

Abstract
Mitochondrial DNA depletion syndrome (MDS) is usually a severe disorder of infancy or childhood, due to a reduced copy number of mtDNA molecules. MDS with only mild, nonspecific clinical manifestations and onset in adulthood has not been reported. A 47-year-old Caucasian female with short stature and a history of migraine, endometriosis, Crohn’s disease, C-cell carcinoma of the thyroid gland, and a family history positive for mitochondrial disorder (2 sisters, aunt, niece), developed day-time sleepiness, exercise intolerance, and myalgias in the lower-limb muscles since age 46y. She slept 9-10 hours during the night and 2 hours after lunch daily. Clinical exam revealed sore neck muscles, bilateral ptosis, and reduced Achilles tendon reflexes exclusively. Blood tests revealed hyperlipidemia exclusively. Nerve conduction studies, needle electromyography, and cerebral and spinal magnetic resonance imaging were noninformative. Muscle biopsy revealed detached lobulated fibers with subsarcolemmal accentuation of the NADH and SDH staining. Realtime polymerase chain reaction revealed depletion of the mtDNA down to 9% of normal. MDS may be associated with a mild phenotype in adults and may not significantly progress during the first year after onset. In an adult with hypersomnia, severe tiredness, exercise intolerance, and a family history positive for mitochondrial disorder, a MDS should be considered.
Key words: depletion syndrome, mitochondrial DNA, genetics, fatigue, polysomnia, exercise intolerance
Introduction
Mitochondrial DNA depletion syndrome (MDS) is usually a severe disorder of infancy or childhood due to a reduced copy number of mtDNA molecules within a mitochondrion.1,2 Depletion of mtDNA results from a replication defect, which may be caused by mutations in at least nine different nDNA-located genes.3 MDS with only mild clinical manifestations and onset in adulthood has not been reported.
Case Report
The patient is a 47-year-old Caucasian female, height 158 cm, weight 60 kg, who developed day-time sleepiness, exercise intolerance, and myalgias in the lower-limb muscles since age 46y. She slept 9-10 hours during the night and 2 hours after lunch daily. Her individual history was noteworthy for a number of previous disorders. In 3/96 an ovarian endometriotic cyst, a follicular ovarian cyst, and a hydatoid cyst of the left Fallopian tube were resectioned and an adhesiolysis carried out. In 10/96 she experienced a gastro-intestinal haemorrhage time-linked to menstruation. Endometriosis was suspected. Since 1997 she was diagnosed with migraine with up to 3-4 non-triggered attacks per month. In 2/99 she experienced a second gastro-intestinal haemorrhage, this time requiring 6 blood transfusions. Colonoscopy did not detect any source of bleeding. In 5/02 a periproctitic abscess developed and was adequately treated. In 3/03 erythema nodosa of the lower legs occurred, preceded by diarrhoea. Upon histological examination of the colonic mucosa, Crohn’s disease was diagnosed and a therapy with steroids (initially aprednisolone, since 10/03 budesonide) and mesalazine initiated, resulting in remission of the enteritis. In 10/06 a chronic anal fistula was diagnosed requiring surgical intervention. In 12/08 budenoside was discontinued. Since 08 she was taking the pill for endometriosis. Cerebral MRI in 5/09, carried out for work-up of headache, was normal. MRI of the cervical and lumbar spine, carried out for dorsal pain, revealed only slight degenerative abnormalities.
The family history was noteworthy for thyroid cancer (grandmother from the mother’s side, sister), gastric cancer (second sister), clinically multisystem mitochondrial disorder [2 sisters, niece, aunt (sister of mother, mother (dilative cardiomyopathy, myopathy, Hashimoto thyroiditis, recurrent synkopes, restless-leg syndrome, tinnitus, endometriosis, easy fatigability)], and sudden cardiac death (mother). Upon a family screening in 4/10 for thyroid carcinoma she, her sister, and her niece were found positive for the rearranged during transfection (RET)-oncogen mutation, which is associated with an increased risk to develop thyroid cancer.4 Following these results the niece underwent prophylactic thyroidectomy. In 7/10 gastrointestinal bleeding relapsed and budesonide was restarted. In 8/10 budesonide was replaced by infliximab (400 mg every 8 weeks) and mesalazine was discontinued. In 10/10 recurrence of the anal fistula required surgical intervention again. In 3/11 a non-specific impaired aggregation of thrombocytes was diagnosed. Clinical exam in 4/11 revealed short stature, sore neck muscles, exaggerated masseter reflex, and reduced Achilles tendon reflexes exclusively. Hyperlipidemia was noted but serum lactate, myoglobin, and thyroid function tests were normal. Nerve conduction studies and needle electromyography were noninformative. Echocardiography and cardiac MRI revealed a bicuspid aortic valve exclusively. In 7/11 a medullary thyroid carcinoma was diagnosed and treated exclusively by resection. For post-operative hypothyroidism she took Lthyroxine. In 3/12 she reported an increase in intensity of migraine attacks, increasing fatigability, and myalgias. She was on a therapy with infliximab (every 8 weeks), L-thyroxin, and the pill for endometriosis.
During thyroid resection in 3/11, a muscle biopsy from the sternocleidomastoid muscle was additionally taken revealing mild to moderate variation in fiber size (Figure 1A), predominance of type 2 fibers, single fibers with eosinophilic cytoplasmic bodies (Figure 1B), and single lobulated fibers exhibiting moderate subsarcolemmal accentuation of SDH (Figure 1C), and NADH (Figure 1D) activity. Biochemical investigations of the muscle homogenate revealed reduced activity of all respiratory chain complexes in relation to non-collagen protein [NADH-CoQ-oxidoreductase: 6.1 U/g NCP (n, 15.8-42.81 U/g NCP), succinate/cytochrome C-oxidoreductase: 5.41 U/g NCP (n, 6.0-25.0 U/g NCP), cytochrome-c-oxidase: 45U/g NCP (n, 112-351 U/g NCP)]. Southern blot and long-range PCR were negative but real-time PCR was indicative of a MDS. Other clinically affected family members did not consent with a genetic investigation so far. The amount of residual mtDNA was reduced to 9% of normal. For funding reasons, neither sequencing of any of the nine candidate genes, nor exomesequencing could be offered.
Figure 1.
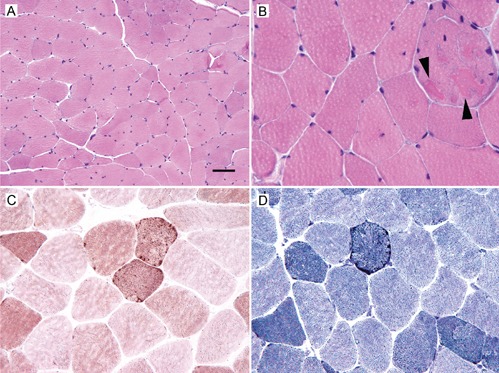
Light microscopy of the sternocleidomastoid muscle showing mild to moderate variation in fiber size (A: Haematoxylin & Eosin staining), a fiber with eosinophilic cytoplasmic bodies (B: arr owheads; Haematoxylin & Eosin staining), moderate subsarcolemmal accentuation of SDH activity C, combined COX/SDH histochemistry), and moderate subsarcolemmal accentuation of NADH activity (D: NADH histochemistry). Bars indicate 50 μm in A, 20 μm in B, and 25 μm in C and D.
Discussion
MDSs are characterised by severe reduction of the mtDNA copy number.1 Residual mtDNA copy levels may be as low as 1-2% of that of normal. In up to 50% of the cases, mtDNA depletion may be caused by mutations in at least nine different genes (POLG1, PEO1, RRM2B, SUCLG1, SUCLA2, DGUOK, MPV17, TK2, TYMP).1-6 Among these, mtDNA depletion is most commonly caused by mutations in the POLG1 gene, encoding the catalytic subunit of DNA polymerase-gamma, the only polymerase to replicate mtDNA.1,7 In the majority of the cases with MDS, however, the underlying genetic defect remains undetected. The phenotypic expression of mutations in these 9 genes is quite variable.2 Usually, infants or children are affected and the cerebrum, the liver, or the skeletal muscles are predominantly affected, alone or in combination (myopathic, encephalo-myopathic, or hepato-cerebral MDS).2,8,9 Patients with POLG1 mutations manifest as non-syndromic hepato-cerebral depletion syndrome, Alpers-Huttenlocher syndrome (AHS), infantile onset spinocerebellar ataxia (IOSCA), non-syndromic encephalomyopathic depletion syndrome, or Leigh-syndrome.9 Whether the MDS in the presented patient was due to long-term treatment with infliximab remains speculative but previous studies have shown that infliximab at least induces apoptosis of monocytes.10 An argument against the presence of a MDS in the presented case is that the family history suggestss a maternal trait of inheritance whereas all other MDS so far reported follow an autosomal recessive trait.
Contrary to previous descriptions, the phenotype in the presented patient was mild. Why mtDNA depletion in the presented patient resulted in only minor abnormalities, remains speculative but could be explained by the low rate of depleted mtDNA in tissues other than the muscle or by a mutation in a gene so far not reported in association with MDS. Probably, mtDNA depletion was absent or only mild in tissues other than the muscle. Possibly, the mild phenotype at onset will turn into a more severe presentation during the disease course, but given the stable presentation during the first year after onset and the benign course in other family members, such a scenario is rather unlikely. Assuming, that any of the nine genes so far associated with MDS was mutated in the presented patient, the ones most likely involved are the TK2, SUCLA2, RRM2B, SUCLG1, POLG, or TYMP genes since they have been found most frequently associated with myopathy.2,8 Among these, mutations in TYMP were found in adult patients with MNGIE.11 Also twinkle mutations were associated adult-onset MDS.5 Exercise intolerance has been reported as a phenotypic feature of DGUOK mutations and migraine or migrainelike headache as a feature of PEO1 mutations. 12,13 Hypersomnia or other sleep disorders and myalgias have not been reported in association with MDS. It must be admitted, however, that the phenotype of the presented patient fitted to none of those previously described in MDS. However, muscle biopsy was taken from the sternocleidomastoid muscle and though it showed some changes, myopathological alterations may vary considerably between muscles.
Whether the C-cell carcinoma was causally related to the MDS remains speculative, but preliminary observations in our cohort of patients with mitochondrial disorders suggest that the prevalence of malignancies is increased among these patients. It remains also unclear if endometriosis was causally related to the mitochondrial disorder. Arguments for a causal relation are that mtDNA polymorphisms were made responsible for the development of endometriosis and that mitochondrial biomarkers are increased in eutopic endometriosis.14,15 It remains also unclear, if Crohn’s disease was causally related to the MDS. Since some of the mitochondrial disorders go along with non-specific colitis,16 it is possible that the gastrointestinal problem was actually a manifestation of the MDS, but missing reports about an association between MDS and enteritis and unequivocal histological abnormalities argue against such an assumption.
Conclusions
In conclusion, MDS may start in adulthood, may be associated with a mild phenotype, and may not significantly progress during the first year after onset. In an adult patient with severe tiredness, exercise intolerance, hypersomnia, and a family history positive for mitochondrial disorder, MDS should be considered. Endometriosis, colitis, or malignancy may be a phenotypic feature of a mitochondrial disorder.
References
- 1.Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child 2009;94:3-5 [DOI] [PubMed] [Google Scholar]
- 2.Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes-many genes, common mechanisms. Neuromuscul Disord 2010;20:429-37 [DOI] [PubMed] [Google Scholar]
- 3.Spinazzola A. Mitochondrial DNA mutations and depletion in pediatric medicine. Semin Fetal Neonatal Med 2011;16:190-6 [DOI] [PubMed] [Google Scholar]
- 4.Romei C, Elisei R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front Endocrinol (Lausanne) 2012;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulton J, Hirano M, Spinazzola A, et al. Collated mutations in mitochondrial DNA (mtDNA) depletion syndrome (excluding the mitochondrial gamma polymerase, POLG1). Biochim Biophys Acta 2009;1792:1109-12 [DOI] [PubMed] [Google Scholar]
- 6.Shaibani A, Shchelochkov OA, Zhang S, et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol 2009;66:1028-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol 2012;47:64-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinazzola A, Invernizzi F, Carrara F, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis 2009;32:143-58 [DOI] [PubMed] [Google Scholar]
- 9.Huang CC, Hsu CH. Mitochondrial disease and mitochondrial DNA depletion syndromes. Acta Neurol Taiwan 2009;18:287-95 [PubMed] [Google Scholar]
- 10.Lügering A, Schmidt M, Lügering N, et al. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology 2001;121:1145-57 [DOI] [PubMed] [Google Scholar]
- 11.Giordano C, Sebastiani M, De Giorgio R, et al. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalo-myopathy is caused by mitochondrial DNA depletion. Am J Pathol 2008;173:1120-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Limongelli A, Vila MR, et al. Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol Genet Metab 2005;84:75-82 [DOI] [PubMed] [Google Scholar]
- 13.Lönnqvist T, Paetau A, Valanne L, Pihko H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain 2009;132:1553-62 [DOI] [PubMed] [Google Scholar]
- 14.Cho S, Lee YM, Choi YS, et al. Mitochondrial DNA polymorphisms are associated with susceptibility to endometriosis. DNA Cell Biol 2012;31:317-22 [DOI] [PubMed] [Google Scholar]
- 15.Ding X, Wang L, Ren Y, Zheng W. Detection of mitochondrial biomarkers in eutopic endometria of endometriosis using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry. Fertil Steril 2010;94:2528-30 [DOI] [PubMed] [Google Scholar]
- 16.Vanderborght M, Nassogne MC, Hermans D, et al. Intractable ulcerative colitis of infancy in a child with mitochondrial respiratory chain disorder. J Pediatr Gastroenterol Nutr 2004;38:355-7 [DOI] [PubMed] [Google Scholar]