages in its backbone instead of deoxyribose and phosphodiester bonds. Due to its completely unnatural chemistry, PMO is hardly recognised by cellular nucleases. It has higher affinity for RNA than for DNA; the Tm value of a hybrid of PMO and RNA is much higher than that of DNA and RNA16. Several papers have reported the local and systemic administration of PMO to mice and dogs. For example, when a high dose of PMO (3 g/kg) was administrated intravenously into the mdx mouse, a mouse model of Duchenne muscular dystrophy, the PMO entered skeletal muscle without any assistive delivery reagent17. However, unlike mdx mice, muscle penetration of Evans Blue dye did not increase in HSALR and wild-type mice18, which indicated a physical barrier to PMO uptake should be grater in HSALR than in mdx. In preceding reports on PMO treatment in HSALR, intravenous administration of CAG25 PMO led no detectable improvements in Serca1 splicing in HSALR mice19, and even when PMO was injected intramuscularly its uptake was limited to the needle track14. In these studies, they used electroporation to administer unmodified PMO intramuscularly, so we investigated a less invasive PMO delivery method to develop PMO treatment for DM1.
Recently, ultrasound exposure has been used for the intracellular delivery of molecules such as dextran, plasmid DNA and siRNA. If ultrasound is sufficiently strong, it will generate microscopic vacuum bubbles in a solution by a process known as inertial cavitation. The bubbles immediately collapse, producing a shock wave, which is believed to transiently increase the permeability of cell membranes in the vicinity. Inertial cavitation is enhanced by using micro bubbles of echo-contrast gas. This method has been applied to gene delivery into various mouse tissues, including skeletal muscle, liver and tumour tissues20,21,22; however, introducing genes into deep tissues with microbubbles is difficult because of their size and instability. To overcome these problems, we previously developed a novel drug delivery reagent coined “Bubble liposomes”, polyethylene glycol-modified liposomes (PEG liposomes) encapsulating echo-contrast gas23. Owing to the stability in serum and uniform microscopic size of PEG liposomes, we successfully delivered genes and siRNA into several tissues24,25,26,27,28. However, does the Bubble liposome-ultrasound delivery system efficiently deliver PMO into skeletal muscles in the HSALR mice? In this study, we examined the ability of the Bubble liposome-ultrasound system to deliver PMO into skeletal muscles of HSALR mice as a treatment for abnormal splicing.
We newly designed antisense PMOs targeting exon 7A of the Clcn1 gene and delivered them into HSALR mice. The PMOs were successfully delivered into skeletal muscles by the Bubble liposome-ultrasound system, which decreased the inclusion of exon 7A in vivo. Furthermore, the injection of PMO ameliorated the myotonic phenotype of the model mice. Our results suggest that Bubble liposomes should be effective for delivering PMOs into muscle tissues and can be applied to PMO treatment of DM1.
Results
We first determined the optimal target sequence of the Clcn1 pre-mRNA to promote skipping of exon 7A. To achieve this, we used a Clcn1 minigene and examined its alternative splicing using a cell culture-based assay. The minigene contains the genomic region from exon 6 to exon 7 of the murine Clcn1 gene. When it was transfected into COS-7 cells, approximately 50% of transcripts contained exon 7A (Fig. 1b, minigene only). To screen for an optimal AON sequence, we used 25-mer phosphorothioate 2′ O-methyl (PS2OMe) RNA, which can regulate alternative splicing by sterically preventing spliceosomal assembly, just like PMO. We examined PS2OMe RNA molecules that covered the whole of exon 7A (1–25, 26–50, 51–75 and 76–90) and the boundary of intron 6 and exon 7A (−10–15). We transfected the minigene together with the PS2OMe RNA into COS-7 cells and analysed the alternative splicing of the minigene. We found that −10–15 and 1–25 PS2OMe significantly reduced the rate of inclusion of exon 7A, with 1–25 PS2OMe being the most effective molecule (Fig. 1b).
Figure 1. AON-mediated exclusion of Clcn1 exon 7A in COS-7 cells.

(a) Locations of the target sites of AONs (thick black lines) along the Clcn1 pre-mRNA. The numbers over exon 7A (rectangle) indicate the positions of nucleotides. (b) Cellular splicing assay to detect the exclusion of exon 7A of the Clcn1 minigene by PS2OMe RNA in COS-7 cells. 1–25 was the most successful AON. A representative result is shown above and the bars indicate mean and s.e.m. (n = 6). (c) The same assay as in (b) except that PMOs were used. 1–25 PMO decreased the inclusion of exon 7A effectively (n = 3). Statistical significance was analysed by Tukey's multi-comparison test (* P < 0.05, ** P < 0.01, *** P < 0.001).
Previously, we identified the 8 nt at the 5′ end of exon 7A as an exonic splicing enhancer (ESE) essential for basal inclusion of the exon29. Given that both −10–15 and 1–25 PS2OMe covered the ESE, and that 1–25 PS2OMe seemed to be more effective at excluding exon 7A, we speculated that another ESE (16–25) would be located in the region +16 to +25, and that 1–25 PS2OMe would not share it with −10–15 PS2OMe. To examine this possibility, we tested whether 16–40 PS2OMe enhanced normal splicing. 16–40 PS2OMe markedly reduced the rate of inclusion of exon 7A of the Clcn1 minigene (Supplementary Fig. S1). As 26–50 PS2OMe did not change the alternative splicing, we conclude that the other ESE (16–25) is important for exon 7A inclusion. Thus, we used 1–25 AON, which targeted both ESEs, in subsequent experiments.
PS2OMe is highly resistant to nuclease-mediated degradation owing to its phosphorothioate linkages; however, it is still degraded slowly and releases monomers that have a toxic, free phosphorothioate group. In contrast, PMO is remarkably resistant to degradation and is not noxious. Therefore, we next investigated whether a PMO with the same sequence as 1–25 PS2OMe also improved the alternative splicing of exon 7A by using the cell culture-based splicing assay (Fig. 1c). The −11–14 PMO we used here had the same sequence as that Wheeler and his colleagues used in a previous study14. RT-PCR analysis showed that both 1–25 and −11–14 PMOs significantly reduced exon 7A inclusion. Although the effect of 1–25 PMO was greater than that of −11–14 PMO, no statistically significant difference was observed between them.
1–25 PMO was so effective at improving alternative splicing of the Clcn1 minigene in cultured cells that we expected it to work well in vivo. To test whether 1–25 PMO could work in vivo, we administrated 60 μg of 1–25 PMO intramuscularly four times at weekly intervals into the tibialis anterior (TA) muscles of HSALR mice. The alternative splicing of the Clcn1 gene was moderately improved, with an approximately 30% decrease in exon 7A inclusion. Electromyography (EMG) with a single needle electrode, however, revealed that the occurrence of myotonia was not altered by PMO injection (Supplementary Fig. S2). Because 60 μg of PMO was quite a high dose for administration into a single muscle, we assumed that an effective delivery system would be required to introduce 1–25 PMO into muscle tissues. We therefore examined the usefulness of ultrasound-enhanced delivery with Bubble liposomes for PMO delivery. We administrated 20 μg of 1–25 PMO three times at weekly intervals into the TA muscles of HSALR mice with or without Bubble liposomes and ultrasound (Fig. 2). RT-PCR analysis revealed that the rate of inclusion of exon 7A decreased to its lowest level when both Bubble liposomes and ultrasound were applied, indicating that use of the combination of Bubble liposomes and ultrasound could enhance PMO delivery efficiency.
Figure 2. PMO delivery by the combination of Bubble liposomes and ultrasound.
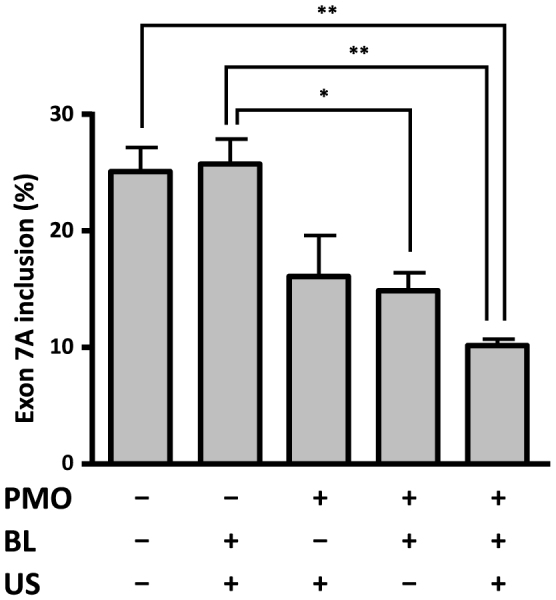
1–25 PMO (20 μg) or saline was locally administrated into TA muscles of HSALR mice with/without Bubble liposomes (BLs) and ultrasound (US). The inclusion rate of exon 7A decreased most when both Bubble liposomes and ultrasound were applied. (n = 3). The bars indicate mean and s.e.m., and statistical significance was analysed by Tukey's multi-comparison test (* P < 0.05, ** P < 0.01).
We next investigated whether 1–25 PMO could cure myotonic symptoms in HSALR mice when delivered using the Bubble liposome-ultrasound system. We administrated 1–25 PMO as described above. Three weeks later, we harvested the injected muscles and conducted RT-PCR and immunohistological analyses. RT-PCR showed that 1–25 PMO decreased the inclusion of exon 7A to a level comparable to that in wild-type FVB/n mice (Fig. 3a). We checked four other alternative splicing events, Cypher (Ldb3) exon 11, Mbnl1 exon 5, Ryr1 exon 70 and Serca1 exon 22, which are known to be abnormally regulated in patients with DM1and HSALR mice30. We found that the alternative splicing of none of them was changed by the injection of 1–25 PMO (Fig. 3b); thus, the effect of the PMO was specific to the alternative splicing of the Clcn1 gene.
Figure 3. In vivo improvement of aberrant splicing of the Clcn1 gene by 1–25 PMO.

(a) The alternative splicing of Clcn1 exon 7A in HSALR and WT mice. 1–25 PMO (20 μg) or saline was locally administrated into TA muscles of HSALR mice. The ratio of the splicing variant containing exon 7A in thevPMO-injected muscles decreased to a level comparable with that in WT muscle (n = 5). The bars indicate mean and s.e.m., and statistical significance was analysed by Tukey's multi-comparison test (*** P < 0.001). (b) Abnormal splicing of other genes in HSALR mice was not affected by 1–25 PMO. (c) Immunofluorescence analysis of transverse sections of TA muscle with an anti-Clcn1 antibody. Injection of 1–25 PMO restored the sarcolemmal localisation of Clcn1 protein in TA muscles of HSALR mice.
The abnormal splicing of Clcn1 is believed to cause myotonia by introducing a premature termination codon into the subsequent exon and by decreasing the expression of the Clcn1 protein via nonsense-mediated mRNA decay. Thus, we examined whether 1–25 PMO restored the expression of Clcn1 protein in HSALR mice. Immunofluorescence analysis of TA muscles showed the sarcolemmal localisation of Clcn1 protein in wild-type muscle, but such a pattern was not detected in saline-injected HSALR mice. The injection of 1–25 PMO clearly restored the sarcolemmal distribution, demonstrating that correction of the abnormal splicing of the Clcn1 gene led to the normal expression of its protein (Fig. 3c).
Finally, we investigated whether injection of 1–25 PMO improved the myotonic phenotype of HSALR mice. Electromyographic analyses showed bursts of action potentials after electrical stimulation (4–8 V) in HSALR mice, but not in wild-type mice (Fig. 4a). Myotonia occurred even in denervated muscles (data not shown), which indicates that it was not caused by hyperactivation of motor neurones, but by increased excitability of the sarcolemma. Myotonic EMG activities continued for 1 to 3 s and their average duration was 1.27 s in saline-treated TA muscles. The injection of 1–25 PMO decreased their duration, but the change was not statistically significant; the integrated EMG (iEMG) was reduced by the PMO administration (P < 0.05). The decreased iEMG indicated that 1–25 PMO mitigated the hyperexcitability of the HSALR muscles.
Figure 4. Reduction of myotonic discharge by 1–25 PMO in HSALR mice.

(a) Representative EMG signals of TA muscle. Tibialis muscle was electrically stimulated 1 s after the EMG recording started. In the HSALR muscle, repetitive discharges that were absent in the WT muscle could be seen. (b)vDuration of myotonic discharge and integrated EMG (iEMG). 1–25 PMO significantly reduced iEMG in HSALR muscle (saline, n = 6; 1–25 PMO, n = 7).The bars indicate mean and s.d., and statistical significance was analysed by Student's t-test (* P < 0.05).
These results suggest that 1–25 PMO could improve the function of the Clcn1 gene in the DM1 model mouse at the RNA, protein and phenotypic levels, and that the Bubble liposome-ultrasound system should be capable of delivering enough PMO to ameliorate myotonia.
Discussion
The greatest advantage of PMO is its remarkable innocuity. According to a report from Gene Tools, administration of a single 700 mg/kg dose of PMO to a mouse did not cause any obvious acute toxicity31, while the 50% lethal dose of phosphorothioate DNA, a first-generation antisense oligonucleotide, was estimated to be 750 mg/kg in mice32. The high pharmaceutical potential of PMO is also supported by the fact that safety issues were not raised following its administration to mice and humans; however, PMO must overcome the low permeability of the cell membranes to achieve a significant effect on alternative splicing. Because of the inefficient cellular uptake of PMO, systemic delivery and functional splicing modification were not successful in a mouse model33. Therefore, for the clinical application of PMO, establishing an effective delivery method is essential.
The major achievement in this study was that we increased the efficiency of PMO delivery using Bubble liposomes and ultrasound. The use of the ultrasound-mediated delivery system with Bubble liposomes improved the alternative splicing of the Clcn1 gene in HSALR mice to a level comparable to that of wild-type mice and decreased myotonic discharges, indicating that the delivery system increased introduction of 1–25 PMO into skeletal muscle. Our results show that the intramuscular injection itself delivered the PMO into muscle to some extent, and so the intramuscular injection might have contributed to the relief of the pathology. However, the Bubble liposome- and ultrasound-mediated enhancement of delivery efficiency suggests that the new delivery system will have a beneficial effect over much less invasive injection, such as intravenous injection, which cannot by itself be expected to promote the entry of PMO into skeletal muscle19.
A fair amount of the Clcn1 splicing variant without exon 7A was expressed even in the HSALR mouse. Because the myotonic discharge of a HSALR mouse was remedied by correcting Clcn1 alternative splicing in a previous study14, abnormal splicing of the gene must be the primary cause of myotonia. The expression levels of the “normal” splicing variant in saline-injected muscles were 57% of that in the wild-type muscles. PMO injection increased the expression about 1.4-fold to 78% of that in wild-type mice. As Clcn1 heterozygous mutant mice did not show a myotonic phenotype34, the myotonia in HSALR mice was unlikely to have been caused by haploinsufficiency of full-length Clcn1 protein. Instead, the truncated protein translated from the exon 7A-countaining mRNA may have dominant-negative activity, since full-length Clcn1 protein functions in a dimeric form. Berg et al. showed that the truncated Clcn1 protein did not function as a chloride channel, but rather disturbed the channel activity of full-length Clcn1 protein35. In this study, expression of the splicing variant containing exon 7A was decreased by 40% in the PMO-administered group. Thus, this may have contributed to the improvement of the pathology, as well as the increased expression of the exon 7A skipping variant. The dominant-negative hypothesis suggests that the AON therapy should completely prevent exon 7A inclusion when used to treat myotonia, in contrast to Duchenne muscular dystrophy, in which the partial restoration of dystrophin expression could lead to the improvement of muscle strength.
In the course of our search for the optimal PMO sequence to correct Clcn1 splicing, we found that 1–25 and 16–40 PMOs suppressed the inclusion of exon 7A well, but that 26–50 PMO did not. The fact that steric blocking of the 16–25 region promoted exon skipping indicates that proteins that bind to this region are essential for exon 7A recognition. Previously, we showed that the 8 nt at the 5′ end of exon 7A serve as an ESE and that an RNA-binding protein, Mbnl1, prevented the inclusion of exon 7A by binding to the ESE29. Unlike the ESE, the sequence of the 16–25 region was pyrimidine-rich and did not contain the Mbnl1-recognition motif, YGCY. It remains to be determined which proteins bind to the region to regulate exon 7A splicing.
In this study, we tried to cure DM1 model mice using a PMO targeted to Clcn1. However, considering that dozens of genes are abnormally spliced in patients with DM1, it might be impractical to treat all symptoms due to mis-splicing of such genes with AONs at the same time. DM1 is caused by expansion of the CTG repeats in the 3′ UTR region of the DMPK gene. Transcripts with these expanded repeats sequester Mbnl proteins, which regulate alternative splicing, leading to global alternative splicing dysfunction36. Thus, expanded CUG repeat-containing RNA must be the most important target of antisense therapy for DM1, and many groups have studied the use of CAG repeat-containing AONs to dissociate Mbnl1 proteins from CUG repeat-containing RNA. Some trials to treat DM1 model mice with CAG AONs were successful19,37, but here again the obstacle to clinical application of the AONs was the lack of an efficient delivery method. Our ultrasound-mediated delivery system with Bubble liposomes must have a beneficial effect on the delivery of CAG-containing AONs.
Methods
AONs
Phosphorothioate 2′ O-methyl RNA oligonucleotides and phosphorodiamidate morpholino oligonucleotides were purchased from IDT (Coralville, IA, USA) and Gene Tools (Philomath, OR, USA), respectively. The sequences of the oligonucleotides are listed in Supplementary Table ST1. Both AONs were dissolved in water.
Construct
The Clcn1 minigene has been described previously29. Briefly, a Clcn1 minigene fragment covering exons 6 to 7 was amplified from mouse genomic DNA by PCR and inserted into the BglII-SalI sites of pEGFP-C1 (Clontech Laboratories, Mountain View, CA, USA).
Cellular splicing assay
COS-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies, Foster City, CA, USA) in a humidified atmosphere containing 5% CO2 at 37°C.
For the splicing assay, COS-7 cells were cultured in 12-well plates and transfected with 0.1 μg of the Clcn1 minigene and AONs (0.1 μmol) at 60–80% confluence. Polyethylenimine and Endo-Porter (Gene Tools) were used for the transfection of PS2OMe RNA and PMOs, respectively. Forty-eight hours later, total RNA was extracted from the transfected cells using a GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich).
Animals
HSALR mice are FVB/n-background transgenic mice that express expanded CTG repeats under the control of the human skeletal actin promoter in skeletal muscle38. Compared with the first established line, the number of the repeat was reduced: the mice used in this study carried 180–200 repeats. All the mutant mice showed persistent contraction of gluteal muscles after they bucked. We used FVB/nJcl mice (Clea Japan, Tokyo, Japan) as wild-type controls.
The present study was approved by the Ethical Committee for Animal Experiments at the University of Tokyo, and was carried out in accordance with the Guidelines for Research with Experimental Animals of the University of Tokyo and the NIH Guide for the Care and Use of Laboratory Animals (NIH Guide, revised 1996).
Bubble liposomes
Bubble liposomes were prepared by previously described methods24. Briefly, PEG liposomes composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (NOF Corporation, Tokyo, Japan) and 1,2-distearoyl-sn-glycero-3-phosphatidyl-ethanolamine-polyethyleneglycol (DSPE-PEG2000-OMe) (NOF Corporation) at a molar ratio of 94:6 were prepared by a reverse phase evaporation method. Briefly, all reagents were dissolved in 1:1 (v/v) chloroform/diisopropyl ether. Phosphate-buffered saline was added to the lipid solution, and the mixture was sonicated and then evaporated at 47°C. The organic solvent was completely removed, and the size of the liposomes was adjusted to less than 200 nm using extruding equipment and a sizing filter (pore size: 200 nm) (Nuclepore Track-Etch Membrane; Whatman Plc, Maidstone, Kent, UK). The lipid concentration was measured using a Phospholipid C test (Wako Pure Chemical Industries, Ltd, Osaka, Japan). Bubble liposomes were prepared from liposomes and perfluoropropane gas (Takachio Chemical Ind. Co. Ltd, Tokyo, Japan). First, 2-ml sterilised vials containing 0.8 ml of liposome suspension (lipid concentration: 1 mg/ml) were filled with perfluoropropane gas, capped and then pressurised with a further 3 ml of perfluoropropane gas. The vial was placed in a bath-type sonicator (38 kHz, 250 W) (SONO-CLEANER CA-4481L; Kaijo Denki, Tokyo, Japan) for 1 min to form Bubble liposomes.
Injection of morpholino oligonucleotides with Bubble liposomes and ultrasound
PMO (20 μg) and the Bubble liposome suspension (30 μl) were injected into the TA muscles of HSALR mice (6 weeks old) using a 30-gauge needle (NIPRO Co., Osaka, Japan). Immediately after injection, ultrasound (frequency, 1 MHz; duty, 50%; intensity, 2.0 W/cm2; time, 60 s) was applied transdermally downstream of the injection site using a 6-mm diameter probe. A SONITRON 1000 device (Rich-Mar, Chattanooga, TN, USA) was used to generate the ultrasound. We administrated the PMO three times at weekly intervals. Three weeks after the last administration, we performed EMG measurements and then killed the mice and harvested the TA muscles for RT-PCR analysis and immunohistochemistry.
Electromyographic recording and electrical stimulation
Implantation of EMG electrodes and stimulating electrodes was carried out under aseptic conditions on mice anaesthetised with 2% vapourised isoflurane in air. Body temperature was measured rectally and was maintained at 37–38°C using a homeothermic heating pad (BioResearch Center, Aichi, Japan). Bipolar wire electrodes (tip distance, 1–2 mm) made of Teflon-insulated stainless steel wire (76 μm diameter bare, 140 μm coated; cat. no. 791000; A-M Systems, Carlsborg, WA, USA) were implanted in the TA and gastrocnemius (GA) muscles to record EMG activity. The electrical stimulation of the TA muscle was achieved using two wire electrodes that were inserted under the skin over the TA muscle and placed along the longitudinal axis of the muscle. After full recovery from the anaesthesia, alert mice were restrained in a cylindrical mouse-sized cage, with their hind limbs out of the cage to maintain their muscles at their resting lengths. The EMG signals were amplified and bandpass filtered (15 Hz–1 KHz; AB-611J; Nihon-Koden, Co., Tokyo, Japan), digitised with an analog-digital converter (PowerLab 16/30, ADInstruments Ltd, Oxford, UK) and recorded (sampling rate 10 kHz) on a computer. Electrical stimulation consisted of repetitive square pulses (train of 20 pulses at 100 Hz, 1 ms duration) delivered by an isolation unit (SS-202J; Nihon-Koden) connected to a pulse generator (SEN-3401, Nihon-Koden). The stimulus intensity was adjusted to evoke ankle dorsiflexion and avoid overt movements and animal discomfort. EMG measurements were recorded in single-blinded manner.
EMG data analysis
Myotonic EMG activities were easily confirmed by visual inspection and analysed using custom-written MATLAB software (MathWorks, Inc., Natick, MA, USA). EMG signals were full-wave-rectified and filtered with a 20 Hz low-pass second-order Butterworth filter. Offset of the EMG signal was defined as a deflection below three standard deviations from baseline. The baseline level was defined as the mean EMG signal in the resting state before stimulation. Duration of myotonic activities was defined as the period from the termination of stimulation to the offset time. Myotonic activities were integrated during the duration of myotonia and calculated by subtracting the baseline level. To quantify EMG activities per unit time, iEMG values were then calculated as the integrated myotonia value divided by corresponding net duration. The EMG data were analysed in a single-blinded manner.
RT-PCR analysis
Total RNA was extracted from TA muscles and cultured cells using TRIzol reagent (Life Technologies) and a GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich), respectively, according to the manufacturers' instructions.
Typically, 0.5–1.0 μg of total RNA was reverse-transcribed with a PrimeScript 1st Strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan) using oligo(dT) primers. PCR reactions were performed using Ex Taq DNA polymerase (Takara Bio). The sequences of the PCR primers are listed in Supplementary Table ST2. The products were electrophoretically resolved on an 8% polyacrylamide gel that was stained with ethidium bromide and analysed using an LAS-3000 luminescence image analyser (FujiFilm, Tokyo, Japan). The ratio of exon 7A inclusion in Clcn1 mRNA was calculated as (7A inclusion)/(7A inclusion + 7A skipping) × 100.
Immunofluorescence
Frozen sections (6 μm thick) of unfixed TA muscles were immunostained with an affinity-purified rabbit polyclonal anti-Clcn1 antibody (dilution 1:50; Alpha Diagnostics International, San Antonio, TX, USA). The secondary antibody was Alexa Fluor 488-conjugated goat anti-rabbit IgG (Life Technologies) used at a dilution of 1:600. Images were collected using an IX70 inverted microscope (Olympus, Tokyo, Japan) equipped with a ×20 objective lens. Exposure time and threshold were identical for all comparisons of antisense and saline controls.
Statistics
A two-tailed Student's t-test or Tukey's multiple comparison test were used for statistical comparison.
Author Contributions
S.I. conceived the project. M.K. designed the experiments. T.K. carried out cell culture-based splicing assay. K.N., M.P.T. carried out the delivery of PMO. R.M., Y.H. and I.N. carried out histochemical staining. H.Y., M.H., M.S. and D.Y. designed and carried out EMG analysis. Y.N. and Y.E.-T. prepared Bubble liposomes.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Prof. Charles A. Thornton (University of Rochester) for providing us HSALR mice. We also thank Dr H. Mitsuhashi for valuable discussions and encouragement. This work was supported in part by the Comprehensive Research on Disability Health and Welfare, from the Ministry of Health, Labour and Welfare Japan, and Intramural Research Grant (23–5) for Neurological and Psychiatric Disorders of NCNP.
References
- Aslanidis C. et al. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 355, 548–551 (1992). [DOI] [PubMed] [Google Scholar]
- Brook J. D. et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808 (1992). [DOI] [PubMed] [Google Scholar]
- Buxton J. et al. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 355, 547–548 (1992). [DOI] [PubMed] [Google Scholar]
- Harley H. G. et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355, 545–546 (1992). [DOI] [PubMed] [Google Scholar]
- Harper P. S. Myotonic dystrophy, 3rd ed. (Saunders, W. B. Philadelphia, 2001). [Google Scholar]
- Philips A. V., Timchenko L. T. & Cooper T. A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741 (1998). [DOI] [PubMed] [Google Scholar]
- Savkur R. S., Philips A. V. & Cooper T. A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29, 40–47 (2001). [DOI] [PubMed] [Google Scholar]
- Fugier C. et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 17, 720–725 (2011). [DOI] [PubMed] [Google Scholar]
- Koebis M. et al. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells 16, 961–972 (2011). [DOI] [PubMed] [Google Scholar]
- Ohsawa N., Koebis M., Suo S., Nishino I. & Ishiura S. Alternative splicing of PDLIM3/ALP, for alpha-actinin-associated LIM protein 3, is aberrant in persons with myotonic dystrophy. Biochem. Biophys. Res. Commun. 409, 64–69 (2011). [DOI] [PubMed] [Google Scholar]
- Charlet B. N. et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 10, 45–53 (2002). [DOI] [PubMed] [Google Scholar]
- Mankodi A. et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 10, 35–44 (2002). [DOI] [PubMed] [Google Scholar]
- Lossin C. & George A. L. Jr Myotonia congenita. Adv. Genet. 63, 25–55 (2008). [DOI] [PubMed] [Google Scholar]
- Wheeler T. M., Lueck J. D., Swanson M. S., Dirksen R. T. & Thornton C. A. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 117, 3952–3957 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R., Krainer A. R. & Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug. Discov. 11, 125–140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein D., Foster E., Huang S. B., Weller D. & Summerton J. A specificity comparison of four antisense types: morpholino, 2′-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev. 7, 151–157 (1997). [DOI] [PubMed] [Google Scholar]
- Wu B. et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 17, 132–140 (2010). [DOI] [PubMed] [Google Scholar]
- Wheeler T. M. et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger A. J. et al. Systemic delivery of a peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 23, 109–117 (2013). [DOI] [PubMed] [Google Scholar]
- Lu Q. L., Liang H. D., Partridge T. & Blomley M. J. Microbubble ultrasound improves the efficiency of gene transduction in skeletal muscle in vivo with reduced tissue damage. Gene Ther. 10, 396–405 (2003). [DOI] [PubMed] [Google Scholar]
- Shen Z. P., Brayman A. A., Chen L. & Miao C. H. Ultrasound with microbubbles enhances gene expression of plasmid DNA in the liver via intraportal delivery. Gene Ther. 15, 1147–1155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag P. et al. Microbubble-enhanced ultrasound to deliver an antisense oligodeoxynucleotide targeting the human androgen receptor into prostate tumours. J. Steroid Biochem. Mol. Biol. 102, 103–113 (2006). [DOI] [PubMed] [Google Scholar]
- Suzuki R. et al. Gene delivery by combination of novel liposomal bubbles with perfluoropropane and ultrasound. J. Control Release 117, 130–136 (2007). [DOI] [PubMed] [Google Scholar]
- Negishi Y. et al. Delivery of an angiogenic gene into ischemic muscle by novel bubble liposomes followed by ultrasound exposure. Pharm. Res. 28, 712–719 (2011). [DOI] [PubMed] [Google Scholar]
- Negishi Y. et al. Local gene delivery system by bubble liposomes and ultrasound exposure into joint synovium. J. Drug Deliv. 2011, 203986 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi Y. et al. Systemic delivery systems of angiogenic gene by novel bubble liposomes containing cationic lipid and ultrasound exposure. Mol. Pharm. 9, 1834–1840 (2012). [DOI] [PubMed] [Google Scholar]
- Sugano M. et al. Gene delivery system involving Bubble liposomes and ultrasound for the efficient in vivo delivery of genes into mouse tongue tissue. Int. J. Pharm. 422, 332–337 (2012). [DOI] [PubMed] [Google Scholar]
- Negishi Y. et al. AG73-modified Bubble liposomes for targeted ultrasound imaging of tumor neovasculature. Biomaterials 34, 501–507 (2013). [DOI] [PubMed] [Google Scholar]
- Kino Y. et al. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res. 37, 6477–6490 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097 (2006). [DOI] [PubMed] [Google Scholar]
- Summerton J. & Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7, 187–195 (1997). [DOI] [PubMed] [Google Scholar]
- Templeton N. S. & Templeton N. S. Gene and cell therapy: therapeutic mechanisms and strategies. 2nd ed. (Marcel Dekker, New York, 2004). [Google Scholar]
- Sazani P. et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 20, 1228–1233 (2002). [DOI] [PubMed] [Google Scholar]
- Chen M. F., Niggeweg R., Iaizzo P. A., Lehmann-Horn F. & Jockusch H. Chloride conductance in mouse muscle is subject to post-transcriptional compensation of the functional Cl- channel 1 gene dosage. J. Physiol. 504, 75–81 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J., Jiang H., Thornton C. A. & Cannon S. C. Truncated ClC-1 mRNA in myotonic dystrophy exerts a dominant-negative effect on the Cl current. Neurology 63, 2371–2375 (2004). [DOI] [PubMed] [Google Scholar]
- Ranum L. P. & Cooper T. A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 29, 259–277 (2006). [DOI] [PubMed] [Google Scholar]
- Wheeler T. M. et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A. et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information