

Abstract
Background and Aims
MicroRNAs (miRNAs) play an important role in the responses and adaptation of plants to many stresses including low nitrogen (LN). Characterizing relevant miRNAs will improve our understanding of nitrogen (N) use efficiency and LN tolerance and thus contribute to sustainable maize production. The objective of this study was to identify novel and known miRNAs and their targets involved in the response and adaptation of maize (Zea mays) to LN stress.
Methods
MiRNAs and their targets were identified by combined analysis of deep sequencing of small RNA and degradome libraries. The identity of target genes was confirmed by gene-specific RNA ligase-mediated rapid amplification of 5′ cDNA ends (RLM-RACE) and by quantitative expression analysis.
Key Results
Over 150 million raw reads of small RNA and degradome sequence data were generated. A total of 46 unique mature miRNA sequences belonging to 23 maize miRNA families were sequenced. Eighty-five potentially new miRNAs were identified, with corresponding miRNA* also identified for 65 of them. Twenty-five new miRNAs showed >2-fold relative change in response to LN. In addition to known miR169 species, two novel putative miR169 species were identified. Deep sequencing of miRNAs and the degradome, and RLM-RACE and quantitative polymerase chain reaction (PCR) analyses of their targets showed that miRC10- and miRC68-mediated target cleavage may play a major role among miR169 families in the adaptation to LN by maize seedlings.
Conclusions
Small RNA and degradome sequencing combined with quantitative reverse transcription–PCR and RLM-RACE verification enabled the efficient identification of miRNAs and their target genes. The generated data sets and the two novel miR169 species that were identified will contribute to our understanding of the physiological basis of adaptation to LN stress in maize plants.
Keywords: Maize, Zea mays, low-nitrate stress, adaptation, miRNA, degradome, combined analysis
INTRODUCTION
Agricultural practices determine world food supply and, to a great extent, the state of the global environment because of their influence on the ecosystem and dependence on the inputs of many nutrients including nitrogen (N) and phosphorus (P) (Tilman et al., 2002). Intensive crop production has resulted in environmental pollution (Ju et al., 2009) and soil acidification (Guo et al., 2010) due to excessive application of N fertilizers. An increased understanding of the physiological and genetic regulation of the plant's response to low-nitrogen (LN) stress may provide insights into the development of crop varieties with increased nutrient-use efficiency or LN tolerance.
MicroRNAs (miRNAs) are small [approx. 21 nucleotides (nt)] endogenous RNAs that can play key regulatory roles in plants and animals by targeting mRNAs for cleavage or translational repression (Bartel, 2004; Mallory and Vaucheret, 2006; Voinnet, 2009). Previous studies have indicated that miRNAs help sense and reduce nutrient stresses such as phosphate, sulfate and copper in higher plants (Fujii et al., 2005; Bari et al., 2006; Chiou et al., 2006; Sunkar et al., 2006, 2007; Chiou, 2007; Gifford et al., 2008; Hsieh et al., 2009). Nitrate is a major form of inorganic N taken up by cereal crop roots. For identification of the miRNAs and their targets in response to LN stress in maize leaves and roots, the known miRNAs were screened by using miRNA microarrays (Xu et al., 2011). Microarray hybridization is a sensitive methods for identification of known miRNAs (Franco-Zorrilla et al., 2009). However, miRNA profiling via next-generation sequencing technologies can be used to identify both novel and known miRNAs (Hsieh et al., 2009; Ju et al., 2009; Pant et al., 2009; Jiao et al., 2011; Wang et al., 2011; Pritchard et al., 2012; Tang et al., 2012). A transcriptome-wide experimental method called ‘degradome sequencing’ or ‘parallel analysis of RNA ends (PARE)’ has been developed to identify directly and globally the remnants of small RNA-directed target cleavage (Addo-Quaye et al., 2008; German et al., 2008; Zhou et al., 2010). High-throughput miRNA profiling combined with degradome sequencing analysis is of great interest because integrated miRNA and mRNA cleavage profiles may provide genome-wide evidence for miRNA-directed mRNA cleavage at a large scale (Addo-Quaye et al., 2008). Using a combined sequencing strategy, a total of 99 new loci belonging to 47 miRNA families were identified by small RNA and degradome sequencing from four small RNA libraries and one degradome of maize seedling exposed to extreme N deficiency, which was actually an N starvation treatment without any N in solution (Zhao et al., 2012). Comparative analysis of identified known miRNAs at LN by microarray (Xu et al., 2011) with identified miRNAs under N-free conditions by sequencing (Zhao et al., 2012) indicates that around two-thirds of them matched each other. In Arabidopsis, different physiological responses were found in the root system under N limitation and N-free conditions (Remans et al., 2006), among different N regimes (Brun et al., 2010) and also in the photosynthesis system (Sun et al., 2002). However, there is limited information available for identification of novel zma-miRNAs associated with LN stress through sequencing. LN stress is more straightforwardly associated with agricultural practices since the primary goal for crop production and breeding is to breed and develop crop plants that adapt to LN stress rather than N-free stress. So far, only a very few studies have reported using the degradome for identification of miRNA targets in maize (Zhao et al., 2012; Zhai et al., 2013). In order to provide more meaningful experimental evidence for small RNAs and their mRNA targets, it is necessary to design new experiments on LN stress with combined small RNA and full degradome sequencing with an established full data set.
Systems biology such as whole-scale miRNA analysis can accelerate the discovery of N-regulatory networks as well as integrated knowledge of plant biology (Gutiérrez, 2012). Furthermore, specific miRNA analysis may increase our understanding of the key signalling components that plants use to sense N and regulate metabolism, physiology, and growth and development (Gifford et al., 2008; Vidal et al., 2010). Interestingly, miR169 had been identified as being involved in the adaptation of Arabidopsis to LN stress (Zhao et al., 2012), drought stress (Li et al., 2008) and salinity stress (Zhao et al., 2009). However, zma-miR169 is one of the large miRNA families, which is composed of 18 identified members with formal registration in maize in miRBase (http://mirbase.org/). These family species targeting the same or different genes are encoded by the corresponding MIR genes, which are independently expressed and regulated (Zhang et al., 2009). Thus, the abundance and differential expression level of known and novel species identified in the miR169 family in response to LN treatment may provide further experimental evidence on which members among the complex families may play a key role in adaptation to stress.
The objectives of this study were to: (1) identify and report the known and new miRNAs along with their targets by joint analysis of small RNAs and the corresponding degradome sequencing at LN; and (2) determine which unique miRNA species within the zma-miR169 family might play key roles in adaptation to LN stress. These reported data and findings will provide fundamental information concerning miRNAs and/or given species within a large miRNA family, and will increase our understanding of the physiological basis for LN tolerance and adaptation in maize.
MATERIALS AND METHODS
Materials and cultures
Maize (Zea mays) inbred line B73 seeds of similar size were surface sterilized in 10 % H2O2 for 30 min, soaked in distilled H2O for 4 h, and then germinated on filter paper moistened with distilled H2O at 28 °C for 2 d in the dark. The seedlings were transferred to coarse silica and placed in a growth chamber at 28 °C/22 °C and a 14/10 h light/dark cycle. After 5 d, uniform seedlings were selected, their endosperms were discarded and the seedlings were then grown hydroponically in a half-strength nutrient solution for 1 d before they were supplied with full-strength solution. The solution contained 4 mm nitrate in the optimal-nitrate (ON) treatment and 0·04 mm nitrate in the low-nitrate (LN) treatment; Ca (NO3)2·4H2O was the nitrate source. The other components of the nutrient solution were described previously (Xu et al., 2011), and the solution was continuously aerated and changed every 2 d. Shoots and roots were harvested 15 d after germination, immediately frozen in liquid nitrogen and stored at –80 °C.
RNA isolation and purification
RNAs were extracted from the four kinds of samples (ON shoots, ON roots, LN shoots and LN roots) using Trizol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Total RNAs were treated with RNase-free DNase I (TaKaRa, Dalian, China) and then purified following the manufacturer's instructions. The RNA concentration was measured with a NANO Drop-2000c spectrophotometer (Thermo Scientific, USA), and RNA quality and integrity were assessed by gel analysis.
Small RNA library construction and sequencing
Low molecular weight RNA was isolated from about 200 µg of total RNA by adding 50 % polyethylene glycol (PEG; mol. wt 8000 Da) to a final concentration of 5 % and 5 m NaCl to a final concentration of 0·5 m. Small RNA libraries were constructed as previously described (Sunkar and Zhu, 2004; Hafner et al., 2008). In brief, small RNAs (18–30 nt long) were purified by PAGE and ligated sequentially to 5′ and 3′ oligonucleotide RNA adaptors. After reverse transcription and polymerase chain reaction (PCR) amplification, PCR products were purified and subjected to SBS sequencing with a Solaxa/Illumina genome analyser at the BGI (Beijing Genomics Institute, Shenzhen, China).
Degradome library construction and sequencing
Approximately 150 µg of total RNA was used for the purification of polyadenylated RNA molecules with oligo(dT) cellulose (Ambion). Degradome libraries were constructed as previously described (German et al., 2008). Briefly, a single-stranded 5′ RNA adaptor was ligated to the cleaved products using T4 RNA ligase (Ambion). The ligated products were isolated again using oligo(dT) cellulose, and first-strand cDNAs were synthesized using Superscript II reverse transcriptase (Invitrogen). Following six cycles of PCR amplification, MmeI digestion (NEB), ligation of a double-stranded adaptor to the 3′ end, and another 21 cycles of PCR amplification, gel-purified PCR products were subjected to SBS sequencing with a Solaxa/Illumina genome analyser at the BGI.
Sequencing data deposition and access
All small RNA and degradome sequencing data analysed herein have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002; Barrett et al., 2011) and are accessible through GEO Series accessions SRX222260, SRX222261, SRX222262, SRX222263, SRX222264, SRX222265, SRX222266 and SRX222267 (Study name SRP018376; http://www.ncbi.nlm.nih.gov/sra/?term=SRP018376).
Small RNA sequencing analysis
The low quality reads were filtered from the raw data. Adaptors were trimmed, and contamination sequences formed by adaptor–adaptor ligation were cleaned up. Tags with <18 nt were then removed to generate clean reads. For primary analysis, the length distribution of clean sequences and common and specific sequences among samples was determined. For standard analysis, clean sequences were aligned to the maize B73 RefGen_V2 genome (www.maizesequence.org). Perfectly matching sequences were identified for further analysis. Non-coding RNAs that were annotated as rRNAs, tRNAs, small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) were identified by alignment and BLAST search against Rfam (9·1) (Gardner et al., 2009) and GenBank databases (Benson et al., 2006). Known miRNAs were then screened by alignment against the miRBase (Release 19) database (Griffiths-Jones et al., 2008), and known miRNAs differentially expressed among the different samples were identified. Unannotated small RNAs were used for prediction of novel miRNAs according to previously reported criteria for miRNA annotation (Nobuta et al., 2008).
Degradome sequencing analysis
Clean reads were generated using the same method as used for small RNA sequencing analysis. The alignments against the maize genome, Rfam (9·1) database and GenBank database were performed sequentially. The filtered reads were then aligned to exons and introns of maize mRNAs to identify the fragments of degraded mRNAs. Target plots, indicating the abundance of each distinct read on the genome, were generated. Next, miRNA–target RNA pairs for both the known miRNAs that were registered in miRBase (http://www.mirbase.org/) and the novel miRNAs that were identified from small RNA sequencing were determined as previously described (Addo-Quaye et al., 2008).
Prediction of small RNA targets
Targets of small RNAs that were identified by small RNA sequencing were predicted using the online prediction programs psRNATarget (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao, 2011) and WMD3 (http://wmd3.weigelworld.org/cgi-bin/webapp.cgi) (Ossowski et al., 2008) and the prediction tool in the UEA plant sRNA toolkit (http://srna-tools.cmp.uea.ac.uk/plant/cgi-bin/srna-tools.cgi) (Moxon et al., 2008). For psRNATarget, the maize cDNA sequences file (http://www.plantgdb.org/download/download/xGDB/ZmGDB/) was selected as a pre-loaded transcript library; for WMD3, Z. mays ZmB73 v5b (MGC) was selected as a genomic library, and other parameters were set as default. Potential targets predicted by all three programs were searched in the degradome data. Analysing the degradome data corresponding to the maize genome sequence, Target-align (http://www.leonxie.com/targetAlign.php?type=blast) (Xie and Zhang, 2010) was used to verify the cleavage site in the corresponding mRNA transcript. PsRobot, a web-based tool, has also been used for identification of small modulating RNAs with stem–loop-shaped precursors and their target genes/transcripts (Wu et al., 2012).
RLM-RACE
To obtain the cleavage site within miRNA targets, RLM-RACE (RNA ligase-mediated rapid amplification of 5′ cDNA ends) was employed using the GeneRacer Kit (Invitrogen) according to the manufacturer's instructions. Briefly, total RNA was extracted from the four samples (ON and LN for shoots and roots) and ligated with GeneRacer RNA Oligo (5′ RNA adaptor). The ligated RNAs were reverse transcribed using the GeneRacer oligo(dT) primer. Two rounds of nested PCR were then performed using GeneRacer 5′ primer/gene-specific primer and GeneRacer 5′ nested primer/nested gene-specific primer, respectively. The final PCR product was gel-purified and cloned into the pMD®18-T Vector (TaKaRa) for sequencing. Each target was confirmed by at least ten clones. All the primers for RLM-RACE as well as their parameters are listed in the Supplementary Data Table S6.
Real-time PCR
Expression of miRNA target genes was quantified using an ABI 7500 real-time PCR system. To estimate accurately the regulation by miRNA of its target gene, forward primer and reverse primers located in both the 5′ and 3′ fragments of the predicted cleavage site were designed with Beacon Designer 7 software. First-strand cDNAs were synthesized using M-MLV reverse transcriptase (Invitrogen) with 5 µg of total RNA per 20 µL of reaction, and the cDNAs were diluted 3-fold. Quantitative reverse transcription–PCR (qRT-PCR) with three technical and three biological replicates was performed with the Power SYBR Green PCR Master Mix (Applied Biosystems), and maize α-tubulin 5 (Tub5) was selected as an endogenous control. A 5 µL volume of cDNA was used in the 25 µL PCR mixture that contained 1× SYBR Green PCR Master Mix and the primers. The mixture was incubated at 94 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 40 s and 72 °C for 40 s (Franco-Zorrilla et al., 2009; Xu et al., 2011). PCR efficiency was determined with a standard curve based on a dilution series (30, 31, 32, 33, 34, 35) of a selected cDNA sample (Ramakers et al., 2003), and 3n dilution of each sample was used for the calculation of the relative expression level. The ΔΔCt method was used to determine the relative expression level of targets among different samples if the difference in amplification efficiency between targets and Tub5 was <5·3 % (Livak and Schmittgen, 2001); otherwise, the Pfaffl method was applied (Pfaffl, 2001). All primers for real-time RT–PCR as well as their parameters and analyzing methods are listed in the Supplementary Data Table S6.
RESULTS
Small RNAs and degradome sequences generated across libraries of different nitrate treatments
Small RNA and degradome sequence data were generated, including >150 million raw reads. Table S1 in the Supplementary Data provides the statistics of generated data on the raw reads, unique reads and clean reads, along with the perfectly matched maize genome sequences of B73 across four small RNA and four degradome sequencing samples. These matched unique sequences were used in further analyses. The overall size distribution of the unique reads from all four sequencing samples showed a quite similar pattern, with the 24 nt class being the most abundant (Supplementary Data Fig. S1). Our sequencing data showed that the abundance of highly conserved miRNAs, such as miR156, miR159, miR164, miR166, miR167, miR168 and miR172, was very high across the four small RNA libraries constructed from shoot and root tissues under different nitrate levels. For example, the lowest abundance in the four libraries for zma-miR156 and zma-miR168 was 59 534·09 reads per million (RPM) and 50 226·89 RPM, respectively. Interestingly, some non-conserved miRNAs, such as zma-miR528, a monocot-specific miRNA (Zanca et al., 2010), were found at an abundance of as much as >20 000 RPM in shoots.
Our small RNA sequencing data showed a good coverage of the known miRNAs. A total of 46 unique mature miRNA sequences belonging to 23 known miRNA families in the current miRBase (release 19·0) were sequenced within at least two of the four libraries with an abundance of > 1 RPM. Most of these miRNAs in the database were identified by a computational method based on sequence conservation using sequences of known miRNAs of other species. All unique miRNA sequences were confirmed by our sequencing effort, while 36 of 46 unique mature miRNA sequences were detected in all four libraries. Two unique miRNA species (zma-miR319a and zma-miR399a,c,h) were not detected in shoot libraries, while eight unique miRNA species (zma-miR166j,k,n, zma-miR169l, zma-miR171l,m, zma-miR172e, zma-miR394a,b, zma-miR397a, zma-miR399e,i,j and zma-miR529) were not detected in root libraries.
Identification of new miRNAs improved the maize miRNA database. Eighty-five potentially new miRNAs, which had not been registered on miRBase (release 19), were identified from four small RNA sequencing libraries (Supplementary Data Table S2). Compared with novel maize miRNAs identified in the recent literature (Jiao et al., 2011; Wang et al., 2011; Ding et al., 2012; Zhang et al., 2012; Zhao et al., 2012; Zhai et al., 2013), 55 out of 85 potentially new miRNAs identified in this study were first reported. The corresponding miRNA*s were also identified for 65 of these potentially new miRNAs. Sixty-five out of 85 potentially new miRNAs could be mapped onto the maize genome to find the corresponding MIR genes encoding their miRNAs. Fifty-six potentially new miRNAs were found in shoots, 15 of them were detected in roots while 14 of them were identified in both shoots and roots (Supplementary Data Table S2).
Novel maize miRNAs in response to LN
Twenty-five potentially new miRNAs were identified with the selection criteria of >2-fold relative change between LN and ON and the normalized sequencing reads >5 RPM (Table 1). The miRNA*, which has been considered as one of most important criteria for miRNA identification (Nobuta et al., 2008), was also identified for 15 of the 25 new miRNAs. Among these novel miRNAs, zma-miRC10 and zma-miRC68 were classified as new members of the miR169 family; zma-miRC40 and zma-miRC73 were classified as belonging to the miR398 and miR528 families, respectively (Table 1). Some novel miRNAs that were detected in two or more small RNA libraries also showed relatively high abundance, and their expression levels changed significantly under LN treatment. For example, zma-miRC69, zma-miRC70 and zma-miRC74 were significantly upregulated under the LN condition (Table 1). These results suggest that some newly identified miRNAs may play important roles in adaptation to LN stress in maize. The results also provide a good resource for further examination of regulatory roles of novel miRNAs in response and adaptation to nitrate limitation.
Table 1.
Identified new species from known miRNAs and some new miRNAs with a 2-fold change in the ratio between LN and ON conditions
| No. | miRNA ID | Sequence | Length (nt) | Locus (B73 V2) | miRNA* | Shoot (RPM) |
Root (RPM) |
Family | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LN | ON | LN/ON | LN | ON | LN/ON | |||||||
| 1 | zma-miRC4 | UUCUCCAGGAGUUGAUGGACAA | 22 | Unkown | Yes | 7·05 | – | ** | 5·98 | – | ** | – |
| 2 | zma-miRC5 | AUGCGGAGAGGCUCUCGAGAGA | 22 | Chr5:92369205–92369360(–) | Yes | 6·75 | 2·57 | 2·63 | – | – | – | – |
| 3 | zma-miRC10 | UAGCCAAGCAUGAUUUGCCCG | 21 | Chr1:298294462–298294550(+) | Yes | 5·41 | 31·72 | 0·17 | – | 34·15 | ** | miR169 |
| 4 | zma-miRC11 | AAUACACAUGGGUUGAGGAGG | 21 | Chr9:17048238–17048335(–) | Yes | – | 4·61 | ** | 1·20 | – | – | |
| 5 | zma-miRC13 | CUUAUUCCCGUCGGUUUCAGG | 21 | Chr2:2093367–2093462(–) | Yes | 156·29 | – | ** | – | – | – | – |
| 6 | zma-miRC15 | UCUAGAUCCAACGGACCAAAA | 21 | Chr8:94301451–94301631(–) | Yes | 8·23 | – | ** | – | – | – | – |
| 7 | zma-miRC16 | GGAGGAGAUUGGAGGGGCUAA | 21 | Chr5:190239698–190239787(–) | Yes | 5·79 | – | ** | – | – | – | – |
| 8 | zma-miRC17 | UGGUCUUGCCACCGUGUCGUCC | 22 | Chr9:143163223–143163312(–) | Yes | 5·34 | – | ** | – | – | – | – |
| 9 | zma-miRC38 | AUUAUUGAAAAUGGUUGUUGA | 21 | Unkown | Yes | – | 6·91 | ** | – | – | – | – |
| 10 | zma-miRC39 | AAUUUCCAGAUCUUUGUGCAUA | 22 | Chr6:100210090–100210253(–) | Yes | – | 5·23 | ** | – | – | – | – |
| 11 | zma-miRC49 | AAGCAAGUUGGGGUAGGCUAGA | 22 | Chr4:116857976–116858025(–) | Yes | – | – | – | 24·46 | – | ** | – |
| 12 | zma-miRC50 | CGGAGGGAAUUGGAGGGGCUA | 21 | Chr5:16432684–16432783(+) | Yes | 45·54 | 9·30 | 4·90 | 15·48 | 7·21 | 2·15 | – |
| 13 | zma-miRC51 | AUCGAAUGAGAUUGGGGGGAU | 21 | Chr7:120201243–120201337(–) | Yes | 113·64 | 26·58 | 4·27 | 11·44 | – | ** | – |
| 14 | zma-miRC59 | UCCCUCUCUCCCUUGAAGGCUC | 22 | Chr2:224138892–224139000(+) | Yes | - | – | – | – | 16·30 | ** | – |
| 15 | zma-miRC60 | UAAAGUGGCUUAUAAUUUGGA | 21 | Chr5:19125908–19126051(+) | Yes | 26·93 | 10·37 | 2·60 | – | 5·66 | ** | – |
| 16 | zma-miRC68 | UAGCCAAGGAUGAGCUGCCUG | 21 | Chr2:192700627–192700741(+) | No | 15·13 | 80·54 | 0·19 | – | – | – | miR169 |
| 17 | zma-miRC69 | AUUGGAGGGGAUUGAGGAGGCU | 22 | Chr1:151286998–151287087(–) | No | 244·93 | 85·42 | 2·87 | – | – | – | miR395a** |
| 18 | zma-miRC70 | ACCGGAGGAGGUUAGAGGAGC | 21 | Chr4:195789318–195789440(–) | No | 119·05 | 32·08 | 3·71 | – | – | – | – |
| 19 | zma-miRC71 | AGAAGAUGUUGUGCUGUUUCCU | 22 | Chr1:181210941–181211128(+) | No | 26·56 | 10·81 | 2·46 | – | – | – | miR167e** |
| 20 | zma-miRC72 | GCUUAUUUUCGGCGGCGUCUG | 21 | Unkown | No | 32·56 | 13·56 | 2·40 | – | – | – | – |
| 21 | zma-miRC73 | UCGAAGGGGAUUGGAGAGGUU | 21 | Chr2:74096511–74096608(+) | No | 19·73 | 8·86 | 2·23 | – | – | – | miR528 |
| 22 | zma-miRC74 | UUGAGAGGGAUUGGAGGGGUUU | 22 | Unkown | No | 124·25 | 42·27 | 2·94 | – | – | – | – |
| 23 | zma-miRC77 | AGUGGAUUAGAGGGGCUAAAA | 21 | Chr10:111704635–111704730(+) | No | 22·25 | 3·99 | 5·58 | 2·09 | 0·88 | 2·39 | miR2275c-5p |
| 24 | zma-miRC78 | UGGAUUAUGGUGGAAGGGUAA | 21 | Chr1:60498265–60498426(–) | No | 18·02 | 3·01 | 5·98 | 2·24 | 0·94 | 2·38 | – |
| 25 | zma-miRC81 | CAAGAUGUGGGCAAAACGACGG | 22 | Unkown | No | 60·45 | 29·68 | 2·04 | – | – | – | – |
**indicates a significant difference due to being detected only in either the LN or ON treatment.
RPM, reads per million.
Two novel maize miR169 family species and their targets identified across small RNA and degradome libraries in response to nitrate stress
Our small RNA sequencing effort detected the following unique mature miRNA sequences: zma-miR169a,b; zma-miR169c,r; zma-miR169f,g,h; zma-miR169i,j,k; zma-miR169l; and zma-miR169p. In addition to known miR169 species, we also identified two novel putative miR169 species, miRC10 and miRC68, which showed high sequence conservation with miR169 family members (Supplementary Data Fig. S2). Interestingly, miRC68 in our effort verified the identified new zma-miR169 species reported in a previous work on N starvation in maize (Zhao et al., 2012). MiR169c,r and miRC10 were the most abundant species under ON conditions and the most downregulated species under LN conditions (Fig. 1), suggesting that they might have a primary role among zma-miR169 species in regulating responses to LN. In addition, miRC10 and miRC68 were the most abundant species in the miR169 family under the ON condition and the most downregulated species under the LN condition in shoots (Fig. 1).
Fig. 1.

Abundance of multiple unique miR169 species in shoots and roots under low- and optimal-nitrate conditions.
We predicted the target genes of miRC10 and miRC68 and confirmed their identities across the four sets of degradome data (Table 2). GRMZM2G165488 was identified as one of the miRC10 targets across the degradome libraries. Degradome data showed that GRMZM2G165488 mRNA degradation products were significantly induced at a predicted miRC10 cleavage site of 3909 (mapped to the genomic DNA site of GRMZM2G165488) in the maize roots under the ON condition. Moreover, this cleavage mediated by miRC10 differed significantly between LN and ON treatments. In this study, tag abundance of the targeting site of miRC10 on the GRMZM2G165488_T02 transcript was the highest, about 1353 tags per 10 million reads (Fig. 2, red arrows), indicating that GRMZM2G165488 belongs to category I according to the three reported grouping categories based on the relative abundance of signatures at the target sites (Addo-Quaye et al., 2008). Under the LN condition, however, the target tag abundance was much lower (only about 16 tags per 10 million reads) (Fig. 2, blue arrows), indicating a category II classification.
Table 2.
Two identified zma-miR169 species (miRC10 and miRC68), their predicted targets and degradome validation evidence
| miRNA | Predicted targets | Degradome evidence | Gene annotations | Conservation across higher plants |
|---|---|---|---|---|
| zma-miRC10 | GRMZM2G078124,GRMZM5G829103, GRMZM2G165488, GRMZM5G853836, GRMZM5G838907 | GRMZM2G165488, GRMZM2G078124 | Nuclear transcription factor Y subunit A-10, Rhodopsin-like receptor | Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Brachypodium distachyon |
| zma-miRC68 | GRMZM2G078124, GRMZM5G857944, GRMZM2G000686, GRMZM2G038303, GRMZM2G040349, GRMZM2G091964 | GRMZM2G091964 | Nuclear transcription factor Y subunit A-1 | Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Brachypodium distachyon |
Fig. 2.
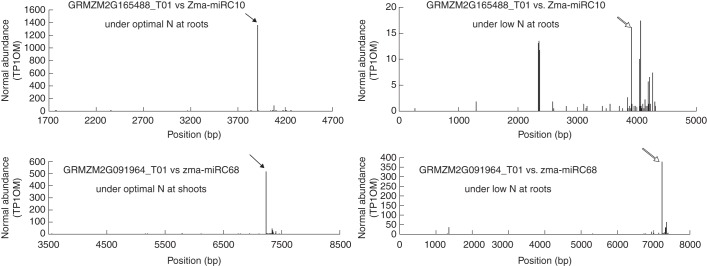
Distribution of the 5′ end of the degradome tags within the full-length target gene sequence. The solid arrows indicate the abundance peaks in the optimal-nitrate condition, while the open arrows indicate the abundance peaks in the low-nitrate condition in maize roots or shoots.
Negative regulation by two novel miR169 family species, miRC10 and miRC68
Our 5′-RACE analysis of GRMZM2G165488 (Fig. 3, top), GRMZM2G078124 and GRMZM2G091964 (Fig. 3, bottom) provides strong evidence that they interact with miRC10. The sequencing result of ten independent RACE fragments of the GRMZM2G165488 3′-untranslated region (UTR) pairing with miRC10 exactly matched the predicted site (Fig. 3, top). With a frequency of 7/29, the RACE sequencing result confirmed the target site of miRC10 on the GRMZM2G078124 pairing region, which is located within the second exon (Fig. 3, middle). The sequencing result of 14 independent RACE fragments of the GRMZM2G091964 3′-UTR pairing with miRC68 also exactly matched the predicted site (Fig. 3, bottom).
Fig. 3.

Experimental validation and diagram of the cleavage site (open arrow) of GRMZM2G165488 (top) and GRMZM2G078124_T01 (middle) with miRC10 and of GRMZM2G091964 with miRC68 (bottom) mediated by miRNA in the gene structure using 5′-RACE assay. Alignment of miRC10 and miRC68 with a portion of their target sequence (top) and distribution of the 5′ end of the degradome tags within the full-length target gene mRNA sequence (bottom) corresponding to their targets. The black arrows indicate the cleavage sites in the gene structure. The open arrow indicates the cleavage site validated by RLM-RACE at the target site. For numbers and slashes, the first number is the frequency of validated events detected in the sequenced tags, and the second number is the total number of events.
We then examined the response of zma-miRC10 targets to LN in shoots and roots by qRT–PCR with specific primers (Fig. 4A). Under the ON condition, the relative expression levels for both GRMZM2G165488 and GRMZM2G078124 were very low. However, the two genes were upregulated >8-fold and >13-fold, respectively, in shoots under the LN condition. Interestingly, GRMZM2G165488 was also highly upregulated in roots under the LN condition. The expression level of GRMZM2G091964, the zma-miRC68 target, was also examined in response to LN in both shoots and roots (Fig. 4B). The relative expression level of GRMZM2G091964 in both tissues was quite low under the ON condition but was upregulated >16-fold in shoots but only 3-fold in roots under the LN condition. These results indicate that the interaction between miRC10 and GRMZM2G165488 in maize may play a key role in the root responses to LN stress and that the interaction between miRC10, GRMZM2G165488 and GRMZM2G078124 may also regulate shoot responses to LN. Moreover, miRC68 may act mainly in shoots and by interacting with GRMZM2G091964.
Fig. 4.
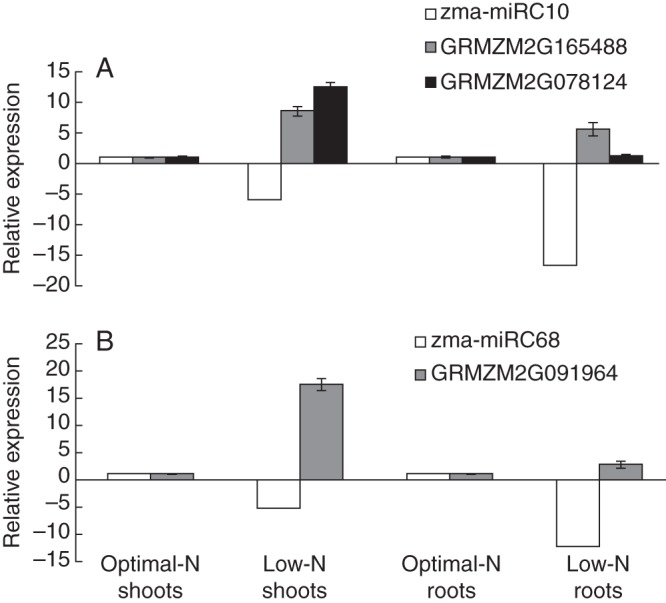
Quantitative RT–PCR analysis of (A) zma-miRC10 and (B) miRC68 targets in response to low nitrate in maize. miRC10 and miRC68 abundances were calculated from the abundance of the small RNA sequences, and therefore the standard deviations are not indicated. GRMZM2G165488 and GRMZM2G078124, both of which are the targets of miRC10, are shown in the top panel. GRMZM2G091964, the target of zma-miRC68, is shown in the bottom panel. The relative expression levels for the target genes are the means of the fold changes and standard deviation from three biological replicates.
DISCUSSION
Sequencing quality
Distinct N deficiency phenotypes of young seedlings were found under LN stress before sequencing (Supplementary Data Fig. S3), including reduced chlorophyll content, reduced biomass, increased anthocyanin accumulation in shoots and early leaf senescence resulting from N remobilization to younger tissues. In addition, the seedlings in the LN treatment allocated more biomass to roots than seedlings in the ON treatment, resulting in a significantly higher root/shoot ratio, as in our previous report (Xu et al., 2011), and developmental changes in primary and lateral roots.
Our small RNA sequencing covered the known miRNA families and species in maize well. The fragment size distribution was consistent with that previously reported for different maize tissues (Jiao et al., 2011; Wang et al., 2011). For example, the 24 nt class represented 38 % of the unique reads from the ON roots and slightly over 30 % of the unique reads from the LN shoots (Supplementary Data Fig. S1). The distribution pattern among tissues might also explain the slight differences in the frequencies of other abundant small RNA classes such as 21 and 22 nt classes.
Validation of the known LN-responsive miRNAs and difference between LN and N-free stresses
Table S3 in the Supplementary Data provides a list of identified differentially expressed known miRNAs with high abundance, their predicted targets and degradome validation across root and shoot libraries. Compared with the previous LN stress microarray study (Xu et al., 2011), most known LN-responsive miRNAs could have been confirmed in this sequencing effort. However, the extents of differential expression for five mature miRNAs under the LN condition were different from those found in the previous report. For example, miR167 and miR172 were induced by 1·95- and 2·97-fold in roots while miR169, miR408 and miR528 were greatly decreased in both roots and shoots in this study. The difference might be due to the different maize lines, as maize B73 was used in this study but Ye478 was used in the previous microarray study (Xu et al., 2011). Importantly, the degradome data have also been available in this study to provide in-depth target information on those miRNAs identified, since degradome analysis can detect 50–80 % of previously validated targets and 15–30 % of predicted targets (Addo-Quaye et al., 2008; German et al., 2008).
Our experiment was designed to identify the miRNAs in response to chronic low N treatment (15 d after low nitrate). We also compared our results with that from an independent test of a transient (2 d) N-free treatment (Zhao et al., 2012). Most of the results on the known miRNAs, including miR169, miR395, miR528, miR398, miR408 and miR164, could be verified against each other (Supplementary Data Table S4). However, the important differences between these two experiments are the N stress regimes and durations of the N stress. In higher plants, different physiological responses were found between N limitation and N-free treatments and among different N regimes in the root system (Remans et al., 2006; Brun et al., 2010) and in the photosynthesis system (Sun et al., 2002). Our system may provide some physiological insight into chronic LN stress, which frequently occurs for major crop plants. The shorter duration but higher N stress (N-free stress) used in the study of Zhao et al. (2012) might be the most important reason for those differences compared with the results in Supplementary Data Table S4.
Two identified novel MIR169 family species
In assigning a new miRNA species to a distinct family, researchers should consider the newly identified paralogous miRNA loci producing identical or nearly identical mature miRNAs; sequence conservation of the new miRNA; and its precise excision from the stem of a stem–loop precursor (Nobuta et al., 2008). The two identified novel miR169 family species showed a potential hairpin secondary structure and could produce a small RNA duplex with a 2 nt overhang at both 3′ ends (Fig. 1), which is the primary criterion of annotation as an miRNA (Ambros et al., 2003; Nobuta et al., 2008). Because read abundances of zma-miRC10-5p and zma-miRC68-5p were both approx. 100 times higher than their -3p counterparts, we may classify them as functional miRNAs and their -3p counterparts as the miRNA*. In addition, there were computationally predicted targets of miRC10, and miRC68 was annotated as one of the nuclear transcription factor Y subunit A proteins (NFYA) with the same annotation of previously reported miR169 targets (Li et al., 2008). All these pieces of evidence support assigning them to miR169 families. One of these two reported miR169 species, miRC68, had also been found in the previous sequencing effort (Zhao et al., 2012). Here we provided in-depth evidence for both newly identified miR169 species, including the degradome, RACE and the differential expression in roots and shoots. Among them, RACE tests provide an unambiguous diagnosis of endonucleolytic cleavage at the presumptive target site within the duplex region between miRNA and mRNA (Llave et al., 2002; Bartel, 2004).
In Arabidopsis, downregulation of miR169 reduces miR169 repression of the target NFYA5 mRNA through an abscisic acid (ABA)-dependent pathway by drought (Li et al., 2008) and high salinity (Zhao et al., 2009). Later, the miR169–NFYA5 interaction was verified also to be involved in the N deficiency response (Zhao et al., 2012). All these reported pieces of evidence suggest that the miR169–NFYA5 module might be conserved in higher plants to help cope with abiotic stresses such as drought, LN and high salinity.
Other small RNAs (non-miRNAs) identified in this research
Although miRNAs play important roles in plant growth, development and stress acclimatization, they are not the only species of functional non-coding small RNAs. Plants also have other types of endogenous small interfering RNAs (siRNAs) that are important in development, and these include heterochromatic siRNAs (hc-siRNAs), trans-acting siRNAs (ta-siRNAs) and natural antisense transcript-derived siRNAs (nat-siRNAs) (Czech and Hannon, 2011). Ta-siRNA is a secondary siRNA that is generated by miRNA-mediated cleavage of TAS1 or TAS3 (Montgomery et al., 2008; Felippes and Weigel, 2009). Nat-siRNAs are usually generated from convergent transcription units present in plant genomes under biotic or abiotic stresses (Borsani et al., 2005; Katiyar-Agarwal et al., 2006; Henz et al., 2007). All these small RNAs, except for the miRNAs mentioned above, play important roles in gene expression, protein modification, and stress response and adaptation. For example, transcriptional silencing caused by hc-siRNA-mediated RdDM (RNA-directed DNA methylation) is an important regulatory pathway in gene expression (Chellappan et al., 2010). Nat-siRNAs and ta-siRNAs can be involved in stress response and adaptation (Khraiwesh et al., 2012). Beside miRNAs, we also identified a list of siRNAs in response to LN treatment (Supplementary Data Table S5). Our small RNA sequencing also detected >0·1 million reads, about 2·5 % of the total small RNA reads, that can be annotated as siRNAs. In the miRNA database, zma-miR390 has been registered but zma-miR173 has not. However, zma-miR390a and zma-miR390b are not differentially expressed under ON vs. LN conditions, and we also failed to detect degraded fragments mediated by miR390 cleavage in the degradome libraries, indicating that miRNA390 mediated ta-siRNA may not be involved in the response of maize to LN. Our siRNA data will be useful for further analysis of siRNA function in response to LN stress.
SUPPLEMENTARY DATA
ACKNOWLEDGEMENTS
This study was supported by a Chinese National ‘863’ program from the China Ministry of Science and Technology (Subprogram of Grant no. 2012AA10A306), the National Science Foundation of China Grant (no. 31171562), and by a Chinese National ‘973’ program from the China Ministry of Science and Technology (Subprogram of Grant no. 2009CB118404) to C.X.
LITERATURE CITED
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Current Biology. 2008;18:758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari R, Datt Pant B, Stitt M, Scheible W-R. PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiology. 2006;141:988–999. doi: 10.1104/pp.106.079707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Research. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Research. 2006;34:D16–D20. doi: 10.1093/nar/gkj157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu J-K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005;123:1279–1291. doi: 10.1016/j.cell.2005.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun F, Richard-Molard C, Pagès L, Chelle M, Ney B. To what extent may changes in the root system architecture of Arabidopsis thaliana grown under contrasted homogenous nitrogen regimes be explained by changes in carbon supply? A modelling approach. Journal of Experimental Botany. 2010;61:2157–2169. doi: 10.1093/jxb/erq090. [DOI] [PubMed] [Google Scholar]
- Chellappan P, Xia J, Zhou X, et al. siRNAs from miRNA sites mediate DNA methylation of target genes. Nucleic Acids Research. 2010;38:6883–6894. doi: 10.1093/nar/gkq590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou TJ. The role of microRNAs in sensing nutrient stress. Plant, Cell and Environment. 2007;30:323–332. doi: 10.1111/j.1365-3040.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- Chiou TJ, Aung K, Lin S-I, Wu CC, Chiang SF, Su CL. Regulation of phosphate homeostasis by microRNA in Arabidopsis. The Plant Cell. 2006;18:412–421. doi: 10.1105/tpc.105.038943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nature Reviews Genetics. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Research. 2011;39:W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Wang Y, Han M, et al. MicroRNA transcriptomic analysis of heterosis during maize seed germination. PLoS One. 2012;7:e39578. doi: 10.1371/journal.pone.0039578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felippes FF, Weigel D. Triggering the formation of tasiRNAs in Arabidopsis thaliana: the role of microRNA miR173. EMBO Reports. 2009;10:264–270. doi: 10.1038/embor.2008.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, Del Toro FJ, Godoy M, et al. Genome-wide identification of small RNA targets based on target enrichment and microarray hybridizations. The Plant Journal. 2009;59:840–850. doi: 10.1111/j.1365-313X.2009.03904.x. [DOI] [PubMed] [Google Scholar]
- Fujii H, Chiou T-J, Lin S-I, Aung K, Zhu J-K. A miRNA involved in phosphate-starvation response in Arabidopsis. Current Biology. 2005;15:2038–2043. doi: 10.1016/j.cub.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, et al. Rfam: updates to the RNA families database. Nucleic Acids Research. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong D-H, et al. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nature Biotechnology. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Gifford ML, Dean A, Gutierrez RA, Coruzzi GM, Birnbaum KD. Cell-specific nitrogen responses mediate developmental plasticity. Proceedings of the National Academy of Sciences, USA. 2008;105:803–808. doi: 10.1073/pnas.0709559105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Research. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JH, Liu XJ, Zhang Y, et al. Significant acidification in major Chinese croplands. Science. 2010;327:1008–1010. doi: 10.1126/science.1182570. [DOI] [PubMed] [Google Scholar]
- Gutiérrez RA. Systems biology for enhanced plant nitrogen nutrition. Science. 2012;336:1673–1675. doi: 10.1126/science.1217620. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landgraf P, Ludwig J, et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44:3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henz SR, Cumbie JS, Kasschau KD, et al. Distinct expression patterns of natural antisense transcripts in Arabidopsis. Plant Physiology. 2007;144:1247–1255. doi: 10.1104/pp.107.100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh L-C, Lin S-I, Shih AC-C, et al. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiology. 2009;151:2120–2132. doi: 10.1104/pp.109.147280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Song W, Zhang M, Lai J. Identification of novel maize miRNAs by measuring the precision of precursor processing. BMC Plant Biology. 2011;11:141. doi: 10.1186/1471-2229-11-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X-T, Xing G-X, Chen X-P, et al. Reducing environmental risk by improving N management in intensive Chinese agricultural systems. Proceedings of the National Academy of Sciences, USA. 2009;106:3041–3046. doi: 10.1073/pnas.0813417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar-Agarwal S, Morgan R, Dahlbeck D, et al. A pathogen-inducible endogenous siRNA in plant immunity. Proceedings of the National Academy of Sciences, USA. 2006;103:18002–18007. doi: 10.1073/pnas.0608258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khraiwesh B, Zhu J-K, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochimica et Biophysica Acta. 2012;1819:137–148. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-X, Oono Y, Zhu J, et al. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. The Plant Cell. 2008;20:2238–2251. doi: 10.1105/tpc.108.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297:2053–2056. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nature Genetics. 2006;38:S31–S37. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- Montgomery TA, Howell MD, Cuperus JT, et al. Specificity of ARGONAUTE7–miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell. 2008;133:128–141. doi: 10.1016/j.cell.2008.02.033. [DOI] [PubMed] [Google Scholar]
- Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24:2252–2253. doi: 10.1093/bioinformatics/btn428. [DOI] [PubMed] [Google Scholar]
- Nobuta K, Lu C, Shrivastava R, et al. Distinct size distribution of endogeneous siRNAs in maize: evidence from deep sequencing in the mop1-1 mutant. Proceedings of the National Academy of Sciences, USA. 2008;105:14958–14963. doi: 10.1073/pnas.0808066105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schwab R, Weigel D. Gene silencing in plants using artificial microRNAs and other small RNAs. The Plant Journal. 2008;53:674–690. doi: 10.1111/j.1365-313X.2007.03328.x. [DOI] [PubMed] [Google Scholar]
- Pant BD, Musialak-Lange M, Nuc P, et al. Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Physiology. 2009;150:1541–1555. doi: 10.1104/pp.109.139139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nature Reviews Genetics. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neuroscience Letters. 2003;339:62–66. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- Remans T, Nacry P, Pervent M, et al. A central role for the nitrate transporter NRT2·1 in the integrated morphological and physiological responses of the root system to nitrogen limitation in Arabidopsis. Plant Physiology. 2006;140:909–921. doi: 10.1104/pp.105.075721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Gibson KM, Kiirats O, Okita TW, Edwards GE. Interactions of nitrate and CO2 enrichment on growth, carbohydrates, and rubisco in Arabidopsis starch mutants. Significance of starch and hexose. Plant Physiology. 2002;130:1573–1583. doi: 10.1104/pp.010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Zhu J-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. The Plant Cell. 2004;16:2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Kapoor A, Zhu J-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. The Plant Cell. 2006;18:2051–2065. doi: 10.1105/tpc.106.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Chinnusamy V, Zhu J, Zhu J-K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends in Plant Science. 2007;12:301–309. doi: 10.1016/j.tplants.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Tang Z, Zhang L, Xu C, et al. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiology. 2012;159:721–738. doi: 10.1104/pp.112.196048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilman D, Cassman KG, Matson PA, Naylor R, Polasky S. Agricultural sustainability and intensive production practices. Nature. 2002;418:671–677. doi: 10.1038/nature01014. [DOI] [PubMed] [Google Scholar]
- Vidal EA, Araus V, Lu C, et al. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proceedings of the National Academy of Sciences, USA. 2010;107:4477–4482. doi: 10.1073/pnas.0909571107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–687. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu H, Li D, Chen H. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genomics. 2011;12:154. doi: 10.1186/1471-2164-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H-J, Ma Y-K, Chen T, Wang M, Wang X-J. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Research. 2012;40:W22–W28. doi: 10.1093/nar/gks554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Zhang B. Target-align: a tool for plant microRNA target identification. Bioinformatics. 2010;26:3002–3003. doi: 10.1093/bioinformatics/btq568. [DOI] [PubMed] [Google Scholar]
- Xu Z, Zhong S, Li X, et al. Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots. PLoS One. 2011;6:e28009. doi: 10.1371/journal.pone.0028009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanca A, Vicentini R, Ortiz-Morea F, et al. Identification and expression analysis of microRNAs and targets in the biofuel crop sugarcane. BMC Plant Biology. 2010;10:260. doi: 10.1186/1471-2229-10-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Liu Z, Zou X, et al. Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiologia Plantarum. 2013;147:181–193. doi: 10.1111/j.1399-3054.2012.01653.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chia J-M, Kumari S, Stein JC, Liu Z, Narechania A, Maher CA, Guill K, McMullen MD, Ware D. A genome-wide characterization of microRNA genes in maize. PLoS Genetics. 2009;5:e1000716. doi: 10.1371/journal.pgen.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lin H, Shen Y, et al. Cloning and characterization of miRNAs from maize seedling roots under low phosphorus stress. Molecular Biology Reports. 2012;39:8137–8146. doi: 10.1007/s11033-012-1661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ge L, Liang R, et al. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Molecular Biology. 2009;10:29. doi: 10.1186/1471-2199-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li W-X. Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One. 2012;7:e29669. doi: 10.1371/journal.pone.0029669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Gu L, Li P, et al. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica) Frontiers in Biology. 2010;5:67–90. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.