

Abstract
Multiple sclerosis (MS) is a chronic disease characterized by multiple areas of central nervous system inflammation, demyelination and axonal loss. Hereditary spastic paraparesis (HSP) is characterized clinically by progressive spasticity and weakness of the lower limbs and pathologically by retrograd axonal degeneration of the corticospinal tracts and posterior columns. We identified a patient with clinical history and investigation findings consistent with the concurrence of both MS and HSP. Laboratory and radiological investigations, cognitive tests were performed. Genetic confirmation for spastin gene mutation has been completed. If this coexistence is not coincidence the mutation in the spastin gene may be a strong susceptibility locus for MS.
Key words: multiple sclerosis, spastin, hereditary spastic paraparesis
Introduction
Hereditary spastic paraparesis (HSP) is a heterogeneous group of inherited disorders, with prevalence ranged from 4.3 to 9.6 cases per 100.000 population.1,2 HSP is characterized clinically by progressive spasticity and weakness of the lower limbs and pathologically by retrograd axonal degeneration of the corticospinal tracts and posterior columns. Harding (1993) divided HSP syndromes into pure and complicated types according to clinical manifestations. 3,4 Pure HSP present with progressive gait disturbance, spasticity of lower limbs, hiperreflexia, extensor plantar responses added to family history.5 Complicated HSP includes spasticity accompanied by muscle amyotrophy, optic atrophy, pigmentary retinopathy, extrapyramidal disease, deafness, dementia, mental retardation, ichthyosis, peripheral neuropathy, epilepsy, ataxic syndrome. 6 Genetically autosomal dominant, autosomal recessive and X-linked mode of inheritance have been described.6 Autosomal dominant inheritance is most commonly associated with püre forms of the disease, whereas autosomal recessive HSP shows greater phenotypic variability.7 Twenty-nine different loci of HSP have been associated with the disease, 11 of the genes (maspardin, paraplegin, atlastin, spastin, spartin, NIPA 1, KIF5A, HSPD1, PLP1, LICAM, BSCL2) known to be involved in the pathogenesis.8 The functions of several of these genes are related to axonal transport or intracellular trafficking. Mostly mutations spastin and atlastin genes identified in ADHSP families.3,4 Around 40% of autosomal dominant HSP, SPG4, is caused by mutations in the spastin gene on chromosome 2p.6,9 HSP shares several clinical features of the progressive phase in Multiple Sclerosis (MS) which is a chronic neurodegenerative disease characterized by multiple areas of central nervous system (CNS) inflammation, demyelination and axonal loss.
We identified a patient with clinical history and investigation findings consistent with the concurrence of both MS and HSP. Three members of the family had a history likely to HSP and three members also had a history of MS. Laboratory and radiological investigations, cognitive tests were performed. Genetic confirmation for spastin gene mutation has been completed.
Case Report
A 34 years old male was referred to our outpatient clinic with one week history of tight, heavy sensation affecting his left arm and leg. Further review of his history revealed tremor at his left leg which spontaneously reverted within 24 hours aged 32 and right eye vision loss with partial resolution without any spesific treatment at the age of 33. On examination, he had bilateral gaze evoked nystagmus, mild spastic gait abnormality, left ankle clonus and hipertonicity of both legs with extensor plantar responses. Pinprick and vibratory sensation was diminished in the feet. Laboratory tests included full blood count, electrolytes, liver function tests, glucose, blood urea, erythrocyte sedimentation rate, thyroid function tests, immunologic study (antinuclear antibodies, anticardiolipin antibodies, anti-neutrophil cytoplasmic antibodies), infectious serology (syphilis, lyme disease), vit B 12, folate were all normal. Electromyography and nerve conduction studies were normal. Visual evoked responses revealed prolonged latency in the right optic pathway. Analysis of CSF could not be performed because the patient did not accept lumbar punction. Cranial MRI showed multipl hyperintense lesions in the deep and periventricular white matter on T2 weighted and FLAIR images (Figure 1). Spine MRI was normal. The patient was treated with intravenous methylprednisolone for 5 days and baclofen 20 mg/d was included in the therapy. On neuropsychological testing show evidence of mild cognitive deficits in verbal function and fluency. Two years later the patient had one more attack. Nystagmus, hypertonicity of both legs with extensor plantar responses, spastic gait abnormality and sensory loss noted distally from the Th 11 level. Spinal MRI demonstrated an inhomogeneous lesion enhancing gadolinium in the segments (Figure 2). All the findings excluding gait abnormality and extensor plantar responses were improved with the methylprednisolone treatment. Aged 39, he was readmitted with weakness in his right leg and arm. Both right hip and arm flexion weakness of 4/5, increased tendon reflexes in the upper and lower limbs, ankle clonus with extensor plantar responses were examined. FLAIR and T2 weighted cranial MRI at this time showed abnormal signal in the periventricular and subcortical white matter. T1 weighted images following contrast showed new enhancements at the centrum semiovale and corona radiata (Figure 3). He treated with ıntravenous methyprednizolone (1000 mg/d) for 10 days. One years later, aged 40, he had a further relapse characterized by right gaze evoked nystagmus, a left isolated VI. cranial nerve palsy, pyramidal signs in all his limbs especially on the left; brisk reflexes in the lower limbs, stiff-legged, unsteady gait. This episode was again treated with pulse steroid for 10 days. Thus, the patient had clinical criteria of time and space dissemination and the diagnosis of MS was additionally corroborated by the presence of Barkhof’s criteria for space dissemination. The decision was made to start interferon beta 1b with the disability EDSS (Extended Disability Status Scale) score of 2.5. Currently the patient is receiving interferon beta 1b and baclofen and had no other relapse, his disability EDSS score is 2.0 after four years of follow up. Family history revealed that the patient’s mother has been recorded with both MS and epilepsy. The patient’s sister has also been diagnosed as MS. The presence of same findings of HSP had been determined in the patient’s aunt who is living in another country. The aunt’s son has also been diagnosed with MS. Four generation of the family pedigree is shown in the Figure 4.
Figure 1.
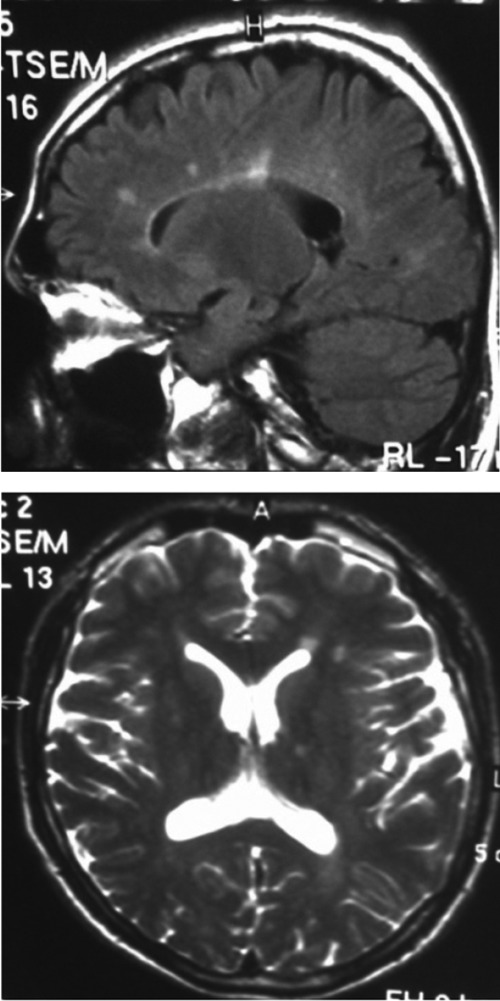
Axial T2 weighted and sagittal FLAIR images show hyperintense lesions in the periventricular and subcortical white matter.
Figure 2.
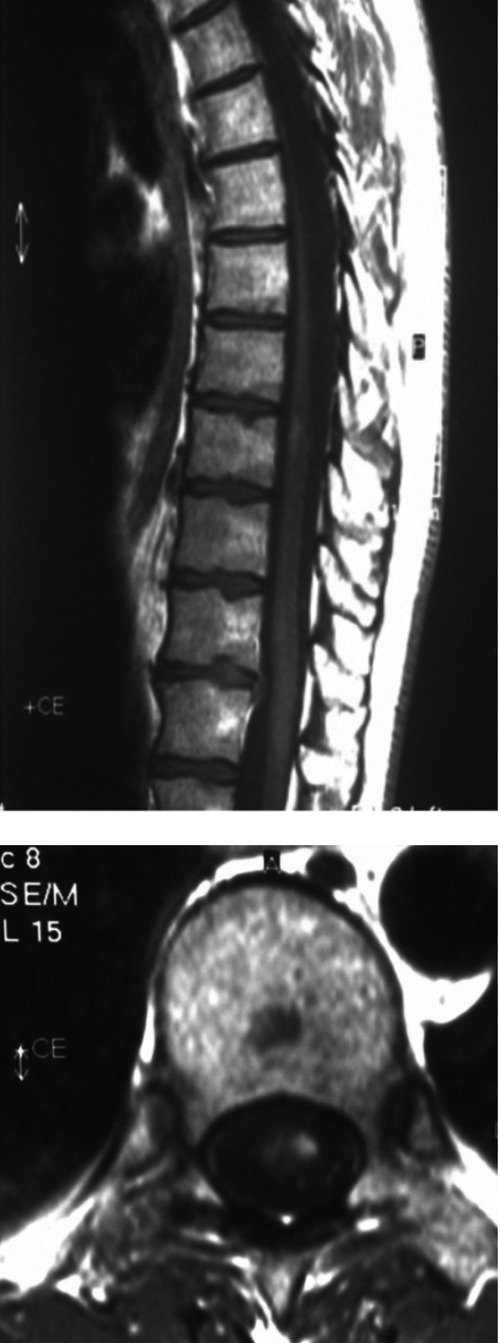
Spinal cord magnetic resonance imaging: sagittal and axial T1 after gadolinium displays contrast enhancement in the lesion T10-11.
Figure 3.
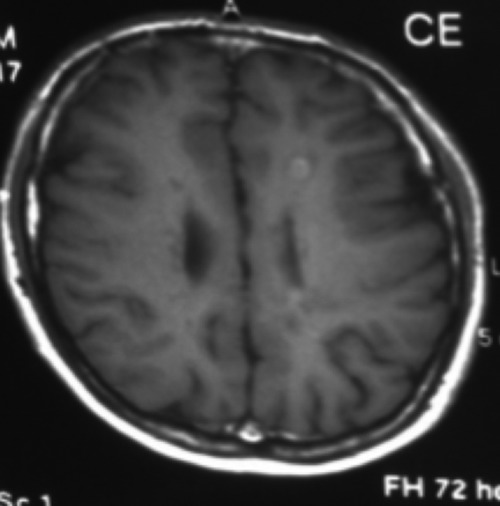
Axial T1 weighted images show contrast enhancement at the centrum semiovale and corona radiata.
Figure 4.
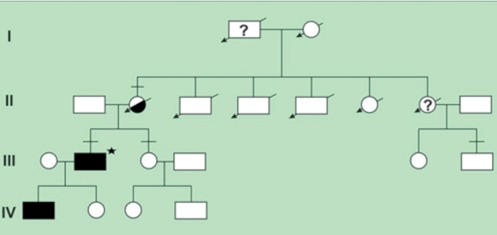
Four generation of the family. Squares indicate men, circles women and diagonal arrows indicate deceased members. Filled symbols indicate spastin gene mutation in the members, question marks indicate that the people definitely or probably affected with HSP. Half solid symbol shows epilepsy in the member. Horizontal bar above signs indicate the people with MS. Asteriks sign indicate the proband.
The patient’s 21-year-old son was affected with the same problems as his father; but he developed spasticity and balance difficulty by the age of 12. On neurological evaluation he presented a severe spasticity and hyperreflexia of the lower limbs with extensor plantar responses. Vibration and proprioception were reduced bilaterally. On detailed neuropsychological examination no cognitive decline was revealed. Magnetic resonance imaging of cranial and spinal cord were all normal. He had been clinically diagnosed as having HSP. Genetically, SPG4 locus was detected and a heterozygous one pair deletion in exon 6 (c.310_311 insA) of the spastin gene has been found. This mutation has been confirmed both in the patient and the son with a clinical phenotype of püre HSP.
Discussion and Conclusions
MS is an neurodegenerative, chronic disorder which pathogenesis is thought to involve an autoimmune process that occurs in genetically susceptible people. HSP is a rare disease with a prevalence ranging between 4.3 and 9.6 cases per 100,000 inhabitants.1,2 We described a case of a 45 year old man who had a clinical findings consistent with the concurrence of both relapsing remitting MS and HSP caused by a deletion in exon 6 of the spastin gene. His son had clinical and investigation findings with a diagnosis of pure HSP. In a European case series of chromosome 2 linked HSP, a 42 year old woman who had HSP disease and clinical, radiological findings of MS was described.10 On other study 11 HSP related genes were screened in 163 MS cases (112 benign MS, 51 malign MS) and 202 control subjects. Any association between paraplegin, HSPD1, atlastin, spartin, PLP1, LICAM, maspardin, BSCL2, spastin genes and MS can not be determined but a small effect of any of these genes in MS pathogenesis can not be denied.11 Also some diseases such as Leber’s hereditary optic neuropathy and neurofibromatosis that attends to MS have also been reported.12,13 The association of HSP and epilepsy was reviewed in several studies.14-20 The inheritance mostly autosomal dominant in these reports. Spastin mutation associated with epilepsy has also been reported twice.21,22 The irreversible clinical disability which are seen both in MS and HSP relates to axonal loss of the long tracts. Genes which are responsible for the maintenance of these axons can play a role in clinical outcome or disease severity of MS. So we need further investigations to search the relation between HSP and development of MS.
References
- 1.Leone M, Bottacchi E, D’Alessandro G, Kustermann S. Hereditary ataxias and paraplegias in Valle d’Aosta, Italy: a study of prevalence and disability. Acta Neurol Scand 1995;91:183-7 [DOI] [PubMed] [Google Scholar]
- 2.Polo JM, Calleja J, Combarros O, Berciano J. Hereditary ataxias and paraplegias in Cantabria, Spain. An epidemiological and clinical study. Brain 1991;114:855-66 [DOI] [PubMed] [Google Scholar]
- 3.Reid E. Science in motion: common molecular pathological themes emerge inthe hereditary spastic paraplegias. J Med Genet 2003;40:81.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fink JK. Hereditary spastic paraplegia. : Rimoin DL, Pyeritz RE, Connor JM, Korf BR, Emory and Rimoin’s principles and practice of medical genetics. 4th ed London: Harcourt UK; 2002. pp 3124-3125 [Google Scholar]
- 5.Fink JK. The hereditary spastic paraplegias: nine genes and counting. Arch Neurol 2003;60:1045-9 [DOI] [PubMed] [Google Scholar]
- 6.McDermott C, White K, Bushby K, Shaw P. Hereditary spastic paraparesis: a review of new developments. J Neurol Neurosurg Psychiatry 2000;69:150-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coutınho P, Barros J, Zemmouri R, et al. Clinical heterogeneity of autosomal recessive spastic paraplegias: analysis of 106 patients in 46 families. Arch Neurol 1999;56:943-9 [DOI] [PubMed] [Google Scholar]
- 8.Klebe S, Azzedine H, Durr A, Bastien P. Autosomal recessive spastic paraplegia (SPG 30) with mild ataxia and sensory neuropathy maps to chromosome 2q37.3. Brain 2006;129:1456-62 [DOI] [PubMed] [Google Scholar]
- 9.Reid E, Grayson C, Rubinsztein DC, et al. Subclinical cognitive impairment in autosomal dominant pure hereditary spastic paraplegia. J Med Genet 1999;36:797-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durr A, Davoine CS, Paternotte C, et al. Phenotype of autosomal dominant spastic paraplegia linked to chromosome 2. Brain 1996;119:1487-96 [DOI] [PubMed] [Google Scholar]
- 11.De Luca GC, Ramagopalan SV, Cader MZ, Dyment DA. The role of hereditary spastic paraplegia related genes in multiple sclerosis. J Neurol 2007;254:1221-6 [DOI] [PubMed] [Google Scholar]
- 12.Harding AE, Sweeney MG, Miller DH, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation. Brain 1992;115:979-89 [DOI] [PubMed] [Google Scholar]
- 13.Ferner RE, Hughes RA, Johnson MR. Neurofibromatosis 1and multiple sclerosis. J Neurol Neurosurg Psychiatry 1995;58:582-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb S, Flanagan N, Callaghan N, Hutchinson M. A family with hereditary spastic paraparesis and epilepsy. Epilepsia 1997;38:495-9 [DOI] [PubMed] [Google Scholar]
- 15.Bruyn GW, Mechelse K. The association of familial spastic paraplegia and epilepsy in one family. Psychiatr Neurol Neurochir 1962;65:280-92 [PubMed] [Google Scholar]
- 16.Gigli GL, Diomedi M, Bernardi G, et al. Spastic paraplegia, epilepsy and mental retardation in several members of a family: a novel genetic disorder. Am J Med Genet 1993;45:711-6 [DOI] [PubMed] [Google Scholar]
- 17.Sommerfelt K, Kyllerman M, Sanner G. Hereditary spastic paraplegia with epileptic myoclonus. Acta Neurol Scund 1991;84:157-60 [DOI] [PubMed] [Google Scholar]
- 18.Skre H. Hereditary spastic paraplegia in Western Norway. Clin Genet 1974;6:165-83 [DOI] [PubMed] [Google Scholar]
- 19.Yih JS, Shuu-Jiun W, Ming-Shung S, et al. Hereditary spastic paraplegia associated with epilepsy, mental retardation and hearing impairment. Paraplegia 1993;31:408-11 [DOI] [PubMed] [Google Scholar]
- 20.Kuroda S. Familial spastic paraplegia with epilepsy. Acta Med Okayama 1985;39:113-7 [DOI] [PubMed] [Google Scholar]
- 21.Mead SH, Proukakis C, Wood N, Crosby AH. A large family with hereditary spastic paraparesis due to a frame shift mutation of the spastin (SPG4) gene: association with multiple sclerosis in two affected siblings and epilepsy in other affected family members. J Neurol Neurosurg Psychiatry 2001;71:788-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinzlef O, Paternotte C, Mahieux F, et al. Mapping of a complicated familial spastic paraplegia to locus SPG4 on chromosome 2p. J Med Genet 1998;35:89-93 [DOI] [PMC free article] [PubMed] [Google Scholar]