

Abstract
Sporadic inclusion body myositis (sIBM) usually manifests with painless weakness of the hand, finger and hip flexors. Absence of symptoms or signs, but mild hyper-CK-emia as the sole manifestation of IBM, has not been reported. We report the case of a 73-year-old male who presented with asymptomatic recurrent hyper-CK-emia ranging from 200 to 1324U/L (n<171U/L), since 10 years. Clinical neurologic investigation, nerve conduction studies and EMG were non-informative. Muscle biopsy surprisingly revealed sIBM. sIBM may be asymptomatic and may manifest with hyper-CK-emia exclusively. So, it has to be included in the differential diagnoses of asymptomatic hyper-CK-emia.
Key words: neuromuscular disorder, myositis, creatine-kinase, cardiomyopathy, muscle biopsy
Introduction
Clinical presentation of sporadic inclusion body myositis (sIBM) is variable, but most frequently characterized by painless weakness of the hand, fingers and hip flexors.1 Creatinekinase (CK) is only mildly elevated in sIBM.1 Absence of symptoms and signs but mild hyper-CK-emia as the sole manifestation of sIBM has not been reported.
Materials and Methods
For assessment of the muscle biopsy routine stainings on frozen sections (hematoxylin and eosin, Gomori-trichrome, PAS, Oil-red-O), enzymehistochemistry (COX/SDH, NADH, ATPase pH 4,2), immunohistochemistry (slow and fast myosin, utrophin-NT, dystrophin and associated proteins, NCAM, vimentin, desmin), a panel of so-called nonsense proteins and neurofilament (SMI-31), and inflammatory markers were performed. The list of antibodies used is detailed in Table 1. We examined in sum nine semithin sections and two were selected for electron microscopy using a Zeiss EM 109 Turbo/DPS (Carl Zeiss Inc., Thornwood, NY, USA).
Table 1.
List of antibodies applied during the histological work-up of the muscle biopsy. Positivity was found for antibodies against CD3, CD8, CD68, NCAM, p62, and SMI31.
| Antibody | Section type | Company | Dilution | Clonality |
|---|---|---|---|---|
| Alpha-B-crystallin | Frozen | Novocastra | 1:250 | Mono |
| Alpha-sarcoglycan | Frozen | Novocastra | 1:200 | Mono |
| Amyloid precursor protein | Paraffin | Chemicon | 1:8000 | Mono |
| Amyloid precursor protein | Frozen | Chemicon | 1:8000 | Mono |
| Amyloid-beta (b-A4) | Paraffin | Dako | 1:100 | Mono |
| Amyloid-beta (b-A4) | Frozen | Dako | 1:100 | Mono |
| Beta-sarcoglycan | Frozen | Novocastra | 1:200 | Mono |
| c5b-9 | Frozen | Dako | 1:4000 | Mono |
| Caveolin | Frozen | Santa Cruz | 1:200 | Mono |
| CD20 | Frozen | Dako | 1:2000 | Mono |
| CD3 | Frozen | Neomarkers | 1:200 | Mono |
| CD4 | Frozen | Dako | 1:100 | Mono |
| CD68 | Frozen | Dako | 1:10000 | Mono |
| CD79a | Frozen | Dako | 1:200 | Mono |
| CD8 | Frozen | Dako | 1:400 | Mono |
| Desmin | Paraffin | Dako | 1:50 | Mono |
| Desmin | Frozen | Dako | 1:50 | Mono |
| Dysferlin | Frozen | Novocastra | 1:10 | Mono |
| Dystrophin 1 | Frozen | Novocastra | 1:20 | Mono |
| Dystrophin 2 | Frozen | Novocastra | 1:20 | Mono |
| Dystrophin 3 | Frozen | Novocastra | 1:20 | Mono |
| Delta-sarcoglycan | Frozen | novocastra | 1:50 | Mono |
| Emerin | Frozen | Novocastra | 1:100 | Mono |
| Gamma-sarcoglycan | Frozen | Novocastra | 1:200 | Mono |
| HLA-ABC | Frozen | Dako | 1:8000 | Mono |
| Leukocyte common antigen | Paraffin | Dako | 1:2000 | Mono |
| Leukocyte common antigen | Frozen | Dako | 1:2000 | Mono |
| Merosin | Frozen | Novocastra | 1:100 | Mono |
| Myosin fast | Frozen | Novocastra | 1:100 | Mono |
| Myosin slow | Frozen | Novocastra | 1:100 | Mono |
| NCAM | Frozen | Monosan | 1:20 | Mono |
| p62 | Paraffin | BD | 1:500 | Mono |
| p62 | Frozen | BD | 1:500 | Mono |
| PrP3F4c | Paraffin | Signet | 1:100 | Mono |
| PrP3F4c | Frozen | Signet | 1:1000 | Mono |
| SMI31 | Paraffin | Sternberger | 1:1000 | Mono |
Results
The patient is a 73-year-old Caucasian male, with a previous history of left bundle-branchblock since age 64 years, recurrent hyper-CK-emia since a border-zone stroke at age 64 ranging between 200 and 1324 U/L (n<171 U/L), hyperlipidemia since age 64 years requiring statin therapy after detection of hyper-CK-emia, ischemic left posterior borderzone stroke at age 72, a 60% stenosis of the left internal carotid artery, and arterial hypertension since age 72. The family history was negative for neuromuscular disorder (NMD). Clinical neurologic examination revealed positional tremor exclusively. CK was 603 U/L. There was mild hypercholesterolemia. Nerve conduction studies and needle electromyography of the right brachial biceps and right gastrocnemius muscles were non-informative. CK remained elevated even after discontinuation of simvastatin.
Muscle biopsy from the right lateral vastus muscle was strongly indicative of IBM (Figure 1). The most obvious muscle pathological alterations were the caliber changes with atrophic fibers (Figure 1A) and many rimmed vacuoles together with endomysial inflammatory infiltrates (Figure 1B and C). In addition, COX negative-fibers (Figure 1D) and a single ragged-red fiber (RRF) and rimmed-vacuoles (Figure 1E and F) were noted in the Gomoritrichrome staining. The inflammatory infiltrates consisted mainly of CD3-positive T-cells, including CD8-positive cytotoxic T-cells with invasion of intact muscle fibers (Figure 1G, H). There were only single B-cells and only a few CD68-immunoreactive monocyte/macrophages. HLA-class I antigen was only focally and weakly upregulated and there was no evidence for the deposition of terminal complement complex (C5b9) in capillaries. Furthermore, there were no necrotic fibers or perifascicular atrophy. SMI-31 (neurofilament) and p62 (Figure 1I) showed immunoreactivity in the vacuoles. Dystrophin and associated proteins (Table 1) did not show alterations and there was a lack of accumulation of desmin (Figure 1J). We noted signs of a neurogenic muscle lesion with groups of atrophic fibers, upregulation of NCAM (Figure 1K) without corresponding vimentin immunoreactivity, and decreased central activity of NADH without typical targets (Figure 1L). Ultrastructural examination revealed fibers with autophagic vacuoles with sequestered cytoplasmic organelles and degradation products (Figure 1M-O). Typical tubulofilamentous inclusions were not detected. In summary, according to the presence of i) invasion of non-necrotic fibers by mononuclear cells, ii) vacuolated muscle fibers, iii) SMI-31 and p62/ubiquitin immunoreactivity of inclusions and vacuoles, IBM was diagnosed. Since there is no established therapy available for sporadic IBM and since symptoms and signs were absent, no specific neurological treatment was introduced.
Figure 1.
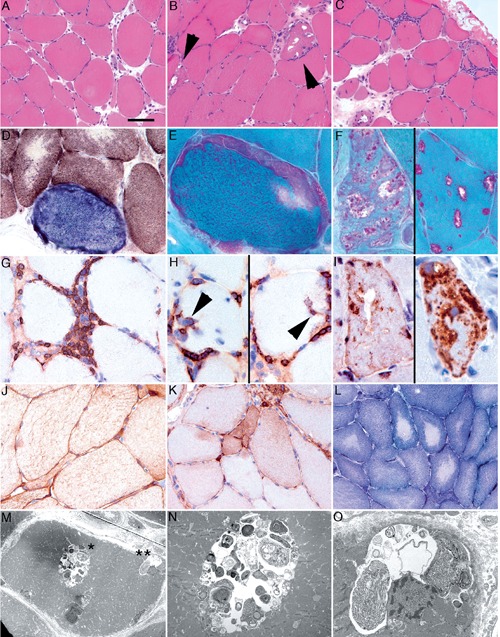
Muscle biopsy from the right lateral vastus muscle showing atrophic fibers with caliber changes (A), many rimmed vacuoles together with endomysial inflammatory infiltrates (B, C), COX negative-fibers (D) and a single RRF with rimmed-vacuoles (E, F). Inflammatory infiltrates consisted mainly of CD3-positive T-cells, including CD8-positive cytotoxic T-cells with invasion of intact muscle fibers (G, H). SMI-31 (neurofilament) and p62 (I) showed immunoreactivity in the vacuoles. Signs of a neurogenic muscle lesion with fiber-type grouping and upregulation of NCAM were noted (K). Ultrastructural examination revealed fibers with autophagic vacuoles with sequestered cytoplasmic organelles and degradation products (M, N, O).
Discussion and Conclusions
The presented patient is interesting for hyper-CK-emia as the sole manifestation of IBM, a poorly understood progressive muscle disease of middle or late life. Contrary to the classical clinical presentation with weakness of the proximal leg and distal arm muscles in an asymmetric distribution,1 our patient presented only with hyper-CK-emia but had not developed muscle weakness so far. In the majority of the cases, sIBM manifests already at onset with muscle weakness of the proximal and distal arm and leg muscles.2 Causes of asymptomatic hyper-CK-emia other than sIBM include calpainopathy, dysferlinopathy, central core disease, multicore disease, McArdle’s disease, metabolic myopathy, polymyositis, sarcoid myopathy, endocrine myopathy, statin myopathy, and rhabdomyolysis.3-5 These differentials were excluded in the presented patient by musclce biopsy and other blood chemical values. Weakness in sIBM predominantly affects the finger flexors, the wrist flexors, or the quadriceps muscle. sIBM has a dual pathology of autoimmunity and unexplained myofiber degeneration or loss of myofibers.6 The diagnosis is established by muscle biopsy showing the typical features of invasion of non-necrotic fibers by mononuclear cells, vacuolated muscle fibers, and either intracellular amyloid deposits or tubulofilaments 15-18 nm in diameter on electron microscopy.6 Contrary to sIBM hereditary inclusion body myopathy (hIBM) may initially affect the quadriceps muscle and may follow an autosomal dominant trait of inheritance,7 may spare the quadriceps muscle but may follow an autosomal dominant transmission,8 may manifest as distal myopathy Nonaka,9 may be associated with Paget’s disease and fronto-temporal dementia,10 or may be linked to the myosin-heavy chain-II locus.11 Patients with hIBM are usually younger than patients with sIBM, are virtually always symptomatic, and there is no inflammation in their biopsies. No reliable effective therapy is currently available, neither for sIBM nor for hIBM.7 The reason why sIBM in the presented patient manifested only with hyper-CK-emia remains speculative but could be explained by a new mutation in one of the genes associated with hIBM. Recently it has been shown that ageing may have an influence on the development of sIBM.12
This case shows that sIBM may be asymptomatic and may manifest with hyper-CK-emia exclusively. sIBM has to be included in the differential diagnoses of asymptomatic hyper-CK-emia.
References
- 1.Dimachkie MM, Barohn RJ. Inclusion body myositis. Semin Neurol 2012;32:237-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenberg SA. Pathogenesis and therapy of inclusion body myositis. Curr Opin Neurol 2012;25:630-9 [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi Y, Takahashi T, Sumi H, et al. [A case of dysferlinopathy asymptomatic for 10 years after an episode of transient muscle weakness]. Rinsho Shinkeigaku 2012;52:495-8 [Article in Japanese.] [DOI] [PubMed] [Google Scholar]
- 4.Schindler C, Thorns M, Matschke K, et al. Asymptomatic statin-induced rhabdomyolysis after long-term therapy with the hydrophilic drug pravastatin. Clin Ther 2007;29:172-6 [DOI] [PubMed] [Google Scholar]
- 5.Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989;12:206-9 [DOI] [PubMed] [Google Scholar]
- 6.Dalakas MC. Sporadic inclusion body myositis - diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol 2006;2:437-47 [DOI] [PubMed] [Google Scholar]
- 7.Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007;6:620-31 [DOI] [PubMed] [Google Scholar]
- 8.Sadeh M, Gadoth N, Hadar H, Ben-David E. Vacuolar myopathy sparing the quadriceps. Brain 1993;116:217-32 [DOI] [PubMed] [Google Scholar]
- 9.Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 1981;51:141-55 [DOI] [PubMed] [Google Scholar]
- 10.Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 2005;65:1304-5 [DOI] [PubMed] [Google Scholar]
- 11.Needham M, Mastaglia FL, Garlepp MJ. Genetics of inclusion-body myositis. Muscle Nerve 2007;35:549-61 [DOI] [PubMed] [Google Scholar]
- 12.Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol 2012;71:680-93 [DOI] [PubMed] [Google Scholar]